
Abstract
Liposomes decorated with tumour-targeting cell-penetrating peptides can enhance specific drug delivery at the tumour site. The TR peptide, c(RGDfK)-AGYLLGHINLHHLAHL(Aib)HHIL, is pH-sensitive and actively targets tumour cells that overexpress integrin receptor αvβ3, such as B16F10 melanoma cells. Liposomes can be modified with the TR peptide by two different methods: utilization of the cysteine residue on TR to link DSPE-PEG2000-Mal contained in the liposome formula (LIPTR) or decoration of TR with a C18 stearyl chain (C18-TR) for direct insertion into the liposomal phospholipid bilayer through electrostatic and hydrophobic interactions (LIPC18-TR). We found that both TR and C18-TR effectively reversed the surface charge of the liposomes when the systems encountered the low pH of the tumour microenvironment, but LIPC18-TR exhibited a greater increase in the charge, which led to higher cellular uptake efficiency. Correspondingly, the IC50 values of PTX-LIPTR and PTX-LIPC18-TR in B16F10 cells in vitro were 2.1-fold and 2.5-fold lower than that of the unmodified PTX-loaded liposomes (PTX-LIP), respectively, in an acidic microenvironment (pH 6.3). In B16F10 tumour-bearing mice, intravenous administration of PTX-LIPTR and PTX-LIPC18-TR (8 mg/kg PTX every other day for a total of 4 injections) caused tumour reduction ratios of 39.4% and 56.1%, respectively, compared to 20.8% after PTX-LIP administration. Thus, we demonstrated that TR peptide modification could improve the antitumour efficiency of liposomal delivery systems, with C18-TR presenting significantly better results. After investigating different modification methods, our data show that selecting an adequate method is vital even when the same molecule is used for decoration.
Similar content being viewed by others
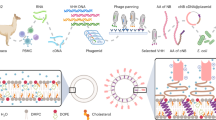


Introduction
Tumours consume more oxygen and other nutrients than the surrounding normal tissues to meet their needs for excessive overgrowth, which ultimately results in the oversecretion of lactic acid and a highly acidic physical microenvironment [1,2,3]. The acidic microenvironment of tumour sites is commonly exploited in cancer therapy to achieve specific drug delivery using pH-responsive strategies [4]. However, an active targeting strategy using particular ligands is another attractive approach to enhance the site-specific delivery of therapeutic drugs [5, 6].
Chemotherapy and surgery are usually used in clinical cancer treatments [7,8,9]. Paclitaxel (PTX) is a widely utilized anticancer chemotherapy drug that has poor solubility in the aqueous phase and lacks tumour targeting ability [10,11,12,13]. Liposomes are a common drug delivery system that can enhance the therapeutic effect of PTX and improve its solubility [14, 15]. To further strengthen the tumour targeting capability of liposomes, decoration with functional cell-penetrating peptides (CPPs) is a useful method [16, 17]. CPPs are simple to synthesize, functionalize and characterize, possess cancer cell penetration abilities without undesirable cytotoxicity, and can be attached to the surface of liposomes in various ways. Because CPPs lack the ability to attach to the membrane to be involved in a linking reaction, additional membrane-binding molecules are necessary [18,19,20,21,22].
The integrin receptor αvβ3 is highly and selectively expressed on the surface membrane of many tumour cells, such as B16F10 melanoma cells [23, 24]. Based on the protonation and deprotonation process of histidine under different pH conditions, a research group fabricated a histidine-rich pH-sensitive peptide TH (AGYLLGHINLHHLAHL(Aib)HHIL-NH2) and connected it to the cyclic integrin receptor αvβ3 targeting peptide [c(RGDfK)] [25]. The whole peptide, named TR, can facilitate tumour targeting and charge conversion for liposomes; hence, TR-modified liposomes could selectively deliver loaded drugs into tumour cells overexpressing integrin receptor αvβ3 and possess pH-responsive cell penetration ability in an acidic microenvironment [25, 26].
There are two totally different methods to effectively anchor the TR peptide to the outer surface of liposomes: linking the cysteine residue on TR [c(RGDfK)-AGYLLGHINLHHLAHL(Aib)HHIL-Cys] to DSPE-PEG2000-Mal contained in the liposome formula (LIPTR), where DSPE-PEG2000-TR is formed on the surface of liposomes [25, 27], or decorating the lysine residue on TR with a C18 stearyl chain [named C18-TR, c(RGDfK)-AGYLLGHINLHHLAHL(Aib)HHIL-Lys-C18] for direct insertion into the liposomal phospholipid bilayer through electrostatic and hydrophobic interactions (LIPC18-TR) [28, 29]. The added C18 chain does not affect peptide function due to its small size.
In this research, we encapsulated PTX into liposomes and anchored the TR peptide onto liposomes using the above methods. We conducted an in vitro cell uptake study and a cytotoxicity study under different pH conditions and an in vivo anticancer assay to investigate the differences between the two methods to modify liposomes with this tumour-targeting peptide to treat melanoma (Scheme 1).

The surface charges of these formulations changed from negative to positive in the acidic tumour microenvironment, where they selectively targeted integrin receptor αvβ3-overexpressing melanoma cells.
Materials and methods
Materials and animals
The C18-TR [c(RGDfK)-AGYLLGHINLHHLAHL(Aib)HHIL-Lys-C18] and TR [c(RGDfK)-AGYLLGHINLHHLAHL(Aib)HHIL-Cys] peptides were synthesized according to standard solid-phase peptide synthesis by ZheJiang Ontores Biotechnologies Co. Ltd. (Hangzhou, China). Paclitaxel was purchased from Ji’anTe Technology Co. Ltd. (Chengdu, China). Soybean lecithin (S100) was obtained from Lipoid Co. Ltd. (Ludwigshafen, Germany). Cholesterol was purchased from A.V.T. Pharmaceutical Co. Ltd. (Shanghai, China). DSPE-PEG2000 and DSPE-PEG2000-Mal were purchased from Laysan Bio, Inc. (Laysan, California, USA). 1,10-Dioctadecyl-3,3,30,30-tetramethylindodicarbocyanine 4-chlorobenzene sulfonate salt (DiD) was obtained from Biotium (Hayward, California, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Biosharp (Shanghai, China). Other chemicals were obtained from commercial sources.
Female C57BL/6 mice were purchased from Chengdu Dashuo Biological Institute (Chengdu, China).
Preparation and characterization of the liposomes
Liposomes decorated with peptides by different methods were prepared using thin-film hydration [30, 31]. The liposomes were composed of S100, cholesterol, DSPE-PEG2000 and DSPE-PEG2000-Mal (3:1:0.33:0.66, w:w:w:w). All lipid materials were dissolved in chloroform, and then the organic solvent was removed through rotary evaporation at 40 °C to form a uniform lipid film. The lipid film was hydrated in HEPES buffer (10 mM, pH 7.4) at 40 °C, dispersed with a probe-type ultrasonicator (JY92-II sonicator, Scientz, Ningbo, China) in an ice bath and further filtered through a 0.22 μm filter to obtain blank liposomes (LIP). PTX-loaded liposomes (PTX-LIP) were prepared with PTX added to the lipid organic solution (PTX: lipids = 1:13, w:w) with the other membrane materials prior to the evaporation step, and then the organic solvent was removed to form the film, hydrated with HEPES buffer and ultrasonicated. Subsequently, to prepare peptide-decorated liposomes through the C18 insertion method (PTX-LIPC18-TR), the C18-TR peptide was incubated with the precooled PTX-LIP at 4 °C for 0.5 h, and C18-TR was inserted into the liposomal phospholipid bilayer [28]. In addition, to prepare peptide-decorated liposomes through the coupling method (PTX-LIPTR), the TR peptide was incubated with PTX-LIP at 37 °C for 4 h, and DSPE-PEG2000-Mal was conjugated to the cysteine residue on the TR peptide [32].
PTX-LIPTR and PTX-LIPC18-TR were obtained by washing with HEPES buffer and using a Sephadex G-75 column to remove free PTX and peptide. In addition, DiD-loaded liposomes (DiD-LIPTR and DiD-LIPC18-TR) were prepared by adding an appropriate amount of DiD to the solution before the evaporation step. Transmission electron microscopy (TEM, H-600, Hitachi, Japan) was used to observe the morphology of PTX-LIPTR and PTX-LIPC18-TR through negative staining with 2% phosphotungstic acid. The particle sizes, size distribution and zeta potentials of PTX-LIPTR and PTX-LIPC18-TR at pH 7.4 and pH 6.3 were measured by a Malvern Zetasizer Nano ZS90 instrument (Malvern Instruments Ltd., U.K.) at 25 °C.
To confirm the successful coupling of TR and DSPE-PEG2000-Mal and the optimal incubation time, a sulfhydryl detection kit (KeyGEN BioTECH, Jiangsu, China) was used, which detects the uncoupled sulfhydryl of the cysteine residue of TR. The absorbance was measured at 412 nm by a microplate reader (Varioskan Flash, Thermo Fisher Scientific, USA). In addition, to confirm the successful insertion of C18-TR, a BCA detection kit (Thermo Fisher Scientific, USA) was used. The peptide conjugation rate was surveyed by the following equation: (PL/PA) × 100%, where PL is the amount of liposomes ultimately decorated with TR or C18-TR, and PA is the total amount of TR or C18-TR added to the liposomes initially. The amount of PTX was measured by HPLC (Agilent 1260, USA) at a wavelength of 227 nm [13]. The entrapment efficiency (EE) was determined by the following equation: WL/WA × 100%, where WL is the amount of drug ultimately loaded into the liposomes collected from the Sephadex G-75 column, and WA is the total amount of drug added to the organic solution initially. The drug loading rate (DL) was calculated by the following equation: WL/(WL + WM) × 100%, where WM is the total amount of lipid material.
In vitro drug release study
The in vitro release of PTX from the different liposomal formulations was determined using a dialysis method under sink conditions [33]. An aliquot of PTX-loaded liposomes (PTX-LIPTR and PTX-LIPC18-TR, 1 mL) was placed into a dialysis bag (MWCO = 8 kDa), which was immersed in release media [PBS containing 0.4% (v/v) Tween 80 (pH 7.4 or pH 6.3)] and incubated at 37 °C with gentle oscillation for 72 h. At predetermined time points (1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h and 72 h), 1 mL of release media was sampled and replaced with an equal volume of fresh release media. Then, each sample was diluted with acetonitrile, and the concentration of PTX was determined by HPLC.
In vitro stability study in serum
Variations in the particle sizes and turbidity of the liposomes were measured to demonstrate the serum stability of different liposomes in the presence of foetal bovine serum (FBS). In brief, PTX-LIPTR and PTX-LIPC18-TR were mixed with an equal volume of FBS at 37 °C with gentle shaking. At predetermined time points, 200 μL of each sample was transferred to a 96-well plate to measure the transmittance (T) at 750 nm with a microplate reader, and absorbance (A) was calculated as follows: A = −LogT. Another 200 μl of each of the PTX-LIPTR and PTX-LIPC18-TR samples was diluted to 1 mL to measure the variation in particle size [13, 34].
Cell line and cell culture
B16F10 cells (a mouse melanoma cell line) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, KeyGEN BioTECH, China) with 10% FBS and 5% streptomycin/penicillin at 37 °C in a humidified 5% CO2 atmosphere. Cells utilized for the assays at pH 6.3 were preadapted to pH 6.3 culture media.
Cellular uptake study
To compare the differences in LIPTR and LIPC18-TR cellular uptake, B16F10 cells were seeded in a 24-well plate and incubated overnight. DiD-LIPTR and DiD-LIPC18-TR in media with different pH values (pH 6.3 and pH 7.4) were added to the plate at a concentration of 400 ng/ml DiD. After incubating at 37 °C for another 2 h, the cells were washed with precooled PBS, trypsinized, and resuspended in PBS. The fluorescence intensity was determined using flow cytometry (Cytomics™ FC 500, Beckman Coulter, USA) with an excitation wavelength of 633 nm and an emission wavelength of 675 nm. To survey the function of the TR peptide in the cellular uptake process, B16F10 cells were seeded in a 6-well plate overnight and incubated with free TR peptide or normal media (as a control group) for 1 h at 37 °C. After incubation, the peptide-containing culture medium or normal medium was replaced by PTX-LIP-, PTX-LIPTR- or PTX-LIPC18-TR-containing media (pH 6.3) for another 2 h of incubation. The content of intracellular PTX was determined by liquid chromatography-mass spectrometry combined with liquid chromatography (LC‒MS/MS).
In vitro cytotoxicity study
The toxicity of different peptide-loaded liposomes to B16F10 cells was evaluated using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) method at different pH values. B16F10 cells were inoculated in 96-well plates, and after overnight incubation, free PTX, PTX-LIP, PTX-LIPTR, and PTX-LIPC18-TR at pH 7.4 or pH 6.3 were placed into each well for another 24 h of incubation. The media were then replaced with 100 μL of MTT solution (1 mg/mL), and the cells were cultured for 4 h. Then, the formazan was dissolved in 150 μL dimethyl sulfoxide. The absorbance was measured by a microplate reader at 490 nm. The cell viability (%) in each well was calculated using the following formula: (Asample − Ablank)/(Acontrol − Ablank) × 100%, where Asample, Acontrol and Ablank represent the absorbance values of the cells treated with the different test solutions and blank culture media and blank wells, respectively. The toxicity of the bare formulations (LIP, LIPTR, and LIPC18-TR) to B16F10 cells was evaluated using the same method at pH 6.3.
Tumour model
All animal experiments were performed in accordance with the experimental guidelines of the Animal Experimentation Ethics Committee of Sichuan University. B16F10 cells were subcutaneously injected into the left flanks of female C57BL/6 mice to establish melanoma tumour-bearing model mice. Tumour volume (mm3) was measured as follows: [length × width2]/2.
Biodistribution study
To investigate the differences in the tumour-targeting abilities of LIPTR and LIPC18-TR in vivo, tumour-bearing mice with an average tumour volume of 200–500 mm3 were randomly assigned to different groups and intravenously (iv) injected with sterile saline, DiD, DiD-LIP, DiD-LIPTR, and DiD-LIPC18-TR at a dose of 200 μg/kg DiD. At 24 h, the mice were sacrificed, and the hearts, livers, spleens, lungs, kidneys and tumour tissues were collected and photographed with an IVIS® Spectrum system (Caliper, USA).
To quantify the effect of peptide modification on the distribution of PTX in vivo, tumour-bearing mice were randomly divided into four groups and iv injected with PTX, PTX-LIP, PTX-LIPTR, and PTX-LIPC18-TR at a dose of 8 mg/kg PTX. At 24 h, the mice were sacrificed, and the hearts, livers, spleens, lungs, kidneys and tumour tissues were collected and weighed. Two volumes of physiological saline were added to each sample, and the mixtures were homogenized with the aid of a homogenizer (Precellys 24, Bertin, France). The distribution of PTX in the different organs was measured by LC‒MS/MS.
Antitumour efficacy
When the mouse tumour volumes reached approximately 50 mm3, the mice were weighed, assigned to different groups, and iv injected with sterile saline, free PTX, PTX-LIP, PTX-LIPTR, or PTX-LIPC18-TR at a dose of 8 mg/kg PTX every other day for a total of 4 injections. The day of tumour treatment was defined as d 1, and tumour volume was measured every other day. The tumour growth inhibition rate was calculated as (1 − Wtreated/Wcontrol) × 100%, where Wtreated and Wcontrol represent the mean tumour weights in the treated groups and the control group, respectively. On d 11, all the mice were sacrificed, and the tumours were collected, photographed and weighed. Additionally, haematoxylin and eosin (HE) staining of paraffin-embedded tumours was conducted.
Statistical analysis
Data are reported as the mean ± SD (standard deviation). Comparisons among multiple groups were evaluated by one-way ANOVA, and *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001, ****P value < 0.0001 were considered statistically significant.
Results
Preparation and characterization of the different liposomes
The particle sizes and zeta potentials of PTX-LIP, PTX-LIPTR, and PTX-LIPC18-TR were determined by dynamic light scattering (DLS), and their shapes were examined by TEM. The sizes of both PTX-LIPTR and PTX-LIPC18-TR were less than 200 nm, and their shapes were spherical (Fig. 1a, c, d). The pH-sensitive and tumour-targeting CPPs C18-TR and TR were synthesized using standard solid-phase peptide synthesis methods. The optimal reaction time between TR and DSPE-PEG2000-Mal was 4 h (Table 1). The peptide conjugation rates of C18-TR and TR were 87.8% ± 1.48% and 85.8% ± 1.67%, respectively. The peptide modification density rates of C18-TR and TR were 36.3% ± 0.6% and 22.6% ± 0.4%, respectively. The zeta potentials of PTX-LIPTR and PTX-LIPC18-TR turned from negative at pH 7.4 to positive at pH 6.3 (Fig. 1b, Table 2), indicating that TR was coupled to PTX-LIP through reaction between the cysteine residue of TR and maleimide of DSPE-PEG2000-Mal [35] and that the C18-TR peptide was successfully inserted into the phospholipid bilayer of the liposomes via electrostatic and hydrophobic interactions [36]. However, PTX-LIPC18-TR exhibited better charge conversion ability, as the zeta potential changed from −6.37 ± 0.61 mV to +8.99 ± 0.18 mV, indicating that the peptide modification method of insertion into the liposomal phospholipid bilayer would promote liposomal entry into cells in the tumour microenvironment by electrostatic adsorption.

a Size distributions of PTX-LIP, PTX-LIPTR and PTX-LIPC18-TR at pH 6.3. b Zeta potential variations of PTX-LIPTR and PTX-LIPC18-TR at pH 7.4 and pH 6.3. TEM images of c PTX-LIPTR and d PTX-LIPC18-TR. Scale bar, 100 nm.
In vitro PTX release profile and stability study
An in vitro PTX release study was conducted to determine whether the drug release properties would be influenced by different peptide-modified liposomes under different pH conditions (Fig. 2). LIP, LIPC18-TR and LIPTR all showed similar sustained release patterns in a pH-independent manner. No initial burst release behaviour was observed. These results showed that pH-sensitive peptide modification and the different peptide modification methods did not affect the release profile of PTX.

Release profiles of PTX from the different PTX-loaded liposomes in PBS over 72 h at a pH 7.4 and b pH 6.3 determined by HPLC (mean ± SD, n = 3). Variations in c transmittance and d particle size of the different PTX-loaded liposomes in 50% FBS (mean ± SD, n = 3).
To investigate the stability of the different liposomes in a biological environment, transmittance and particle size variations were measured in 50% FBS, which mimics conditions in vivo. These two important parameters of LIP, LIPTR and LIPC18-TR displayed little alteration after 72 h (Fig. 2c, d), indicating that they were all stable and would not aggregate in vivo, which may be due to the protective ability provided by PEGylation that reduced the interactions between serum proteins and the liposomes [25, 32, 37].
In vitro cell uptake study and cytotoxicity study
The entrapment efficiencies of DiD by LIPTR and LIPC18-TR were 95.8% ± 1.6% and 96.2% ± 2.4%, respectively. B16F10 cells exhibited higher uptake of both DiD-LIPTR (2.6-fold higher) and DiD-LIPC18-TR (8.5-fold higher) than DiD-LIP at pH 7.4. At pH 6.3, DiD-LIPTR (5.2-fold) and DiD-LIPC18-TR (13.5-fold) displayed better uptake efficiency than DiD-LIP (Fig. 3a). Moreover, DiD-LIPC18-TR maintained higher efficiency than DiD-LIPTR under different pH conditions, indicating that C18-TR-decorated liposomes could deliver more drugs into B16F10 cells than TR-decorated liposomes; however, uptake of both of these liposomes occurred in a pH-sensitive manner. To explore the active targeting capacity of the TR peptide, free peptide was employed to block the integrin receptors highly expressed on melanoma cells. The results (Fig. 3b) demonstrated that the internalization efficiency of both LIPTR and LIPC18-TR was influenced by the free TR peptide. The inhibition rates of the LIPTR- and LIPC18-TR-treated groups were 28.79% and 40.87%, respectively.

a Cellular uptake of DiD-loaded liposomes by B16F10 cells at pH 7.4 and pH 6.3 (mean ± SD, n = 3). **** indicates P < 0.0001 vs. DiD-LIPTR or DiD-LIPC18-TR. b The ability of the TR peptide to actively target B16F10 cells at pH 6.3 (mean ± SD, n = 3). *** indicates P < 0.001 vs. the control groups. c The toxicity of blank carriers to B16F10 cells at pH 6.3. The toxicity of PTX, PTX-LIP, PTX-LIPTR and PTX-LIPC18-TR to B16F10 cells at d pH 7.4 and e pH 6.3 (mean ± SD, n = 3). **** indicates P < 0.0001 vs. LIPC18-TR.
The toxicity of the different liposomal formulations to B16F10 cells was examined using MTT assays (Fig. 3c–e, and Table 3). At a concentration of 24 μM PTX, the cell viability in the PTX-LIP-, PTX-LIPTR- and PTX-LIPC18-TR-treated groups was 59.0%, 39.0% and 36.5% at pH 7.4 and 56.9%, 27.7% and 17.2% at pH 6.3, respectively, showing that the in vitro antiproliferative activities of PTX-LIPTR and PTX-LIPC18-TR increased in a pH-dependent fashion, while C18-TR modification strengthened the antitumour toxicity more effectively than that of TR. At pH 7.4, the half-maximal inhibitory concentration (IC50) values of PTX-LIPTR and PTX-LIPC18-TR were 1.8-fold and 1.9-fold lower than that of PTX-LIP, respectively. When the pH decreased from 7.4 to 6.3, the IC50 values of PTX-LIPTR and PTX-LIPC18-TR were also reduced and were 2.1-fold and 2.5-fold lower than that of PTX-LIP, respectively.
These results illustrated that both TR and C18-TR exhibited stronger pH-dependent cellular uptake efficiency and tumour cell killing ability in the acidic tumour microenvironment than the unmodified liposomes, and PTX-LIPC18-TR presented a higher drug delivery ability and greater cytotoxicity than PTX-LIPTR.
Biodistribution study
Insufficient accumulation of therapeutic anticancer drugs at tumour sites may hinder cancer therapy [38, 39]. To investigate the drug delivery ability of the different peptide-modified liposomes in vivo, sterile saline (as a negative control), free DiD, DiD-LIP, DiD-LIPTR and DiD-LIPC18-TR were injected into B16F10 tumour-bearing C57BL/6 mice. Compared with the free DiD, DiD-LIP and DiD-LIPTR groups, DiD-LIPC18-TR showed the greatest accumulation in the tumour sites in the in vivo images (Fig. 4a, b), illustrating that decoration with C18-TR could provide more assistance than TR, as the average fluorescence intensity after treatment with DiD-LIPC18-TR was 1.3 times higher than that after DiD-LIPTR administration. Ex vivo images of tumour tissues also showed similar results (Fig. 4c, d). The semiquantified data of fluorescence strength showed that the drug delivery efficiency of LIPC18-TR was 1.4-fold and 1.7-fold higher than that of LIPTR and LIP, respectively. These results suggested that LIPC18-TR could accumulate more precisely at tumour sites and deliver more loaded drugs into tumours than LIPTR.

a In vivo whole-body fluorescence images of B16F10 tumour-bearing mice 24 h after intravenous injection of different DiD-loaded formulations. b The semiquantitation of the in vivo fluorescence intensity of the mouse tumours 24 h after injection of different DiD-loaded formulations (mean ± SD, n = 3). *, *** and **** indicate P < 0.05, P < 0.001 and P < 0.0001 vs. DiD-LIPC18-TR. c Ex vivo fluorescence images of (i) major organs and (ii) tumours from mice 24 h after intravenous injection of different DiD-loaded formulations. d The semiquantitation of the ex vivo fluorescence intensity of the mouse tumours 24 h after injection of different DiD-loaded formulations (mean ± SD, n = 3). *, ** and **** indicate P < 0.05, P < 0.01 and P < 0.0001 vs. DiD-LIPC18-TR. Quantification of PTX levels in e tumours and f major organs 24 h after injection of different PTX formulations measured by LC‒MS/MS (mean ± SD, n = 4). *, *** and **** indicate P < 0.05, P < 0.001 and P < 0.0001 vs. PTX-LIPC18-TR.
Encouraged by the above results, the differences in PTX distribution in the tumours and major organs influenced by peptide modification were further examined by LC‒MS/MS. Tumour-bearing mice were euthanized to collect the tumours and major organs for measurement of the PTX concentrations (Fig. 4e, f). Both LIPC18-TR and LIPTR delivered more PTX to the tumour area and reduced nonspecific delivery to the major organs than traditional liposomes. However, the PTX concentration in the tumour site after administering LIPC18-TR was 1.5-fold higher than that after LIPTR treatment, demonstrating that the former peptide modification, achieved by insertion into the liposomal phospholipid bilayer, had better tumour targeting ability and could exert a better tumour inhibition effect.
Antitumour study
To demonstrate the therapeutic antitumour activity of the different peptide-modified liposomes, B16F10 tumour-bearing C57BL/6 mice were intravenously injected with saline, free PTX, PTX-LIP, PTX-LIPTR and PTX-LIPC18-TR. On d 11, the mice were euthanized, and the tumours were collected (Fig. 5a). Changes in body weight during treatment reflect systemic toxicity and side effects [40]. The body weight indices of all groups showed a slight increase (Fig. 5b), indicating that the formulations exhibited low systemic toxicity [41, 42]. The mouse tumour volumes were effectively impaired after PTX-LIPTR and PTX-LIPC18-TR treatment compared with the control group, where the tumour weights were 2.51 ± 0.26 g and 1.81 ± 0.21 g and the tumour reduction ratios were 39.4% and 56.1%, respectively (Fig. 5a, c–e). Moreover, mice treated with PTX-LIP displayed only weak tumour inhibition, in which the tumour weight was 3.27 ± 0.36 g and the tumour reduction ratio was 20.8%. The HE images showed that PTX-LIPC18-TR caused more necrosis and stronger antiproliferative effects in vivo than the PTX-LIP and PTX-LIPTR (Fig. 5f). Cell necrosis in the control group was attributed to ischemia and hypoxia caused by tumour overgrowth [43]. These data revealed that both TR and C18-TR modification could enhance the tumour inhibition ability of liposomes compared with unmodified liposomes, while the addition of C18-TR could suppress tumour growth to a greater extent than the addition of TR.

a Images of tumours at the end of treatment (mean ± SD, n = 5). b The body weight changes and c tumour volume curves of B16F10 tumour-bearing mice during treatment (mean ± SD, n = 5). d The tumour weights and e tumour inhibition rates of different formulation-treated mice at the end of treatment (mean ± SD, n = 5). f HE staining of tumour tissues after treatment. Scale bar, 50 μm. *, ** and **** indicate P < 0.05, P < 0.01 and P < 0.0001 vs. PTX-LIPC18-TR or PTX-LIPTR.
Discussion
In this study, the performances of two different peptide modification methods were investigated. The model molecule applied was the pH-sensitive TR peptide, which actively targets tumours overexpressing integrin receptors and improves cellular internalization. The TR peptide was either conjugated to liposomes containing DSPE-PEG2000-Mal via its cysteine residue or directly inserted into the liposomal phospholipid bilayer by electrostatic and hydrophobic interactions with the addition of a C18 chain to TR (C18-TR). Moreover, the DSPE-PEG2000 chains in the liposomes may also reduce the nonspecific binding of biomolecules in the tumour microenvironment [44]. We found that the modified liposomes displayed better charge reversal ability in the acidic tumour microenvironment, and higher internalization and tumour suppression were observed with the latter method (LIPC18-TR). We suggest that both the C18 stearyl chain and the DSPE-PEG2000-Mal spacer could move the peptide away from the liposomal surface; however, the DSPE-PEG2000-Mal chain is longer and more flexible than C18. Therefore, DSPE-PEG2000-Mal may reduce the influence of TR on the surface charge of the liposomes, thus leading to weaker charge reversal ability and cell internalization efficiency. In addition, the long stearyl chain from the C18 linker inserted into the liposomal lipid bilayer could decrease the permeability and leakage of the entrapped drug in vivo; hence, more antitumour drugs would be delivered to the tumour site to exert its tumour suppression effect [45, 46]. Moreover, the membrane-binding molecule C18 can anchor more functional peptides onto liposomes than DSPE-PEG2000-Mal, which would further strengthen its charge reversal and tumour targeting abilities [47]. Decoration with alkyl chains could even enhance the stabilization, biocompatibility, membrane permeability and secondary structure of CPPs [48,49,50,51]. However, the detailed mechanism of these differences involves multiple factors and requires further investigation. Nonetheless, both types of TR-modified liposomes improved tumour suppression efficiency and reduced the in vivo side effects and toxicity of the delivery system. Therefore, surface modification is an effective way to improve liposomal delivery systems, but the exact modification method requires evaluation for better outcomes.
References
Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–65.
Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–84.
Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm. 2011;8:2032–8.
Tian L, Bae YH. Cancer nanomedicines targeting tumor extracellular pH. Colloids Surf B Biointerfaces. 2012;99:116–26.
Béduneau A, Saulnier P, Benoit JP. Active targeting of brain tumors using nanocarriers. Biomaterials. 2007;28:4947–67.
Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41:147–62.
Vugrin D, Whitmore WF Jr, Sogani PC, Bains M, Herr HW, Golbey RB. Combined chemotherapy and surgery in treatment of advanced germ-cell tumors. Cancer. 1981;47:2228–31.
Tormey DC. Combined chemotherapy and surgery in breast cancer: a review. Cancer. 1975;36:881–92.
Morise Z, Sugioka A, Tokoro T, Tanahashi Y, Okabe Y, Kagawa T, et al. Surgery and chemotherapy for intrahepatic cholangiocarcinoma. World J Hepatol. 2010;2:58–64.
Markman M, Mekhail TM. Paclitaxel in cancer therapy. Expert Opin Pharmacother. 2002;3:755–66.
Rowinsky EK, Donehower RC. Paclitaxel (taxol). N. Engl J Med. 1995;332:1004–14.
Spencer CM, Faulds D. Paclitaxel. Drugs. 1994;48:794–847.
Wang LY, Zhou BJ, Huang SQ, Qu MK, Lin Q, Gong T, et al. Novel fibronectin-targeted nanodisk drug delivery system displayed superior efficacy against prostate cancer compared with nanospheres. Nano Res. 2019;12:2451–9.
Yang T, Cui FD, Choi MK, Cho JW, Chung SJ, Shim CK, et al. Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm. 2007;338:317–26.
Koudelka Š, Turánek J. Liposomal paclitaxel formulations. J Control Rel. 2012;163:322–34.
Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385–93.
Juliano RL, Alam R, Dixit V, Kang HM. Cell-targeting and cell-penetrating peptides for delivery of therapeutic and imaging agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:324–35.
Copolovici DM, Langel K, Eriste E, Langel Ü. Cell-penetrating peptides: design, synthesis, and applications. ACS Nano. 2014;8:1972–94.
Torchilin VP. Cell-penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers. 2008;90:604–10.
Snyder EL, Dowdy SF. Cell-penetrating peptides in drug delivery. Pharm Res. 2004;21:389–93.
Torchilin VP, Lukyanov AN. Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov Today. 2003;8:259–66.
Fonseca SB, Pereira MP, Kelley SO. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev. 2009;61:953–64.
Mondal G, Barui S, Chaudhuri A. The relationship between the cyclic-RGDfK ligand and αvβ3 integrin receptor. Biomaterials. 2013;34:6249–60.
Sakurai Y, Hatakeyama H, Sato Y, Hyodo M, Akita H, Ohga N, et al. RNAi-mediated gene knockdown and anti-angiogenic therapy of RCCs using a cyclic RGD-modified liposomal-siRNA system. J Control Rel. 2014;173:110–8.
Shi KR, Li JP, Cao ZL, Yang P, Qiu Y, Yang B, et al. A pH-responsive cell-penetrating peptide-modified liposomes with active recognizing of integrin αvβ3 for the treatment of melanoma. J Control Rel. 2015;217:138–50.
Song WT, Tang ZH, Zhang DW, Zhang Y, Yu HY, Li MQ, et al. Anti-tumor efficacy of c (RGDfK)-decorated polypeptide-based micelles co-loaded with docetaxel and cisplatin. Biomaterials. 2014;35:3005–14.
Wang LY, Qu MK, Huang SQ, Fu Y, Yang LQ, He SS, et al. A novel α-enolase-targeted drug delivery system for high efficacy prostate cancer therapy. Nanoscale. 2018;10:13673–83.
Jiang TY, Mo R, Bellotti A, Zhou JP, Gu Z. Gel–liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater. 2014;24:2295–304.
Jiang TY, Wang T, Li T, Ma YD, Shen SY, He BF, et al. Enhanced transdermal drug delivery by transfersome-embedded oligopeptide hydrogel for topical chemotherapy of melanoma. ACS Nano. 2018;12:9693–701.
Song X, Wan ZY, Chen TJ, Fu Y, Jiang KJ, Yi XL, et al. Development of a multi-target peptide for potentiating chemotherapy by modulating tumor microenvironment. Biomaterials. 2016;108:44–56.
Zhang XM, Zhang Q, Peng Q, Zhou J, Liao LF, Sun X, et al. Hepatitis B virus preS1-derived lipopeptide functionalized liposomes for targeting of hepatic cells. Biomaterials. 2014;35:6130–41.
Zhang QY, Tang J, Fu L, Ran R, Liu YY, Yuan MQ, et al. A pH-responsive α-helical cell-penetrating peptide-mediated liposomal delivery system. Biomaterials. 2013;34:7980–93.
Yang T, Choi MK, Cui FD, Kim JS, Chung SJ, Shim CK, et al. Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Rel. 2007;120:169–77.
Qu MK, Lin Q, He SS, Wang LY, Fu Y, Zhang ZR, et al. A brain targeting functionalized liposomes of the dopamine derivative N-3, 4-bis (pivaloyloxy)-dopamine for treatment of Parkinson’s disease. J Control Rel. 2018;277:173–82.
Huang SQ, Deng L, Zhang HM, Wang LY, Zhang YC, Lin Q, et al. Co-delivery of TRAIL and paclitaxel by fibronectin-targeting liposomal nanodisk for effective lung melanoma metastasis treatment. Nano Res. 2022;15:728–37.
Khalil IA, Kogure K, Futaki S, Hama S, Akita H, Ueno M, et al. Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther. 2007;14:682.
Jang B, Park JY, Tung CH, Kim IH, Choi Y. Gold nanorod-photosensitizer complex for near-infrared fluorescence imaging and photodynamic/photothermal therapy in vivo. ACS Nano. 2011;5:1086–94.
Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Rel. 2012;161:175–87.
Bigini P, Previdi S, Casarin E, Silvestri D, Violatto MB, Facchin S, et al. In vivo fate of avidin-nucleic acid nanoassemblies as multifunctional diagnostic tools. ACS Nano. 2014;8:175–87.
Gao YJ, Zhou YX, Zhao L, Zhang C, Li YS, Li JW, et al. Enhanced antitumor efficacy by cyclic RGDyK-conjugated and paclitaxel-loaded pH-responsive polymeric micelles. Acta Biomater. 2015;23:127–35.
Al-Jamal KT, Al-Jamal WT, Wang JT, Rubio N, Buddle J, Gathercole D, et al. Cationic poly-L-lysine dendrimer complexes doxorubicin and delays tumor growth in vitro and in vivo. ACS Nano. 2013;7:1905–17.
Xu ZH, Wang YH, Zhang L, Huang L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8:3636–45.
Huang SQ, Zhang YC, Wang LY, Liu W, Xiao LY, Lin Q, et al. Improved melanoma suppression with target-delivered TRAIL and Paclitaxel by a multifunctional nanocarrier. J Control Rel. 2020;325:10–24.
Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137–43.
Spinks CB, Zidan AS, Khan MA, Habib MJ, Faustino PJ. Pharmaceutical characterization of novel tenofovir liposomal formulations for enhanced oral drug delivery: in vitro pharmaceutics and Caco-2 permeability investigations. Clin Pharmacol. 2017;9:29.
Lainé AL, Gravier J, Henry M, Sancey L, Béjaud J, Pancani E, et al. Conventional versus stealth lipid nanoparticles: Formulation and in vivo fate prediction through FRET monitoring. J Control Rel. 2014;188:1–8.
Xiao D, Jia HZ, Ma N, Zhuo RX, Zhang XZ. A redox-responsive mesoporous silica nanoparticle capped with amphiphilic peptides by self-assembly for cancer-targeting drug delivery. Nanoscale. 2015;7:10071–7.
Yu YC, Tirrell M, Fields GB. Minimal lipidation stabilizes protein-like molecular architecture. J Am Chem Soc. 1998;120:9979–87.
Forns P, Lauer‐Fields JL, Gao S, Fields GB. Induction of protein-like molecular architecture by monoalkyl hydrocarbon chains. Biopolymers. 2000;54:531–46.
Han J, Huang X, Sun LD, Li Z, Qian H, Huang WL. Novel fatty chain-modified glucagon-like peptide-1 conjugates with enhanced stability and prolonged in vivo activity. Biochem Pharmacol. 2013;86:297–308.
Zhang HY, Schneider SE, Bray BL, Friedrich PE, Tvermoes NA, Mader CJ, et al. Process development of TRI-999, a fatty-acid-modified HIV fusion inhibitory peptide. Org Process Res Dev. 2008;12:101–10.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81690261, 82022070 and 81872824) and the Medico-Engineering Cooperation Programme from the Med-X Center for Materials, Sichuan University (MCM202103).
Author information
Authors and Affiliations
Contributions
ZRZ and LZ designed the research; SQH, HMZ and YCZ performed the research; ZRZ and LZ contributed new reagents or analytical tools; SQH and LYW analysed the data, and SQH and LZ wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, Sq., Zhang, Hm., Zhang, Yc. et al. Comparison of two methods for tumour-targeting peptide modification of liposomes. Acta Pharmacol Sin 44, 832–840 (2023). https://doi.org/10.1038/s41401-022-01011-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-01011-4
