
Abstract
Anticancer drug discovery has yielded unprecedented progress in recent decades, resulting in the approval of innovative treatment options for patients and the successful implementation of personalized medicine in clinical practice. This remarkable progress has also reshaped the research scope of pharmacological research. This article, as a tribute to cancer research at Shanghai Institute of Materia Medica in celebration of the institute’s 90th birthday, provides an overview of the conceptual revolution occurring in anticancer therapy, and summarizes our recent progress in the development of molecularly targeted therapeutics and exploration of new strategies in personalized medicine. With this review, we hope to provide a glimpse into how antitumor pharmacological researchers have embraced the new era of personalized medicine research and to propose a future path for anticancer drug discovery and pharmacological research.
Similar content being viewed by others
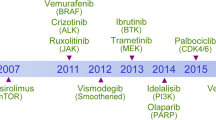

Anticancer drug discovery has experienced unprecedented progress in the past decades, especially in the development of molecularly targeted anticancer therapeutics that have profoundly altered the conceptual overview of cancer treatment and promoted a paradigm shift from a population-based “one-size-fits-all” treatment approach to a personalized medicine [1]. These advancements have significantly benefited cancer patients and also reshaped the research scope of antitumor pharmacology. Apart from conventional studies addressing the therapeutic potential and mechanism of action of new anticancer agents, the pharmacological research in the new era has focused on identifying biomarkers for tailoring drug treatment in preclinical development and clinical practice and understanding molecular mechanisms that confer synergy between different anticancer modalities, thereby providing useful insights for clinical decision-making in personalized medicine.
Antitumor pharmacological research at the Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences, was initiated in the 1950s when anticancer drug discovery was new in China. Cancer research at SIMM has progressed remarkably in recent decades due to the collective efforts of several generations of scientists. Here, we summarize the progress made by the antitumor pharmacological group over the past decade to provide a glimpse into how pharmacological researchers have embraced the excitement of the new era and evolved to address challenges in cancer therapy.
Targeted anticancer drug discovery: exploring oncogene-addiction for therapeutic opportunities
Since the 1990s, anticancer drug discovery worldwide has experienced a conceptual evolution from conventional chemotherapy to molecularly targeted therapy. Protein kinases were among the first exploited drug targets and had rapidly dominated the field due to the remarkable clinical success obtained with the leading compounds. Despite a debate regarding the preference for highly selective or multitargeted kinase inhibitors [2, 3], the focus of kinase inhibitor development has gradually developed into selectively targeting clinically prevalent oncogenic drivers. The rationale behind this approach is a well-established paradigm known as oncogene addiction whereby a single oncogenic alteration, despite a diverse array of coinciding mutations alongside, dominates tumor growth and provides a therapeutic vulnerability (Fig. 1). This notion has paved the way for the genomics-guided development of kinase inhibitors and accelerated their clinical translation for treating molecularly-defined subsets of cancer patients [4]. To date, over 70 kinase inhibitors targeting 21 kinase families have been approved by the US Food and Drug Administration (FDA), and kinase inhibitors are considered as the most successful class of oncology drugs developed over the past two decades. Notably, most approved kinase inhibitors, including next-generation drugs targeting both canonical activating and common resistance mutations, are associated with the concept of oncogene addiction [5,6,7].

For tumors harbor oncogenic alterations, genomics-guided targeted therapy could provide an effective approach. For non-oncogene addicted tumors, targeted therapy often generates new therapeutic vulnerabilities that could support synthetic lethality or combination therapy.
Recently, the rapid growth of large-scale sequencing of the cancer genome, as sponsored by various projects, such as The Cancer Genome Atlas (TCGA), has illustrated the genomic landscape of the most common cancer types, further expanding our knowledge of oncogene addiction. A good example is the genomic sequencing of acute myeloid leukemia (AML), which identified a recurrent mutation in isocitrate dehydrogenase (IDH) and ignited the enthusiasm for pursuing new treatment options. It was later discovered that this mutation confers a neomorphic enzyme activity and creates an addiction to oncometabolite 2-hydroxyglutarate. Together, these findings led to the ultimate approval of IDH1/2 inhibitors for treating IDH1/2-mutant AML [8,9,10,11]. On the other hand, some of the most prevalent oncogenic drivers that used to be intractable, such as KRAS, became targetable through the use of covalent inhibitors of KRAS (G12C) mutant [12,13,14]. It is predictable that there are still substantial unexplored opportunities for interfering with oncogene addiction in cancer therapy.
Anticancer drug development at SIMM has followed a similar path. The efforts initiated from different classes of kinase inhibitors [15], among them a multi-targeted angiogenic kinase inhibitor lucitanib (AL3810/E-3810) was the first to proceed to clinical testing and showed the encouraging response in advanced breast and nasopharyngeal carcinoma patients [16,17,18]. Growing insight into oncogene addiction has significantly accelerated drug development, bringing a pipeline of kinase inhibitors from the bench to the bedside. SCC244, a highly selective c-Met inhibitor, shows robust efficacy and encouraging intracranial antitumor activity in non-small cell lung cancer (NSCLC) patients harboring MET exon 14-skipping (METex14) mutations [19, 20]. A new drug application (NDA) has been filed for this treatment in China. CYH33, a selective PI3Kα inhibitor, has demonstrated an encouraging preliminary antitumor activity and a manageable safety in patients with advanced solid tumors harboring PIK3CA mutations [21, 22]. Meanwhile, next-generation kinase inhibitors designed to overcome acquired resistance have also been actively explored. For example, the third-generation epidermal growth factor receptor (EGFR) inhibitor limertinib (ASK120067) exhibits promising efficacy for treating EGFR T790M-mutated NSCLC patients and an NDA for this drug has been filed in China [23, 24]. A next-generation ROS1/ALK inhibitor, SAF-189s, is active against both oncogenic ROS1 fusions and mutations related to crizotinib resistance. SAF-189s is currently being evaluated in clinical trials for NSCLC patients with ROS1 alterations who experienced relapse after crizotinib treatment (NCT04237805) [25]. Recently, we have been seeking new therapeutic opportunities in emerging areas, such as cancer epigenetics and metabolism. HH2853, a dual inhibitor of EZH1/2 with potent antitumor activities, is currently in phase I clinical development [26]. HH2301, a selective inhibitor of mutant IDH1, shows promising efficacy for treating IDH1-mutant cholangiocarcinoma in preclinical models and has received permission for clinical testing in China and the United States [27]. Along with their therapeutic potential, these drug candidates have also transformed our research and allowed us to address the challenges encountered in clinical treatment.
Fine-tuning the personalized medicine with a dual-biomarker strategy
The enormous success in exploring oncogene addiction has led to a paradigm shift toward personalized medicine that tailors the anticancer drugs to tumors characterized by oncogenic alterations. These genetic alterations, also known as “predictive/diagnostic biomarkers”, guide the effective selection of subgroup patients and maximize the chance of drug responses, exemplified most clearly in NSCLC [28, 29]. Nevertheless, even within tumors bearing predictive biomarkers, the treatment is frequently challenged by de novo resistance. The development of acquired drug resistance is also inevitable and is the most frequent cause of clinical treatment failure. The reason behind is that diminishing oncogenic signaling often causes a context-dependent feedback activation of compensatory pathways, enabling cancer cells to escape the original addiction.
To overcome this challenge, we propose a solution to identify a common substrate or effector of oncogenic pathways that may allow monitoring of the biological outcome of oncogene-addicted tumors during treatment [1]. Taking c-Met inhibitor as a representative, we identified the transcription factor c-Myc as a key downstream effector of c-Met signaling that mediates the biological outcome of c-Met inhibition in cancer cells harboring MET alterations. In both c-Met addicted cancer and its derived resistance due to the compensatory activation of different upstream kinases, c-Myc converges the upstream kinase signaling to drive cell growth [30]. This unique position allows c-Myc to serve as a “response biomarker” to assist the decision-making during c-Met-targeted therapy. Specifically, in MET-altered tumors, measuring c-Myc levels could identify the de novo resistance, monitor the emergence of acquired resistance during the long-course treatment, and assess the effectiveness of alternative therapies. Of interest, the application of this strategy to fibroblast growth factor receptor (FGFR) inhibitors also identified c-Myc as a response indicator, suggesting it may sit at the convergence of multiple kinase signaling pathways [31]. Moreover, the molecular linkage between FGFR and hexokinase 2 (HK2) also inspired the adoption of 18F-fluorodeoxyglucose (18F-FDG) for assessing the response to FGFR-targeted therapies [32]. Based on these findings, we have proposed a dual-biomarker strategy that emphasizes an additional “response biomarker” following the tumor stratification using predictive biomarkers.
The response of cancer cells to targeted therapy is shaped by their close interaction with the tumor microenvironment (TME). In addition to intrinsic tumor alterations, the integration of microenvironmental status could provide a more powerful biomarker. For example, in IDH1-mutant tumors, the vascular endothelial expression of SLC1A1, a Na+-dependent glutamate transporter, is required for the therapeutic benefit of mIDH1 inhibitors in solid tumors, due to a mechanism of 2-hydroxyglutarate-mediated crosstalk between IDH1-mutant tumor cells and vascular endothelial cells. While IDH1-mutant cholangiocarcinoma does not necessarily respond to IDH1 inhibitors, integration of endothelial SLC1A1 expression may substantially increase the chance of tumor responses [27].
All these findings suggest that a dual-biomarker approach that integrates both tumor alterations and microenvironment status will provide a possible solution to fine-tune the personalized medicine to further improve the clinical benefit of targeted therapies (Fig. 2).

Biomarkers integrating tumor alterations and microenvironment status could be categorized into predictive and response biomarkers. Predictive biomarkers are used to enrich responsive patients prior to the treatment. Response biomarkers are measurable molecular alterations that are closely associated with the drug response during the treatment and therefore could be used to differentiate de novo resistance, indicate the development of acquired resistance and facilitate the selection of alternative therapies including combination therapy.
Rationalizing combination therapy to defeat non-oncogene addiction
In addition to oncogenic drivers, the tumorigenic state depends on the activities of a wide variety of genes and pathways, many of which are not inherently oncogenic or undergoing oncogenic mutations in tumors, termed as non-oncogene addiction [33, 34]. Non-oncogene addiction can be tumor intrinsic and tumor extrinsic, where tumor-intrinsic targets support the oncogenic state of the tumor cell in a cell-autonomous manner, whereas tumor-extrinsic targets function in the tumor microenvironment to provide support for the tumor. Many actively exploited anticancer targets belong to this class, including those involved in pathways in maintaining genomic stability, metabolic hemostasis and cell death regulation. As it often lacks a clear stratification strategy for selecting responsive tumor subset for non-oncogene addiction targets, the outcome of therapeutic interference is unpredictable. Of note, it is increasingly noted that targeting non-oncogene addiction pathway could induce a stress state and yield synthetic lethality with other targets, providing a rationale for combination therapy (Fig. 1).
To date, various drug combinations have been shown to be beneficial in experimental models, yet very few combinations have been approved for clinical use. To understand the reason behind this, we take epigenetic therapy as a representative as this class of drugs is increasingly appreciated for its potential in synergy with other anticancer therapies in solid tumors [35]. In our findings using patient-derived preclinical models encompassing a broad spectrum of solid tumors, epigenetic therapies synergize with different targeted therapies, yet these combinations are only effective in a subset of tumors, likely reflecting the situation in the clinic. For example, the combination of histone deacetylase (HDAC) and JAK signaling inhibitors is effective against triple-negative breast cancer, due to HDAC inhibition-induced feedback activation of leukemia inhibitory factor receptor, yet it is only effective in a subset of cancer models [36]. Further studies of enhancer of zeste homolog 2 (EZH2) inhibitors allowed us to develop a personalized combination strategy. In tumors across different tissue types, the response to EZH2 inhibitors is determined by the expression of mixed-lineage leukemia 1 (MLL1), a methyltransferase that facilitates the conversion of histone H3 lysine 27 (H3K27) methylation to acetylation and renders tumors resistant to EZH2 inhibition, supporting the rationale of concurrent intervention of H3K27 methylation and acetylation. However, in a small subset of tumors, this regimen could cause feedback activation of the ERK pathway and further jeopardize the tumor response. A triple-combination of EZH2, bromodomain containing 4 (BRD4) and ERK inhibition displays robust efficacy with tolerable toxicity in this subset of tumors [37]. Based on these findings, we proposed a stepwise combination therapy guided by specific molecular alterations that could potentially be applicable to other epigenetic targets (Fig. 3).

For non-oncogene addicted tumors with EZH2 high expression, EZH2 inhibitors are only effective in a small proportion of tumors with low MLL1 expression. The combination of EZH2 and BRD4 inhibitors is recommended for those with high levels of MLL1 expression, among which a subset of tumors will experience a feedback activation of ERK signaling and require a triple-combination of EZH2, BRD4 and ERK inhibitors.
Rational drug combinations provide a solution for non-oncogene addicted tumors. With the increasing number of targeted drugs available, more combinations associated with diverse molecular mechanisms will arise and benefit a broad spectrum of cancer patients. For example, we discovered that a PI3Kα inhibitor synergizes with the angiogenesis inhibitor AL3810 in hepatocellular carcinoma [38], with an EZH2 inhibitor in esophageal cell squamous carcinoma [39], with a CDK4/6 inhibitor in KRAS-mutant NSCLC [40], and with the fatty acid metabolism inhibitor in breast cancer via promoting the antitumor immunity [41].
Moreover, rational drug combinations also provide a venue for emerging new targeted therapies. For example, targeting Ack1, a non-receptor tyrosine kinase, effectively overrides the acquired resistance of the third-generation EGFR inhibitors [23]. Inhibitors of phosphoglycerate mutase 1, a relatively new target in cancer metabolism, synergize with poly(ADP-ribose) polymerase (PARP) in triple-negative breast cancer [42]. The combination of DOT1L and SHP2 inhibitors, both of which are still under exploration for possible indications, is an effective treatment specific for a subset of KRAS mutant cancers [43]. Whether these combinations discovered in preclinical models could eventually benefit the patients requires clinical testing. However, it is foreseeable that in complex clinical settings, combination therapies guided by proper molecular alterations are likely a solution. Rational combinations considering key factors, including patient selection, biomarker evaluation, drug scheduling and response assessment, are future directions for personalized combination therapy.
Perspectives
We are experiencing a new era of personalized medicine in cancer therapy, which will likely continue to dominate the field of cancer research in the coming decade. This new era imposes challenges and meanwhile provides an exciting stage for antitumor pharmacological research to flourish. In collaboration with multidisciplinary scientists, pharmacological research aims to bring innovative drugs to the clinic and to uncover new approaches of personalized treatment.
Leveraging innovative technologies to empower next-generation targeted therapy
The past two decades have witnessed the immense progress in anticancer drug discovery. The low-hanging fruit has been mostly harvested. The current drug discovery landscape highlights the need to explore new avenues for therapeutic opportunities and gaining the ability to tackle challenging targets. In the years to come, the enduring progress in successive generations of therapies will rely on the successful implementation of new technologies. The integration of genome-wide functional screens, deep sequencing, large-scale proteomics, advanced imaging approaches and artificial intelligence in data mining will offer extensive ground for the discovery of new targets. In terms of rational drug design, the paradigm of absolute selectivity is probably not critical. Selective modulation of desired multitargets exhibiting compensatory functions, such as dual- or trifunctional drugs, will have a better chance to secure a therapeutic benefit. Moreover, with the emergence of new-generation interventions, such as targeted degraders using proteolysis-targeting chimeric molecules (PROTACs) and molecular glues, the development and refinement of efficient drug screening and profiling technologies to meet the needs of these inhibitors will also be important. Furthermore, as is already deeply appreciated, preclinical disease models better recapitulating tumor heterogeneity and clinical therapeutic response are needed to comprehensively and precisely understand the molecular mechanisms of drug responses in different cancer contexts, so as to provide useful information for precisely tailoring the new therapeutic interventions.
Exploring new strategies for rational combination with immunotherapy
The approval of immunotherapy, in particular immune checkpoint blockades, has changed the landscape of cancer therapy. However, due to the limited overall response rates, combinations with different types of therapeutic modalities have been intensively explored both preclinically and clinically [44]. For this purpose, revisiting the immune modulatory effect of different classes of clinically used drugs could be an efficient approach, as most of these drugs were initially developed for targeting cancer cells and their immune-modulatory effects have been largely neglected, in particularly considering that they were mostly evaluated in immune-deficient preclinical models. For example, conventional chemotherapeutics have been developed based on their ability to preferentially kill malignant cells, yet the clinical activity of various chemotherapies is now known to involve the stimulation of anticancer immunity either by initiating the release of immunostimulatory molecules from dying cancer cells, by mediating off-target effects on immune cell populations or by altering whole-body physiology [45, 46]. Likewise, accumulating evidence indicates that kinase inhibitors enhance T cell infiltration or cause an immunogenic cell death phenotype [5]. Therefore, understanding the precise immunological mechanisms that underlie the efficacy of approved therapies has the potential to identify superior biomarkers of response and provide a rationale for combination therapy.
References
Huang M, Shen A, Ding J, Geng M. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci. 2014;35:41–50.
de Jonge MJ, Verweij J. Multiple targeted tyrosine kinase inhibition in the clinic: all for one or one for all? Eur J Cancer. 2006;42:1351–6.
Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol. 2006;33:407–20.
Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–4.
Cohen P, Cross D, Janne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. 2021;20:551–69.
Paliouras S, Pearson A, Barkalow F. The most successful oncology drug portfolios of the past decade. Nat Rev Drug Discov. 2021;20:811–2.
Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO Mol Med. 2011;3:623–36.
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–98.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66.
Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207–17.
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–23.
Herbst RS, Schlessinger J. Small molecule combats cancer-causing KRAS protein at last. Nature. 2019;575:294–5.
Miao ZH, Feng JM, Ding J. Newly discovered angiogenesis inhibitors and their mechanisms of action. Acta Pharmacol Sin. 2012;33:1103–11.
Dienstmann R, Andre F, Soria JC. Significant antitumor activity of E-3810, a novel FGFR and VEGFR inhibitor, in patients with FGFR1 amplified breast cancer. 2012 ESMO Congress 2012; Abstract 3190: Presented October 1, 2012.
Zhou Y, Chen Y, Tong L, Xie H, Wen W, Zhang J, et al. AL3810, a multi-tyrosine kinase inhibitor, exhibits potent anti-angiogenic and anti-tumour activity via targeting VEGFR, FGFR and PDGFR. J Cell Mol Med. 2012;16:2321–30.
Zhang Y, Luo F, Ma YX, Liu QW, Yang YP, Fang WF, et al. A phase Ib study of lucitanib (AL3810) in a cohort of patients with recurrent and metastatic nasopharyngeal carcinoma. Oncologist. 2022;27:e453–62.
Ai J, Chen Y, Peng X, Ji Y, Xi Y, Shen Y, et al. Preclinical evaluation of SCC244 (glumetinib), a novel, potent, and highly selective inhibitor of c-Met in MET-dependent cancer models. Mol Cancer Ther. 2018;17:751–62.
Lu S, Yu Y, Zhou J, Goto K, Li X, Sakakibara-Konishi J, et al. Abstract CT034: phase II study of SCC244 in NSCLC patients harboring MET exon 14 skipping (METex14) mutations (GLORY study). Cancer Res. 2022;82(Suppl 12):CT034.
Xiang HY, Wang X, Chen YH, Zhang X, Tan C, Wang Y, et al. Identification of methyl (5-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinopyrrolo[2,1-f][1,2,4]triazin-2-yl)-4-(trifluoromethyl)pyridin-2-yl)carbamate (CYH33) as an orally bioavailable, highly potent, PI3K alpha inhibitor for the treatment of advanced solid tumors. Eur J Med Chem. 2021;209:112913.
Wei XL, Xu RH, Zhao H, Zhang Y, Zou BY, Wang F, et al. A first-in-human phase I study of CYH33, a phosphatidylinositol 3-kinase (PI3K) α selective inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2020;38(15_Supplement):e15645.
Zhang T, Qu R, Chan S, Lai M, Tong L, Feng F, et al. Discovery of a novel third-generation EGFR inhibitor and identification of a potential combination strategy to overcome resistance. Mol Cancer. 2020;19:90.
Shi Y, Li B, Wu L, Pan Y, Pan Z, Liu Y, et al. Efficacy and safety of limertinib (ASK120067) in patients with locally advanced or metastatic EGFR Thr790Met-mutated NSCLC: a multicenter, single-arm, phase 2b study. J Thorac Oncol. 2022;17:1205–15.
Xia ZJ, Ji YC, Sun DQ, Peng X, Gao YL, Fang YF, et al. SAF-189s, a potent new-generation ROS1 inhibitor, is active against crizotinib-resistant ROS1 mutant-driven tumors. Acta Pharmacol Sin. 2021;42:998–1004.
Chen XX, Shen QQ, Zhao Z, Fang YF, Yang JY, Gao YL, et al. Abstract 5436: HH2853 is a selective small molecular dual inhibitor of EZH1/2 with potent anti-tumor activities. Cancer Res. 2022;82(12_Supplement):5436.
Wang X, Chen Z, Xu J, Tang S, An N, Jiang L, et al. SLC1A1-mediated cellular and mitochondrial influx of R-2-hydroxyglutarate in vascular endothelial cells promotes tumor angiogenesis in IDH1-mutant solid tumors. Cell Res. 2022;32:638–58.
Beckman RA, Clark J, Chen C. Integrating predictive biomarkers and classifiers into oncology clinical development programmes. Nat Rev Drug Discov. 2011;10:735–48.
Kelloff GJ, Sigman CC. Cancer biomarkers: selecting the right drug for the right patient. Nat Rev Drug Discov. 2012;11:201–14.
Shen A, Wang L, Huang M, Sun J, Chen Y, Shen YY, et al. c-Myc alterations confer therapeutic response and acquired resistance to c-Met inhibitors in MET-addicted cancers. Cancer Res. 2015;75:4548–59.
Liu H, Ai J, Shen A, Chen Y, Wang X, Peng X, et al. c-Myc alteration determines the therapeutic response to FGFR inhibitors. Clin Cancer Res. 2017;23:974–84.
Jiang Y, Zeng Q, Jiang Q, Peng X, Gao J, Wan H, et al. (18)F-FDG PET as an imaging biomarker for the response to FGFR-targeted therapy of cancer cells via FGFR-initiated mTOR/HK2 axis. Theranostics. 2022;12:6395–408.
Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37.
Francies HE, McDermott U, Garnett MJ. Genomics-guided pre-clinical development of cancer therapies. Nat Cancer. 2020;1:482–92.
Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol. 2020;17:91–107.
Zeng H, Qu J, Jin N, Xu J, Lin C, Chen Y, et al. Feedback activation of leukemia inhibitory factor receptor limits response to histone deacetylase inhibitors in breast cancer. Cancer Cell. 2016;30:459–73.
Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, et al. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 2018;175:186–99.e19.
Xie Q, Chi S, Fang Y, Sun Y, Meng L, Ding J, et al. PI3Kalpha inhibitor impairs AKT phosphorylation and synergizes with novel angiogenesis inhibitor AL3810 in human hepatocellular carcinoma. Signal Transduct Target Ther. 2021;6:130.
Xing H, Gao M, Wang Y, Zhang X, Shi J, Wang X, et al. Genome-wide gain-of-function screening identifies EZH2 mediating resistance to PI3Kalpha inhibitors in oesophageal squamous cell carcinoma. Clin Transl Med. 2022;12:e835.
Wang Y, Li X, Liu X, Chen Y, Yang C, Tan C, et al. Simultaneous inhibition of PI3Kalpha and CDK4/6 synergistically suppresses KRAS-mutated non-small cell lung cancer. Cancer Biol Med. 2019;16:66–83.
Sun P, Zhang X, Wang RJ, Ma QY, Xu L, Wang Y, et al. PI3Kalpha inhibitor CYH33 triggers antitumor immunity in murine breast cancer by activating CD8+ T cells and promoting fatty acid metabolism. J Immunother Cancer. 2021;9:e003093.
Qu J, Sun W, Zhong J, Lv H, Zhu M, Xu J, et al. Phosphoglycerate mutase 1 regulates dNTP pool and promotes homologous recombination repair in cancer cells. J Cell Biol. 2017;216:409–24.
Liu Z, Liu Y, Qian L, Jiang S, Gai X, Ye S, et al. A proteomic and phosphoproteomic landscape of KRAS mutant cancers identifies combination therapies. Mol Cell. 2021;81:4076–90.e8.
de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38:326–33.
Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725–41.
Lian Q, Xu J, Yan S, Huang M, Ding H, Sun X, et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res. 2017;27:784–800.
Acknowledgements
The authors sincerely thank all the individuals who have contributed to the work that has been reviewed in this article, including all the members who were either directly or indirectly involved in the Division of Antitumor Pharmacology at SIMM, as well as all our collaborators. This work is supported by the National Natural Science Foundation of China for Innovation Research Group (No. 81821005).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
M.H. was a consultant of Haihe Biopharma and J.D. is the Chairman of Board of Haihe Biopharma.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, M., Geng, My. & Ding, J. Antitumor pharmacological research in the era of personalized medicine. Acta Pharmacol Sin 43, 3015–3020 (2022). https://doi.org/10.1038/s41401-022-01023-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-01023-0
Keywords
This article is cited by
-
APS celebrates the 90th anniversary of SIMM
Acta Pharmacologica Sinica (2022)
