
Abstract
C1q/tumor necrosis factor (TNF) related proteins (CTRPs) is a newly discovered adipokine family with conservative structure and ubiquitous distribution and is secreted by adipose tissues. Recently, CTRPs have attracted increasing attention due to the its wide-ranging effects upon inflammation and metabolism. To-date, 15 members of CTRPs (CTRP1-15) with the characteristic C1q domain have been characterized. Earlier in-depth phenotypic analyses of mouse models of CTRPs deficiency have also unveiled ample function of CTRPs in inflammation and metabolism. This review focuses on the rise of CTRPs, with a special emphasis on the latest discoveries with regards to the effects of the CTRP family on inflammation and metabolism as well as related diseases. We first introduced the structure of characteristic domain and polymerization of CTRPs to reveal its pleiotropic biological functions. Next, intimate association of CTRP family with inflammation and metabolism, as well as the involvement of CTRPs as nodes in complex molecular networks, were elaborated. With expanding membership of CTRP family, the information presented here provides new perspectives for therapeutic strategies to improve inflammatory and metabolic abnormalities.
Similar content being viewed by others
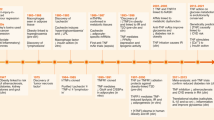


Introduction
Inflammation underlies a wide variety of physiological and pathophysiological processes. Infection and tissue injury, the classic instigators for inflammation, trigger the recruitment of leukocytes and plasma proteins to damaged tissue sites, followed by abundant production of pro-inflammatory cytokines. Complement 1q (C1q)/ tumor necrosis factor (TNF) related proteins (CTRPs), members of the adipokine superfamily secreted by adipose tissues, have drawn recent attentions [1, 2]. Adiponectin is the first identified member of the CTRPs family [3]. In addition to adiponectin, 15 members (CTRP1–15) were later successively reported. CTRPs share a characteristic structure, including four distinct domains with one homologous to adiponectin. Growing evidence has indicated a key regulatory role for CTRP in inflammation or metabolism-related pathological processes, such as immunity, insulin resistance, and fibrosis. The close associations between CTRPs and inflammation or metabolism suggest that CTRPs plays a pharmacologically targeted role in inflammation-related diseases such as diabetes [4, 5], obesity [6, 7], and atherosclerosis [8, 9].
This review summarizes the latest progress with regards to the role of CTRPs in inflammation and metabolism-related diseases. We will first introduce the characteristic domains and polymerization of CTRPs to reveal its pleiotropic physiological functions. Then, we will elucidate the close relationship between CTRPs and inflammation or metabolism before a systematic discussion of the current literature on CTRPs in diseases associated with inflammation and metabolism. Given the prominent role of inflammation as the prime culprit of vast diseases, information presented here should help the audience to put this into a broader range of disease disciplines.
Brief “CTRPs” story
CTRPs and its C-terminal globular domain
Adipose tissues secrete various bioactive molecules, collectively known as adipokines. In 2004, Wong and coworkers cloned a protein family named CTRPs based on its sequence homology with adiponectin [2]. CTRPs (except CTRP4) share four different domains: a signal peptide at the N terminus, a short variable region, a collagenous domain with various lengths of Gly–X–Y repeats, and a globular C-terminal domain homologous to the complement component 1q (C1q) (Fig. 1) [2]. Notably, CTRP4 is structurally unique and comprises two C-terminal globular domains connected by a short linker while lacks a collagenous domain [10]. The C-terminal globular domain forms trimers are composed of A, B and C chains [11, 12] and is highly conserved in a variety of molecules [2]. The complement protein C1q represents the first subcomponent of C1 complex and serves as a target recognition protein for classical signaling cascades [13]. C1q binds with a variety of ligands mainly through its globular C1q domain while it regulates immune cells through collagenous domain, thus offering an important role in immunity (such as pathogen clearance and apoptosis) [13]. Besides, the C-terminal region of TNF homology domain (THD) is nearly identical to gC1q domain. Interestingly, CTRPs is designated to C1q/TNF-related proteins given that the family members have gC1q domain homologous to C1q and TNF [14].

a Protein structure of CTRP1 with the number of Gly-X-Y repeats. b The protein structure of CTRP3A with the number of Gly-X-Y repeats. c The protein structure of CTRP9 with the number of Gly-X-Y repeats. d The common structure of CTRPs.
Tissue expression
CTRP family is a class of chemical hormones mainly secreted by adipose tissue. It is also widely expressed in various organs and tissues and exerts corresponding biological functions [12]. Although from the same family, different members have slightly different tissue expression. CTRP1 mRNA is expressed in heart, placenta, liver, muscle, kidney, prostate, and ovary, and adipose tissue [1, 15, 16]. CTRP2 mRNA is expressed in stromal vascular, myocardial tissue and adipose tissue [1, 17]. CTRP3 mRNA is expressed in adipose tissue, cartilage tissue, fibrous tissue, placenta, osteosarcoma, chondroblastoma, colon, small intestine, pancreas, brain, kidney, thymus, ovary [1, 18, 19]. CTRP4 mRNA is expressed lowly in several tissues and cells including hepatoma cells and colon cancer cells [20]. CTRP5 mRNA is expressed in stromal vascular, adipose tissue, basement membrane, retinal pigment epithelium, ciliary epithelium, myocytes, brain [1, 21]. CTRP6 mRNA is expressed in VSMCs, human synoviocytes, adipose tissue, placent, hepatocellular carcinoma and human submandibular glands (SMGs) [22,23,24]. CTRP7 mRNA is expressed in stromal vascular, adipose tissue and lung. CTRP8 mRNA is expressed in brain, placenta and eye [1]. CTRP9 mRNA is expressed in stromal vascular cells, cardiac cells and adipose tissue [25, 26]. CTRP10 mRNA is expressed in brain, placenta and eye [1]. CTRP11 mRNA is expressed in testes, adipose tissue, heart, brain, kidney, lung, prostate, pancreas and skeletal muscle [27]. CTRP12 mRNA is expressed in adipose tissue [28, 29]. CTRP13 mRNA is expressed in rat liver sinusoidal endothelial cells (rLSECs), stromal vascular cells in adipose tissue [30, 31].
The polymerization
Adiponectin (also known as Acrp30 and AdipoQ) was the first member of the CTRPs to be identified. It carries both anti-inflammatory and insulin-sensitizing effects mediated by two isoforms of adiponectin receptors respectively, adiponectin receptor (AdipoR)1 and AdipoR2 [32]. In serum, different isoforms of adiponectin exist including: trimeric [low molecular weight (LMW)], hexameric [middle molecular weight (MMW)], and multimeric [high molecular weight (HMW)] adiponectin isoforms. The forms of MMW and HMW induce nuclear factor kappa-B (NF-κB) activation in C2C12 myocytes and myotubes, while the trimer form triggers AMP-activated protein kinase (AMPK) activation in muscles [32,33,34,35]. In addition to adiponectin, other members of the CTRPs form trimers as their basic structural units, including homotrimers (CTRP1, CTRP2, CRPP3, CTRP4, CTRP5, CTRP6, CTRP7, CTRP 8, CTRP9, CTRP10, and CTRP13) and heterotrimers (adiponectin and CTRP2, adiponectin and CTRP9, CTRP1 and CTRP6, CTRP2 and CTRP7, and CTRP10 and CTRP13) [12].
CTRP1 and CTRP6 are produced in stromal vascular cells (SVC), and usually form oligomers [1, 12]. CTRP2 and CTRP7 mRNAs are also found in SVC, capable of forming intracellular complexes. Besides, CTRP2 form heterotrimeric complex with adiponectin [12]. In particular, there are two alternatively spliced CTRP3 isoforms (CTRP3A and CTRP3B) in humans, with distinct lengths and degrees of glycosylation [36, 37]. CTRP3B contains an additional 73 amino acids at the N terminus in comparison with CTRP3A. Thus, CTRP3B is much longer and less stable, and the two isoforms form hetero-oligomers in the absence of disulfide bond to protect CTRP3B from protein solubilization. Indeed, CTRP3A interacts with CTRP3B only under colocalization in the same cell [36]. In mammalian cell lines, recombinant CTRP3 forms homotrimers and higher-order oligomers via disulfide bonding mediated by N-terminal cysteine residues. CTRP3 forms stable homotrimeric protein structures in the baculovirus system [1, 12, 37], while acts a HMW form to circulate in the peritoneal fluid around human plasma and visceral adipose tissues [12]. CTRP5 circulates in the blood and forms homotrimers and higher-order oligomers through disulfide bond through the N-terminal cysteine residue [1]. CTRP6 can form homotrimers and higher-order oligomers, and also forms heteromeric complexes with CTRP1. In addition to homotrimer formation, CTRP8 also forms heterogeneous complexes with C1q-related factors (CRF) [12]. Of all the CTRPs paralogs, CTRP9 exhibits the highest degree of amino acid identity to adiponectin in its globular C1q domain, so CTRP9 forms trimers predominantly and circulates in serum similar to adiponectin. When adiponectin and CTRP9 were simultaneously expressed in 3T3-L1 adipocytes and HEK293T cells, the two proteins were co-immunoprecipitated in the supernatant [36], which indicates that adiponectin and CTRP9 are possibly form heterotrimers. Human CTRP9 consists of two isoforms, CTRP9A and CTRP9B. The two isoforms share 98% amino acid identity, although they are encoded by distinct genes with the latter absent in mouse genome [38]. CTRP9A is a secreted polyprotein, while CTRP9B requires physical association with CTRP9A or adiponectin for its secretion [38]. However, whether adiponectin and CTRP9 heterooligomers induce distinct signaling pathway in cells and tissues compared to adiponectin or CTRP9 alone remains unknown. Association between CTRPs is a possible mechanism to generate an expanded repertoire of functionally distinct ligands with altered receptor specificity or kinds [38].
In sum, CTRPs form a variety of polymers, and combine within themselves or other proteins to yield various complexes. Different polymers possibly bind to different receptors and thus exhibit different signaling and functional properties. This pleiotropy is one of the major features for CTRPs, further broadening regulatory pathways in different physiological and pathological conditions.
The relationship of CTRPs with inflammation and metabolism
Alterations in cellular metabolism impact the cell inflammatory state. Multiple studies have noted the role of CTRP family as key nodes in inflammatory and metabolic signaling (REF). Several common members of CTRPs with a close relevance to inflammation and metabolism are described in the following sections.
CTRP1
Lipopolysaccharide (LPS), as a pro-inflammatory medium, does not directly induce CTRP1 in isolated adipocytes, although LPS upregulates expression of CTRP1 in vivo by inducing inflammatory cytokines TNF and interleukin-1β (IL-1β) [12]. Clinically, the level of CTRP1 is higher in patients with obesity, hypertension and adiponectin deficiency, probably related to higher inflammatory cytokine level in these patients [39]. Kim and colleagues found that the level of CTRP1 is highly expressed in adipose tissues of LPS-challenged rats, similar to CTRP1 levels in obese rats, suggesting that CTRP1 is closely related to the low-grade chronic inflammation status of adipose tissues [39]. In addition, insulin level increases in CTRP1 gene knockout mice in fasting, which enhances insulin resistance and promotes hepatic glucogenase expression to decrease glucose conversion [7].
CTRP2
CTRP2 promotes uptake of glucose in muscles and reduces blood sugar levels [12]. CTRP2 produced by bacteria induces phosphorylation of AMPK, acetyl-coenzyme A carboxylase (ACC), and p42/44 mitogen-activated protein kinase (MAPK) in C2C12 myotubes, thus increasing glucose absorption, glycogen accumulation and fatty acid oxidation, causing oxidative stress and chronic inflammation [2].
CTRP3
CTRP3 is a potential anti-inflammatory mediator, and its level is inversely correlated with levels of TNF, IL-6, C-reactive protein (CRP), circulating leptin, and insulin resistance [36, 40]. Meanwhile, overexpression of CTRP3 alleviates systemic inflammation and insulin resistance by reducing levels of TNF-α and improving insulin sensitivity in the CTRP3 transgenic mice with diet‐induced obesity [41]. Kopp et al. used fusion molecules composed of toll-like receptor 4 (TLR4) and myeloid differential protein-2 (MD-2), a receptor for LPS, to probe the specific anti-inflammatory mechanism of CTRP3. They demonstrated that CTRP3, as an endogenous LPS antagonist, exerts anti-inflammatory activity by interacting with LPS to block the binding of LPS to TLR4/MD-2 [42]. In addition, CTRP3 is also closely related to glucose metabolism via extracellular signal-regulated kinase 1/2 (ERK1/2), Akt and NF-κB signaling cascade. The ERK1/2 pathway is a key path to regulate several cellular functions, including proliferation, differentiation, and transformation. CTRP3 phosphorylates the ERK1/2 signaling pathway in the mouse liver [36], although the biological consequence of CTRP3-evoked ERK1/2 activation remains unclear in the liver. ERK1/2 can regulate transcription factors including forkhead box protein O1 (FOXO1) [43], a key transcriptional factor promoting gluconeogenic enzymes, to enhance glucose production. Thereby CTRP3 potentially links ERK1/2 pathway either directly or indirectly with the transcriptional suppression of hepatic glucose output [44]. Meanwhile, CTRP3 significantly lowers blood glucose levels and suppresses expression of gluconeogenic genes G6Pase and PEPCK through activation of Akt in the liver. Besides, recombinant CTRP3 attenuates NF-κB p65 function, and reduces glucose output in cultured rat hepatoma cells [45]. It is noteworthy that the effects of CTRP3 on blood sugar, such as inhibition of glucose gene expression and glucose output, are independent of insulin. In the absence of insulin, CTRP3 still activates Akt [36]. Therefore, CTRP3 is a potential new direction for diabetes research.
CTRP6
CTRP6 regulates local inflammation and glucose metabolism by targeting macrophages and adipocytes. CTRP6 upregulates the expression and production of TNF-α. Loss of CTRP6 decreases the number of macrophages in adipose tissue and the level of circulating inflammatory cytokines, thereby reducing inflammatory response [46]. However, overexpression CTRP6 attenuates Akt phosphorylation and glucose uptake evoked by insulin stimulation in adipocytes, thus annihilating the response of peripheral tissues to glucose and insulin.
CTRP9
CTRP9 improves multiple organ and tissue injury induced by the inflammation. Huang and coworkers reported that CTRP9 overexpression reverses hypoxia/reoxygenation (H/R)-induced upregulation in the expression of TNF-α and IL-6 as well as the H/R-induced reduction in the levels of the anti-inflammatory cytokine IL-10 by inhibiting the TLR4/MyD88/NF-κB signaling pathway [47]. In addition to heart, CTRP9 overexpression activates PI3K/Akt pathway, in order to improve endothelial cell function and inflammation of pulmonary artery [48]. Besides, CTRP9 protects against monosodium iodoacetate (MIA)-induced inflammation and knee cartilage damage by deactivating the MAPK/NF-κB pathway in rats with osteoarthritis [49]. AMPK and Akt signaling pathways are major regulators of insulin-mediated glucose uptake. Recombinant CTRP9 activates AMPK and Akt to stimulate glucose uptake in rat cardiomyocytes [17]. CTRP9 not only enhances the above two pathways, but also stimulates p42/44 MAPK to reduce blood sugar in C2C12 myotubes [26]. Additionally, CTRP9 improves cholesterol efflux through upregulating the expression of proteins associated with cholesterol efflux in foam cells, such as LXRα, CYP27A1, ABCG1 and ABCA1 [50].
In conclusion, CTRPs family members play a vital role in linking inflammation, insulin resistance, and metabolic disorders. As a node of inflammation and metabolism, CTRPs have a strong potential for promising therapeutic targets to treat various inflammation-related diseases.
Downstream targets of CTRPs
Courtesy of their wide distribution and important physiological roles, CTRPs participate in multiple cardinal intracellular signaling pathways. As irreplaceable central molecules in the regulatory network, CTRPs can respond to multiple stimuli or molecules, such as inflammation, transcription factors. Then, CTRPs mediate phosphorylation of a variety of substrates, including Akt, AMPK, MAPK, S1P/cAMP, TGF-β, and ERK1/2 (Table 1). Here we briefly discuss those molecules with particular importance in the treatment of inflammation-related diseases.
Akt
Akt, a serine/threonine kinase known as protein kinase B (PKB), is activated by phosphatidylinositol 3-kinase (PI3K) or phosphoinositide-dependent kinases (PDK) as well as growth factors, and DNA damage at Thr308 or Ser473 phosphorylation [51]. Akt phosphorylates a variety of downstream protein substrates such as mammalian target of rapamycin (mTOR), glycogen synthase kinase 3 beta (GSK3β), or Foxo1 to regulate numerous cellular functions [51]. Multiple members of CTRPs phosphorylate Akt or its upstream molecule PI3K [51].
Autophagy is regulated by a variety of molecular mechanisms, such as the Akt/mTOR pathway and ERK pathway [52]. Akt/mTOR pathway reduces neuronal apoptosis and inflammatory response by inhibiting calcium-dependent pathways. CTRP1 inhibits autophagy and inflammatory response of microglia by regulating Akt/mTOR pathway in BV2 microglia [53]. Downregulation of CTRP1 significantly reduces the phosphorylation levels of Akt and mTOR in BV2 microglia. In addition, A6730 (Akt inhibitor) reverses the inhibition of microglia autophagy by recombinant CTRP1. Knockdown of CTRP1 further enhances autophagy in the oxygen and glucose deprivation and reperfusion injury (OGD/R)-stimulated BV2 cells. Therefore, CTRP1 inhibits autophagy by activating the Akt/mTOR pathway, thereby weakening OGD/R-induced BV2 cell damage [53].
Through activation of the PI3K/Akt/eNOS signaling, CTRP3 attenuates inflammation and then inhibits endothelial dysfunction [54, 55]. Overexpression of CTRP3 inhibits inflammation, elevates cell activity, and decreases lactated hydrogenase release in endothelial cells, accompanied by a marked reduction in cell apoptosis induced by oxidative low density lipoprotein (ox-LDL). Mechanically, phosphorylation levels of PI3K, Akt and eNOS are upregulated following CTRP3 overexpression, while LY294002 (PI3K inhibitor) lifted the protective effect of CTRP3. Similarly, CTRP9 overexpression also activates PI3K/Akt signaling, improves endothelial NOS expression and reduces secretion of endothelin-1 (ET-1) and matrix metalloproteinase-2 (MMP-2). On the contrary, pretreatment with LY294002 antagonizes these phenomenon [48].
In addition, CTRP6 also activates PI3K/Akt signaling to protect against oxidative damage induced by H/R in HK-2 cells, thus reducing renal ischemia-reperfusion (I/R) injury [56]. CTRP15 inhibits TGF-β1-induced Smad3 activation and increases insulin receptor (IR), insulin receptor substrate-1 (IRS-1) and Akt phosphorylation, leading to suppressed differentiation in cardiac fibroblasts (CFs). However, blocking IR/IRS-1/Akt pathway reverses the inhibitory effect of CTRP15 [57].
AMPK
AMPK is a key controller that regulates biological energy metabolism to balance nutrient supply and energy demand and aberrant levels of AMPK are noted in various pathological settings such as diabetes mellitus (DM), cancer and neurological diseases [58]. It is activated by abundant stimuli, including metabolic stress, drugs, xenobiotics, and oxidative stress. These stimuli then catalyze phosphorylation of Thr-172 on its α-subunit [59]. Once activated, AMPK promotes ATP production by increasing the activity or expression of catabolic proteins to modulate energy metabolism in cells [60].
CTRP1 controls fatty acid metabolism by activating AMPK [61]. Activated AMPK phosphorylates its downstream target ACC at Ser-79, leading to the inactivation of ACC, the decrease in malonyl-CoA concentration, and the enhancement of the oxidation of long-chain fatty acids in skeletal muscle of CTRP1 transgenic mice [62, 63]. However, no significant changes of metabolic enzyme genes (such as Acox1, Cpt1, Lcad, and Mcad) and adipokines are observed in skeletal muscle of CTRP1 transgenic mice, suggesting that the activation of AMPK/ACC is the main mechanism of CTRP1 to enhance fat oxidation in skeletal muscle and to alleviate fat accumulation and vasospasm [61, 64]. Therefore, CTRP1 regulates fatty acid oxidation and promotes systemic energy consumption via activating AMPK/ACC axis. Remarkably, there was little hyperphosphorylation of AMPKα and ACC in the liver of CTRP1 transgenic mice, hinting that the activation of AMPK mediated by CTRP1 is possibly muscle-specific [61]. Park and coworkers found that the full-length and globule domains of human CTRP5 also activated AMPK/ACC [65].
Remarkably, CTRP5 has no effect on IRS-1 and Akt that is an important insulin-induced hypoglycemic pathway in the muscle cells, so CTRP5 is the agonist of AMPK but independent of the insulin pathway. Meanwhile, CTRP5 stimulates plasma membrane consolidation of glucose transporter 4 (GLUT4) by inducing phosphorylation of AMPK, and increases glucose uptake in muscle cells [65].
CTRP9 also regulates cholesterol metabolism by enhancing AMPK phosphorylation. Liu and his associates found that CTRP9 promotes AMPK activation in vascular smooth muscle cells (VSMCs) and upregulates the expression of several downstream proteins important for cholesterol efflux such as LXR, ABCA1 and ABCG1 in vivo and in vitro [66]. To the contrary, knockdown of AMPK α1 abolishes CTRP9-offered protective effect. Furthermore, CTRP9 has been also shown to retard development of atherosclerosis through AMPK-NLRP3 inflammasome pathway [67].
P38 MAPK
Protein 38 MAPK (p38), one branch of MAPKs, can be activated and phosphorylated under pressure-load stimulation, and participates in various cardiac hypertrophy [68, 69]. The cAMP response element-binding protein (CREB) is a downstream signal molecule of p38 and can regulate multiple cellular responses by interacting with DNA and regulating gene transcription. Various forms of pathological cardiac hypertrophy are attenuated by inhibiting the p38/CREB pathway [70].
CTRP3 protects from pathological heart remodeling and left ventricular dysfunction induced by pressure overload. On the one hand, CTRP3 deficiency exaggerates phosphorylation of p38 and CREB. Reversely, CTRP3 supplementation inhibits p38 and CREB activation [71]. On the other hand, CTRP3 alleviates ERS induced by p38 through significantly reducing the expression of the main markers of ERS (GRP78 and its downstream molecules eIF2α, CHOP, IRE1, and ATF6) [71].
Extracellular signal-regulated kinase (ERK)
ERK is a subfamily of MAPK, as well as a downstream effector of AMPK pathway. ERK1/2 (a.k.a., p42/p44) pathway plays a vital role in CTRP1-mediated physiological response in endochondral ossification. CTRP1 is predominantly distributed in the reserved and proliferative chondrocytes of fetal and postnatal growth plates. CTRP1 promotes proliferation of immature proliferating N1511 chondrocytes in a dose-dependent manner through activation of ERK1/2 signaling, whereas CTRP1 inhibits maturation of N1511 chondrocytes through suppression of ERK1/2 [72].
Recombinant CTRP3 has been shown to trigger phosphorylation of both Akt and ERK1/2 in mouse livers (without any significant effects on AMPK signaling) [36]. Akt activation in liver potently suppresses hepatic gluconeogenesis by phosphorylating the transcription factor Foxo1 [73], and ERK1/2 potentially also phosphorylates Foxo1 [43]. Phosphorylated Foxo1 is thus excluded from the nucleus, preventing the transcription of gluconeogenic genes [36, 74]. Conversely, CTRP3 also acts as an effective inhibitor of LPS through mitigating LPS-induced ERK1/2 phosphorylation, resulting in suppressed LPS-induced systemic inflammation [75]. Intravenous application of CTRP3 is insufficient to inhibit LPS-induced cytokine level or mRNA expression, suggesting CTRP3 alone might not directly evoke ERK1/2 phosphorylation. However, intraperitoneal administration of CTRP3 significantly reduces LPS-induced ERK1/2 phosphorylation level in inguinal adipose tissue. Furthermore, CTRP9 overexpression activates ERK5/GATA4 signaling to promote hypertrophic remodeling and dysfunction induced by pressure overload of mice [25]. In conclusion, CTRP3 inhibits or activates ERK1/2 through different mediators, thereby exerting discrepant biological effects.
S1P/cAMP
Sphingosine-1-phosphate (S1P), mediated by S1P1–5 receptors and synthesized from sphingosine phosphorylation, is a lysophospholipid mediator with diverse biological function, such as angiogenesis, endothelial protection, myocardial ischemia-reperfusion injury, inhibition of adhesion molecule expression, and immune function [76, 77].
CTRP1 attenuates myocardial cell apoptosis and inflammatory response through S1P/cAMP axis, thus protecting against myocardial ischemic injury [78]. On the one hand, pretreatment of cardiomyocytes with VPC23019 (an antagonist of S1P1 and S1P3) significantly reverses CTRP1-induced inhibition of apoptosis. On the other hand, sphingosine kinase 1 knockout or pretreatment with VPC23019 both eliminated the inhibitory effect of CTRP1 on LPS-stimulated pro-inflammatory cytokines such as TNF, IL-6 and IL-1β in cardiomyocytes [78]. S1P was previously shown to promote the accumulation of cAMP through S1P receptor [79], suggesting a role of cAMP in the protective effects of CTRP1. Treatment with CTRP1 in cardiomyocytes leads to increased cAMP levels, the effect of which are reversed by VPC23019. The adenylate cyclase inhibitor SQ22536 inhibits cAMP activation and simultaneously reverses CTRP1-elicited inhibitory effect on cardiomyocyte apoptosis. Therefore, through activation of S1P/cAMP axis, CTRP1 effectively attenuates myocardial cell apoptosis and inflammatory response.
TGF-β
Transforming growth factor-β (TGF-β) is the prototype of the TGF-β superfamily of growth and differentiation factors. Hofmann and group revealed that CTRP3 inhibits the TGF-β-CTGF-collagen I pathway and antagonizes LPS to reduce secretion of IL-8, exerting a powerful anti-inflammatory and anti-fibrotic effects in primary colonic lamina propria fibroblasts (CLPF) [80]. CTRP3 also slows down progression of IgA nephropathy by inhibiting TGF-β-Src-Smad3 signaling to alleviate mesangial cell activation and inflammatory response [81].
Thrombospondin (THBS) family, such as THBS1, THBS2 and THBS3, belongs to a class of calcium-related glycoproteins and is widely involved in biological processes such as angiogenesis, cell movement and apoptosis, cytoskeletal composition, and ECM reaction, through interactions with various target proteins [82, 83]. It has been reported that CTRP9 regulates TGF-β1/Smad2/3 pathways by THBS1 to protect H9c2 cells from apoptosis, inflammation, and fibrosis induced by coxsackievirus B3 (CVB3) [84]. Mechanically, CTRP9 interacts with THBS1 to decrease THBS1 levels in children with viral myocarditis (VMC) induced by CVB3. Subsequently, downregulation of THBS1 inhibits CVB3-induced apoptosis, inflammation, and fibrosis in H9c2 cells. In addition, adenovirus vectors containing full-length rat CTRP9 suppresses the expression of TGF-β1 and p-Smad2/3 in CVB3-infected H9c2 cells, while THBS1 annihilates this effect [84].
Roles of CTRPs in diseases associated with inflammation and metabolism
Atherosclerosis
Atherosclerosis is the main pathological basis of cardiovascular disease and involves vascular endothelial dysfunction [85]. Dysfunction in vascular endothelial cells serves as the main initiator of and springboard for atherosclerosis, together with accumulation of lipids, adhesion of inflammatory cells, and proliferation of VSMCs [86, 87]. Excessive proliferation and migration of abnormal VSMCs are the major causes for the development of cardiovascular diseases, including atherosclerosis [88, 89]. Macrophages adopt different functional phenotypes according to atherosclerotic environment [88, 90]. M1 is a pro-inflammatory phenotype that leads to atherosclerosis, while M2 is an anti-inflammatory phenotype that maintains the stability of atherosclerotic lesions. Lipid-loaded macrophages are involved in almost all stages of plaque development and progression, which expressing scavenger receptors, taking up high ox-LDL particles and leading to foam cell formation [91, 92]. CTRPs possess a major influence on a variety of atherosclerosis-related cells including endothelial cells, macrophages, and VSMCs. However, different members have opposite effects on atherosclerosis.
CTRP1 regulates low-grade chronic inflammation and lipid metabolism caused by coronary artery disease (CAD) [55]. PPAR-γ mediates macrophage differentiation and lipid metabolism. Wang et al. found that CTRP1 is upregulated in a PPAR-γ-dependent manner under the condition of ox-LDL following differentiation of primary human macrophages from peripheral blood mononuclear cells [82]. Meanwhile, CTRP1 is abundantly induced by inflammatory cytokines, which itself causes concentration-dependent expression of adhesion molecules and inflammatory markers in human endothelial cells, human peripheral blood mononuclear cells, and THP-1 cells. Additionally, CTRP1 induces p38-dependent monocyte endothelial adhesion and leukocyte recruitment to C57BL/6 mouse mesenteric venules in vitro, while CTRP1 knockdown in vivo reduces vascular adhesion molecule p38 phosphorylation in mouse plaques as well as macrophages infiltration [93]. Thus, CTRP1 is involved in lipid metabolism and inflammation of macrophages as a pro-atherogenic factor [9]. Likewise, CTRP5 promotes progression of atherosclerosis, especially during early stages. Permeation of LDL is a key driver in the early development of atherosclerosis. Through upregulation of 12/15-LOX (a key enzyme that mediates LDL transportation and oxidation), CTRP5 acts as a pro-atherogenic cytokine to promote accumulation of oxidized LDL in endothelial cells [8]. Furthermore, CTRP5 promotes the growth, migration and inflammation of vascular endothelial cells by activating Notch1, TGF-β and hedgehog pathway, in order to aggravate in-stent restenosis under CAD [55, 94]. Moreover, vascular remodeling is considered to be one of the key factors leading to luminal stenosis and subsequent atherosclerosis and CTRP12 prevents the development of pathological vascular remodeling through attenuating macrophage inflammatory response and VSMC growth and proliferation to improve atherosclerosis [95].
CTRP3, CTRP9 and CTRP13 display an inhibiting impact against atherosclerosis. CTRP3 delays atherosclerosis progression by promoting PI3K/Akt/eNOS pathway, which inhibits ox-LDL-induced endothelial inflammation [54]. Clinically, patients with acute coronary syndrome and stable angina pectoris often exhibit low CTRP3 levels, which may be used as a predicting biomarker for assessment of CAD risk [96]. CTRP9 exerts a significant protective effect against atherosclerosis, involving multiple factors and pathways. First and the foremost, CTRP9 improves atherosclerotic plaque stability through AMPK activation. CTRP9 also suppresses level of adhesion molecules, such as intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 [55, 97], and secretion of TNF-α and MCP-1 in heat hardening plaques. The aforementioned processes would retard VSMCs proliferation, promote vascular dilation, and inhibit inflammatory response of macrophages [55, 97]. Second, CTRP9 down-regulates expression and activity of NLRP3 in ox-LDL-activated macrophages by activating AMPK to protect against progression of atherosclerosis [67]. On the one hand, CTRP9 directly inhibits the activity of NLRP3 inflammasome in the ox-LDL activated macrophages. On the other hand, CTRP9 activates AMPK [38, 98, 99], which subsequently inhibits NLRP3 inflammasome [100]. CTRP9 enhances cholesterol efflux of macrophage foam cell through activation of AMPK/mTOR signaling pathway to induce autophagy in fatty medium, thus increasing levels of cholesterol transport receptors [such as ATP binding cassette transporter A1 (ABCA1) and ABCG1]. Moreover, apoptosis of foam cells leads to instability of atherosclerotic plaques. CTRP9 enhances cholesterol efflux through the AdipoR1/AMPK pathway on foam cells to alleviate apoptosis. These studies strongly suggest that CTRP9 accelerates cholesterol efflux of foam cells and regulates lipid metabolism, thus inhibiting progression of early atherosclerosis [55, 97]. On the other hand, CTRP13 plays a protective role via inhibition macrophage inflammation and cholesterol metabolism in atherosclerosis [101]. Cluster of differentiation 36 (CD36) expression and foam cell formation are believed to potentially contribute to atherosclerosis [102]. Specific, CTRP13 reduces CD36 levels, ox-LDL uptake in primary peritoneal macrophages in vitro, and formation of foam cells [103]. CTRP13 hydrolyzes cholesterol droplets stored in macrophages, which attenuates cholesterol influx and promotes outflow of cholesterol, thus inhibiting the formation of foam cells and progression of atherosclerosis [104, 105].
In addition, atherosclerosis is considered to be the most common etiology of abdominal aortic aneurysm (AAA). In AAA patients, the arterial wall is structurally damaged so that it cannot withstand the pressure of blood flow shocks, resulting in localized or extensive permanent dilation or bulge. Macrophage-induced inflammation can promote the formation of AAA [106, 107]. Xu et al. identified nicotinamide phosphoribosyl transferase 1 (NAMPT1) could be as a novel target of CTRP13, and demonstrated that CTRP13 reduced aortic macrophage infiltration, levels of inflammatory factors (e.g., IL-6, TNF-α, MCP-1) and apoptosis of vascular smooth muscle cells [108]. Targeting CTRP13 will be an effective treatment strategy for preventing abdominal aortic aneurysm formation in the future.
Taken together, multiple CTRPs members exert important roles in atherosclerosis. Of note, several CTRPs, such as CTRP3 and CTRP9 inhibit the progression of the disease and offer a protective action. On the other side of the coin, CTRP1 and CTRP5 may contribute to the progression of atherosclerosis. The reason will be discussed later in the discussion.
Heart diseases
Factors such as inflammation or ischemia interfere with myocardial metabolism and trigger myocardial injury. Various members of CTRPs are involved in variant physiological processes in cardiomyocytes, such as inflammation, apoptosis, fibrosis and oxidative stress, and offer a vital impact in several cardiac disease such as myocarditis and myocardial infarction (MI).
CTRP1, CTRP3, CTRP12, and CTRP13 protect against myocardial I/R injury although through distinct mechanisms. CTRP1 reduces cardiomyocyte apoptosis and inflammation to improve ischemic heart disease through activating SIRT1 signaling and S1P/cAMP signaling [78, 109]. Besides, CTRP3 knockout mice display cardiac systolic dysfunction, myocardial cell enlargement and myocardial fibrosis. To the contrary, recombinant CTRP3 reduces pathological myocardial remodeling after acute infarction through inhibiting fibrosis and enhances the survival and regeneration of ischemic cardiomyocytes via a PI3K/Akt-dependent mechanism [110]. CTRP12 inhibits apoptosis, oxidative stress, and inflammation to protect against H/R-induced cardiomyocyte injury through Nrf2 signaling [111]. Furthermore, CTRP13 overexpression improves viability of H9c2 cells exposed to H/R, and inhibits H/R-induced oxidative stress by suppressing reactive oxygen species (ROS) and increasing superoxide dismutase (SOD) and catalase (CAT) function [112].
MI is common in coronary heart disease, leading to heart failure and even sudden death. During this period, the immune system primarily drives the monocyte/macrophage phenotype switch. Monocytes are recruited to necrotic sites and differentiate into subpopulations with distinct functions, to participate in the digestion of infarcted tissue and the clearance of necrotic cell debris. Excessive pro-inflammatory factors, such as TNF-α, IL-1β, myeloperoxidase (MPO), and matrix metalloproteinases (MMPs), released from classical monocyte subsets can hinder intrinsic immune repair process [113]. Zhu et al. found that CTRP3 is a “coordinator” of excessive inflammatory response after acute myocardial infarction (AMI). On the one hand, CTRP3 supplement induces an intermediate switch of monocyte subsets which alleviated inflammation. On the other hand, CTRP3 antagonizes the increased IL-6 expression in monocytes induced by LPS through inhibiting ERK1/2 and p38 MAPK pathways [114]. Therefore, CTRP3 contributes to cardiac recovery in AMI patients. Phenotypic transition of M1 to M2 macrophages is important for improving tissue damage and cardiac function in patients with MI. Liu and colleagues demonstrated overexpression CTRP9 promotes the M1 to M2 macrophage transition by inhibiting the TLR4/MD2/MyD88 and AMPK‐NF‐κB signaling, thus reducing inflammatory response and improving early cardiac function after myocardial infarction [115]. In addition to CTRP9, CTRP12 also participates in the development of myocardial infarction. Epicardial fat thickness (EFT) echocardiography is an independent measure of visceral fat, and increased EFT causes inflammation and abnormal lipid metabolism [116]. CTRP12 is a kind of anti-inflammatory cytokines and EFT of patients with acute MI was independently and negatively correlated with CTRP12 levels in clinic [117], hinting that decreased CTRP12 induced by inflammation and lipoprotein metabolism alteration may influence the process. Indeed, these results only reveal the possible direction for further research, and more precise mechanism studies are needed. In addition to ischemic heart disease and MI, CTRP9 overexpression attenuates the apoptosis, inflammation and fibrosis of H9c2 cardiomyocytes induced by CVB3 to relieve myocarditis [84].
Taken together, multiple members of CTRPs have protective effects on several heart diseases, especially in MI. Multiple CTRPs ameliorate inflammation through participating in the phenotypic switch of monocytes and macrophages. Several clinical studies have previously evaluated CTRPs as innovative biomarkers for cardiovascular disease [118], therefore they may have great potential as therapeutic targets for heart diseases.
Metabolic diseases
Metabolism consists of a series of reactions that occur within cells to sustain life and metabolic perturbations underlying many human diseases [119]. The utilization of glucose or fatty acids occurs within the cytosol and mitochondria to provide the majority of cellular energy in animals [119]. Here we will discuss the roles of CTRP family members in metabolic processes, such as insulin resistance [120], glucose metabolism [17], lipid metabolism [61], aldosterone synthesis [121] and bone metabolism [122], and the diseases related to metabolic disorders, including diabetes [123] and obesity [124].
DM is one of the most common metabolic diseases and is mainly attributed to decreased insulin sensitivity. Several members of CTRPs directly improve insulin sensitivity. In type 2 diabetes, CTRP1 and CTRP3 improves insulin sensitivity of adipocytes by improving insulin signal transduction and inhibiting inflammation [61, 125]. CTRP3 also inhibits TLR and NF-κB signaling to reduce secretion of inflammatory cytokines, thereby dampening chronic systemic anti-inflammatory responses associated with insulin resistance and obesity [41, 75]. Peterson et al. reported that recombinant CTRP3 suppressed gluconeogenic gene expression through Akt/PKB signaling, leading to improved insulin sensitivity [36]. Moreover, CTRP12 inhibits glucose generation by activating PI3K/Akt signaling and improving insulin sensitivity [126]. Therefore, CTRP1, CTRP3, CTRP12 are negative regulators of DM, but not CTRP6. Recombinant CTRP6 in adipocytes impaired the response of peripheral tissues to glucose and insulin by reducing phosphorylation and glucose uptake of insulin-stimulated Akt activation. Besides, downregulation of CTRP6 alleviates HG-induced oxidative stress, inflammation and mesangial extracellular matrix accumulation by inhibiting Akt/NF-κB pathway, thus inhibiting the progression of DM [127]. Hence, future therapeutic strategies to antagonize CTRP6 may confer beneficial clinical outcomes in the context of DM [46].
Notably, diabetes causes several complications including diabetic nephropathy (DN) and diabetic retinopathy (DR), in which CTRP3 and CTRP9 play therapeutic roles [120, 128,129,130]. DN is a major complication of diabetes that progresses to end‐stage renal disease. Hu and colleagues found that CTRP3 overexpression inhibits MCs proliferation, ROS level, and ECM production in HG‐stimulated MCs through inhibition of JAK2/STAT3 [128]. DR is one of the most common diabetic complications. CTRP3 overexpression suppressed the HG-induced oxidative stress and apoptosis via Nrf2/HO-1 pathway [130]. Besides, CTRP3 inhibits high glucose high lipid (HGHL)-induced VCAM-1 expression in an AMPK-dependent manner in human retinal microvascular endothelial cells (HRMECs), suggesting CTRP3 may play a role in the inflammatory processes of DR [129]. CTRP9 inhibits inflammation and protects the blood-retina barrier in the retina of db/db mice. In terms of mechanism, on the one hand, CTRP9 inhibits the expression of IL-1β, TNF-α, MCP-1, and adhesion molecules and balances the levels of pigment epithelial-derived factor and vascular endothelial growth factor. On the other hand, CTRP9 prevents the decomposition of the retinal blood-retinal barrier and the downregulation of tight junction protein, thus relieving the retinal vascular leakage in the early stage of diabetes [131].
Obesity is evoked by a variety of metabolic abnormalities, such as abnormal glucose metabolism and lipid metabolism, and endoplasmic reticulum (ER) stress [132]. Multiple CTRP members participate in these pathophysiological processes mentioned above. CTRP1 increases glucose utilization through stimulating glycolysis and GLUT4 translocation toward the plasma membrane, leading to enhanced glucose uptake in C2C12 myotubes [133]. In addition, CTRP3 regulates hepatic glucose output through activating Akt and ERK1/2 pathways in the liver, thereby inhibiting the expression of gluconeogenic enzymes G6Pase and PEPCK [36]. Some members also play an important role in lipid metabolism. ACC is known as the rate limiting enzyme of fatty acid synthesis and inactivated by p-AMPK. CTRP1 activates p-AMPK and inhibits ACC to increase fatty acid utilization rather than synthesis, together with the stimulation of fatty acid oxidation in C2C12 myotubes [7, 133]. CTRP3 deficiency reduces the size of lipid droplets and triglyceride (TG) in adipocytes, indicating that CTRP3 affects adipocytes adipogenesis and differentiation [42]. CTRP6 acts a bridge between obesity and energy expenditure via targeting adipocytes and macrophages, and then promotes the progress of obesity. Lei et al. reported that CTRP6 expression was markedly upregulated in adipose tissues from obese and diabetic humans and rodent models [46]. Further study found that loss of CTRP6 enhances insulin-stimulated Akt activation, metabolic rate, and energy expenditure in adipocytes [46]. CTRP9 is a key component of the metabolic network. First, CTRP9 promotes AMPK activation, skeletal muscle fat oxidation and mitochondrial biogenesis, indicating that CTRP9 enhances energy expenditure in vivo. Second, CTRP9 decreases hepatic lipid accumulation and prevents HFD-induced insulin resistance [99]. And third, CTRP9 relieves ER stress via the AMPK-mediated induction of autophagy to alleviate hepatic steatosis, thus mitigating obesity by reversing ER stress-suppressed energy expenditure [98]. Additionally, CTRP1, CTRP5, and CTRP13 have been found to function as lipid opsonins to bind lipids in synthetic liposomes, LDL, biological cell membranes, and plasma, low-density lipoprotein, cell membranes, and plasma, likely promoting unwanted lipids clearance [134].
Bone metabolism is also an important part of metabolic activities. CTRP1, CTRP3, and CTRP4 derived from adipocytes are important circulating regulators of bone metabolism. CTRP1 regulates chondrocyte proliferation and maturation via activating ERK1/2 signaling pathway [72]. Likewise, CTRP3 contributes to promotion of osteosarcoma cell proliferation via the same way as CTRP1 [135]. CTRP4 is also involved in bone metabolism. CTRP4 interference significantly inhibits the expression of osteogenic marker (Alp and Bglap), indicating CTRP4 promotes osteoblast differentiation [122]. At present, there are very few studies on CTRPs members and bone metabolic diseases.
Sepsis
Sepsis, a life-threatening organ dysfunction, is caused by a dysregulated host response to infection. Sepsis induces excessive inflammation at the early stage, then enter the stage of immune suppression, and eventually develop into multiple organ failure. A series redundant factors are involved in sepsis, such as inflammation, oxidative stress [136], and metabolic disturbance [137]. Recently, researchers found that some of CTRPs participated in the different progression of sepsis.
Biomarkers have been evaluated for several applications in patients with sepsis including diagnosis of infection, prognostication, and therapeutic guidance. Procalcitonin (PCT), CRP and IL-6 are the most widely used sepsis biomarkers [138]. CTRPs also have potential as biomarkers for sepsis. Yagmur and colleagues observed that CTRP1 in critical patients is significantly increased, and the level of circulating CTRP1 is correlated with IL-6, PCT and CRP [5]. Besides, low CTRP3 levels are correlated with overall mortality of critically ill patients. If CTRP3 level is lower than 620.6 ng/mL, the risk of death in ICU patients is significantly increased [139].
Host response to infection is involved in various cell types, especially immune cells and endothelial cells [140]. Pattern recognition receptors (PRRs) activates the innate immunity. The most studied PRRs are the TLRs, particularly TLR4. TLRs are expressed on antigen-presenting cells, especially on macrophages [141]. Cao and associates showed that macrophages expressing CTRP4 represent the essential cell types in inhibiting LPS-induced septic shock. Mechanically, CTRP4-deficient macrophages promote fatal and prolonged pro-inflammatory cytokine storms to amplify the lethal effect of septic shock. However, treatment of macrophages with exogenous CTRP4 inhibits TLR4-MyD88 pathway, eliminating the generation of pro-inflammatory cytokine storms [142]. Therefore, CTRP4 may be an effective strategy targeting macrophages for the treatment of septic shock in the future. Endothelium is responsible for interfacing between circulating blood and the parenchymal cells, functioning as a vascular barrier, vascular regulator, transcellular signaling transmitter, and hemostasis component [140]. Chen et al. demonstrated CTRP3 remarkably downregulates inflammatory cytokines levels and reduces cell apoptosis, efficiently inhibiting the inflammatory response and endothelial dysfunction [54]. CTPR9 was found to significantly reduce ROS production and increase the activities of endogenous antioxidant enzymes to protect against endothelial oxidative damage [143]. Oppositely, CTRP1 and CTRP5 aggravate endothelial injury. CTRP1 increases endothelial permeability to promote endothelial barrier dysfunction under disturbed flow. CTRP5 activates the mitochondrial apoptotic signal of endothelial cells mediated by Nox1 [144].
Targeting pathogens to retard the process of invading host immune cells is also the focus of research in critical infectious diseases such as sepsis. CTRP3 is a potent antagonist of proinflammatory nucleotide-binding oligomerization domain-containing protein 1 (NOD1) activation in adipocytes and monocyte-like cells [145]. NOD1 recognizes bacterial peptidoglycan fragments and promotes the production of pro-inflammatory cytokines. It also stimulates the production of chemokines and antimicrobial molecules and the recruitment of neutrophils to enhance immune defense. Schmid and colleagues found that, CTRP3 not only directly inhibited LPS-induced NOD1 expression in adipose tissue to suppress inflammation but also inhibited peptidoglycan-induced NOD1-dependent inflammation in adipose tissue [145]. These results suggest the ability of CTRP3 as a good potential and candidate for the treatment of acute inflammatory diseases caused by bacteria, especially Gram-negative bacteria. Furthermore, cathelicidin antimicrobial peptide, CAMP has been confirmed by ample researches in sepsis treatment. It is a class of endogenous substances that are crucial in mammalian innate immunity and has a defensive effect on microbial infections recognized by a variety of TLRs (such as TLR1/2 and TLR2/6-recognized gram-positive bacteria, TLR4-recognized gram-negative and TLR3-recognized virus) [146,147,148]. Thomas et al. found that CTRP3 could inhibit the TLR-mediated mechanism of pro-inflammatory activation of CAMP genes in adipocytes [149]. CTRP6 binds to a variety of microbial and endogenous ligands and plays an “adapter” molecule in the innate immune system to mediate complement activation [150]. CTRP6 interacts with collectin-11 (CL-11) and recruits it to acetylated BSA (AcBSA), which mediates downstream activation of the complement cascade. L-fucose, enteroaggregative Escherichia coli (EAEC) and Pseudomonas aeruginos are also ligands for CTRP6 [150]. The above results demonstrate the importance of CTRP6 in complement recognition and innate immunity, together with provide potential directions for the treatment of critical infectious diseases such as sepsis.
Notably, heart is one of the organs which are most vulnerable to sepsis. Jiang et al. first reported that CTRP1 effectively prevents sepsis-induced myocardial inflammation and oxidative damage via Sirt1-dependent pathways [109]. Additionally, overexpression of CTRP3 and CTRP12 remarkably attenuated inflammation, alleviated myocardial dysfunction and suppressed cardiac apoptosis, in order to protect against sepsis-induced cardiac injury [151, 152].
Neuroinflammation
Intracerebral hemorrhage (ICH) is a common hemorrhagic stroke with high mortality. In adult male CD1 mice with ICH, recombinant CTRP9 reduces neuroinflammatory response after cerebral hemorrhage through AdipoR1/AMPK/NF-κB signaling [153]. Thus, CTRP9 may provide a potential therapeutic strategy for alleviating neuroinflammation after ICH.
Experimental autoimmune encephalomyelitis (EAE) is a T-helper (Th) cell-mediated autoimmune disease characterized by T-cell and monocyte infiltration in the central nervous system (CNS) associated with local inflammation [154]. AdipoR2 is expressed in Th17 cells (a highly pro-inflammatory subtype of Th cells) and mainly mediates PPARα signaling pathway [155]. Transcription factor RORγt is a master regulator of Th17 cells, and is induced by STAT3 activation [156]. Masanori et al. showed that CTRP3/AdipoR2/PPARα axis suppresses Th17 cell differentiation through inhibition of the STAT3/RORγt cascade [157]. Hence, as an endogenous regulator of Th17 differentiation, the CTRP3-AdipoR2 axis hopefully becomes the target for the treatment of Th17 cell-mediated diseases such as experimental autoimmune encephalomyelitis (EAE).
Other diseases
Several CTRPs also play the critical roles in other organ injury-related diseases, such as liver disease, kidney disease, arthritis, respiratory diseases, and tendinopathy.
Nonalcoholic fatty liver disease (NAFLD) is a chronic liver disease with a high incidence and is closely associated with metabolic diseases such as diabetes, obesity and cardiovascular disease [158]. Notably, CTRP1 reduces blood glucose and improves insulin sensitivity, as well as stimulating glycolysis and fatty acid oxidation in NAFLD [133, 158]. Besides, CTRP3 suppresses oxidative stress and inflammation, together with lipid metabolism, thus reducing liver injury in particular fatty liver disease [159, 160]. These findings indicate the promises of CTRP1 and CTRP3 as a pharmacologic target in liver diseases.
CTRP3, CTRP6 and CTRP9 have potential therapeutic effects in arthritis. CTRP3 deficiency significantly exacerbated inflammation in a mouse with rheumatoid arthritis [41]. Furthermore, recombinant human CTRP6 (rhCTRP6) into intraarticular space was found to treat arthritic mice [23]. Masanori and coworkers performed daily injections of rhCTRP6 into the articular cavity of the knee joints of rheumatoid arthritis (RA) mice. They found that rhCTRP6, with the assistance of C3, decreases mRNA expression of Il1b, Tnf-α and F4/80, to improve RA [23]. Recombinant CTRP9 also alleviates osteoarthritis induced by monosodium iodoacetate (MIA) [49]. Notably, studies have shown that inappropriate mechanical stress is a possible cause of inflammatory arthritis, and tendinopathy is the most common musculoskeletal disease caused by overuse of mechanical stress [161]. It is characterized by progressive disorganization and alteration of the tendon matrix, culminating in tendon tearing and rupture, and its healing process is accompanied by inflammation and extracellular matrix (ECM) remodeling [161]. A recent study showed that CTRP3 is a booster for tendinopathy which exacerbates its progression [162]. On the one hand, it activates cartilage gene program in tendons, enhances tendon stem/progenitor cell differentiation, and alters EMC. On the other hand, CTRP3 overexpression promotes tendinopathy progression through accumulating cartilage proteoglycans and degenerating collagen fibers in mouse tendons [162].
Chronic obstructive pulmonary disease (COPD) and asthma are both respiratory diseases. The former is often along with local and systemic inflammation, while the latter is accompanied by airway inflammation. CTRP9 significantly inhibits the expression of pro-inflammatory factors TNF-α, IL-1β and IL-6 in peripheral blood mononuclear cells (PBMCs) and sputum cells of asthma patients in vitro, and reduces airway inflammation in asthma reshape [163].
Tendinopathy is a chronic tendon disease characterized by progressive disturbance and alteration of the tendon matrix. The healing process is accompanied by inflammation and extracellular matrix (ECM) remodeling [164]. CTRP3 aggravates tendinopathy, possibly contributing to the activaion of cartilage gene program in tendons, the enhancement of tendon stem/progenitor cell differentiation, as well as the accumulatation of cartilaginous proteoglycans and degenerates collagenous fibers [162].
Prospects and discussion
In addition to adiponectin, there are currently 15 family members (CTRP1–15) with homologous and highly conserved structures. Hence, CTRPs are likely derived from proliferation and differentiation of a common ancient precursor. CTRPs are also widely expressed in humans and mice, and are closely related to physiological and pathophysiological processes, such as inflammation, metabolism, and immunity (Fig. 2). Furthermore, recent new studies uncovered that several aspects of the emerging biology of CTRPs make CTPs attractive therapeutic targets (Table 2).

Herein, we described the positive or negative roles of CTRPs in the multiple organs and tissues by regulating inflammation, autophagy, apoptosis, fibrosis, oxidation, cholesterol metabolism, lipid metabolism, and endoplasmic reticulum stress (ERS).
As discussed above, CTRP1 is not only a pro-inflammatory mediator, but also an anti-inflammatory molecule. Meanwhile, CTRP1 not only promotes atherosclerosis and disrupt endothelial function, but also prevents neointima formation by inhibiting VSMCs growth after arterial injury. This apparent contradiction may be due to a number of facts as follows. First, biological effects of a specific molecule may strictly depend on the genetic background, inflammatory background, disease setting or gender. For instance, all CTRPs are released as glycoproteins, while female mice of different genetic backgrounds have significantly different levels of circulating CTRPs [1, 165]. And also, male and female mice have comparable levels of CTRP5 transcripts, but female mice have significantly higher circulating serum levels of CTRP5 than male mice [1]. Second, post-transcriptional regulation, such as alternative splicing variants of pre-mRNA, also has a significant impact on the biological function of CTRPs proteins. As mentioned above, different mice express comparable levels of CTRP5 transcripts, but exhibit different CTRP5 circulating serum levels, suggesting a potential posttranscriptional regulation of CTRP5 mRNA [1]. Third, recombinant proteins produced in mammalian cells are likely to possess native posttranslational modifications and higher-order structures to confer its different biological activities. For example, GXKG (E/D) motifs are present in the collagen domain of all CTRPs except CTRP4, CTRP12, and CTRP15 [2]. Therefore, potential influence of posttranslational modifications on assembly of higher-order CTRP structures underscores the importance of functional studies using recombinant CTRPs produced in mammalian expression systems [165]. Fourth, CTRPs may bind with each other to form a variety of homoplasmons or heterplasmons and different molecular weight forms. The existence of different molecular weights and heterogeneous binding between proteins may cause differences in effects or biological activities. Finally, the cloned expression of CTRPs may make it more diverse to some extent, such as multiple isoforms production.
Targeting therapy is currently developing rapidly and is widely used in various diseases. Once entering the host, the drug recognizes a target (usually a membrane-specific receptor protein on abnormal cells). After the binding of receptor and ligand, it directly acts on the target organ, tissue or cell, resulting in the specific death of abnormal cells without affecting the surrounding normal tissue cells. Recently, several studies on CTRP family have also focused on receptor-ligand binding. For example, Li and coworkers used ligand-receptor glycocapture technology with TriCEPS™-based ligand-receptor capture to identify putative receptors for CTRP3 in the H4IIE rat hepatoma cell line. They successfully identified lysosomal associated membrane protein 1 (LAMP-1) and lysosomal membrane protein 2 (LIMP II) as putative ligands of CTRP3 [166]. However, further investigations are needed to identify the results. Fortunately, specific ligands for CTRP1 and CTRP8 have been identified. CTRP1 is a novel ligand of receptor for advanced glycation end products (RAGE). By combining with each other, the two up-regulate arginase-1 and compete to catalyze L-arginine, resulting in the reduction of physiological NO synthesis and ultimately endothelial cell-dependent relaxation dysfunction [167]. Additionally, the GPCR relaxin/insulin-like family peptide receptor 1 (RXFP1 receptor) acts as a novel active receptor in human glioblastoma (GB). CTRP8 enhances the motility and matrix invasion of glioblastoma [168]. Physically, Glogowska et al. delineated that the N-terminal region of the globular C1q region of CTRP8 and the leucine-rich repeat units 7 and 8 of RXFP1 mediate this new ligand-receptor interaction [169]. Currently, the identified ligands are scarce, and more in-depth research on the receptor-ligand of this family is needed, which will help advance the development of highly effective targeting therapy.
Ample clinical and experimental findings have implicated a gender difference in CTRP family-associated biological responses in various organ and disease settings. Loss of CTRP4 has been shown to impair glucose tolerance in male but not female mice fed LFD. However, in female mice fed an obesogenic HFD, CTRP4 deficiency reduced body weight and adiposity [170]. Sex difference in diseases susceptibility and outcomes is often noted, and it may be due to the reasons that 1) sex hormones affect metabolic physiology by different effects on metabolic tissues; 2) sex may lead to specific differences in sympathetic activation of adipose tissue lipolysis [171]; 3) differences in plasma adipokine levels (e.g., leptin and adiponectin) between sexes may be another factor. Hence, gender differences in plasma adipokines levels may affect individual performance. In view of this, it is intriguing to discern whether other CTRPs member also have sex differences in regulation of the diseases.
Lipids have irreplaceable functions such as energy storage, energy supply, important components of biological membranes, information transmission, and conversion to generate important organic compounds (cholesterol, steroid hormones, and vitamin D3). Lipid metabolism disorder is the main cause of death from a large number of cardiovascular diseases. A recent study suggested adiponectin and CTRPs may function not only as hormones, but also as lipid opsonins. Ye and her coworkers observed that adiponectin selectively binds with several anionic phospholipids and sphingolipids through the C1q domain in an oligomerization-dependent fashion, including phosphatidylserine, ceramide-1-phosphate, glucosylceramide, and sulfatide. Otherwise, CTRP1, CTRP5, CTRP13 also bind with similar lipids and function as lipid opsonins in liposomes, low-density lipoproteins, cell membranes, and plasma [134]. In a word, CTRPs not only induce lipid clearance through this physical binding, but also act as hormones to chemically regulate lipid metabolism, such as fatty acid β-oxidation and cholesterol efflux [50]. This novel discovery denotes that adiponectin and CTRPs promote lipid clearance and limit complement-mediated inflammation. Therefore, continuing work in this area will be instrumental in building a clearer understanding of extracellular lipid handling and its role in inflammation-related and lipid metabolism diseases.
Taken together, an increasing number of studies on CTRPs in multiple inflammation-related diseases have been surfaced over the last years. CTRPs can directly or indirectly regulate insulin signal transduction, energy metabolism and inflammation, thus reflecting profound biological potency in multiple cell and tissue types (Fig. 3). However, few CTRPs members have been examined clinically to-date. More large-scale clinical studies are needed to evaluate the translational values of CTRPs in the diagnosis and treatment of human diseases.

CTRP1-mediated pathways: CTRP1 increases ERK1/2 phosphorylation, thus contributing to the proliferation of immature chondrocytes; CTRP1 promotes the binding of S1P to its receptor, thereby accelerating the accumulation of cAMP in cardiac fibroblasts; CTRP1 phosphorylates AMPK at Thr-17 and activated AMPK phosphorylates ACC at Ser-79, then AMPK/ACC signaling pathway decreases the concentration of malonyl-CoA, thus enhancing the oxidation of long-chain fatty acids in skeletal muscle. CTRP3-mediated pathways: CTRP3 inhibits phosphorylation of p38 and CREB, and reduces the downstream molecules to attenuate p38-induced ER stress in the heart; CTRP3 exerts anti-inflammatory and anti-fibrotic effects by inhibiting TGF-β production and the expression of CTGF and collagen I in primary human CLPFs; CTRP3 reduces TGF-β and inactivates Src and Smad3, alleviating fibrosis and inflammatory response in mesangial cells. CTRP6-mediated pathways: CTRP6 activates PI3K/Akt signaling to mitigate the increased production of ROS and MDA in HK-2 cells, thus reducing renal I/R injury. CTRP9-mediated pathways: CTRP9 activates the PI3K/Akt pathway and increases eNOS expression, thus attenuating inflammation and improving endothelial cell survival and function; CTRP9 enhances AMPK phosphorylation and upregulates the expression of LXR, ABCA1 and ABCG1 in VSMCs, thus regulating cholesterol metabolism.
References
Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J. 2008;416:161–77.
Wong GW, Wang J, Hug C, Tsao TS, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci USA. 2004;101:10302–7.
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–9.
Choi KM, Hwang SY, Hong HC, Yang SJ, Choi HY, Yoo HJ, et al. C1q/TNF-related protein-3 (CTRP-3) and pigment epithelium-derived factor (PEDF) concentrations in patients with type 2 diabetes and metabolic syndrome. Diabetes. 2012;61:2932–6.
Yagmur E, Buergerhausen D, Koek GH, Weiskirchen R, Trautwein C, Koch A, et al. Elevated CTRP1 plasma concentration is associated with sepsis and pre-existing type 2 diabetes mellitus in critically Ill patients. J Clin Med. 2019;8:661.
Barbieri D, Goicoechea M, Sánchez-Niño MD, Ortiz A, Verde E, Verdalles U, et al. Obesity and chronic kidney disease progression-the role of a new adipocytokine: C1q/tumour necrosis factor-related protein-1. Clin Kidney J. 2019;12:420–26.
Rodriguez S, Lei X, Petersen PS, Tan SY, Little HC, Wong GW. Loss of CTRP1 disrupts glucose and lipid homeostasis. Am J Physiol Endocrinol Metab. 2016;311:E678–e97.
Li C, Chen JW, Liu ZH, Shen Y, Ding FH, Gu G, et al. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis. 2018;278:197–209.
Wang XQ, Liu ZH, Xue L, Lu L, Gao J, Shen Y, et al. C1q/TNF-related protein 1 links macrophage lipid metabolism to inflammation and atherosclerosis. Atherosclerosis. 2016;250:38–45.
Vester SK, Beavil RL, Lynham S, Beavil AJ, Cunninghame Graham DS, McDonnell JM, et al. Nucleolin acts as the receptor for C1QTNF4 and supports C1QTNF4-mediated innate immunity modulation. J Biol Chem. 2021;296:100513.
Kishore U, Gaboriaud C, Waters P, Shrive AK, Greenhough TJ, Reid KB, et al. C1q and tumor necrosis factor superfamily: modularity and versatility. Trends Immunol. 2004;25:551–61.
Schäffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab. 2012;23:194–204.
Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology. 2000;49:159–70.
Shanaki M, Shabani P, Goudarzi A, Omidifar A, Bashash D, Emamgholipour S. The C1q/TNF-related proteins (CTRPs) in pathogenesis of obesity-related metabolic disorders: Focus on type 2 diabetes and cardiovascular diseases. Life Sci. 2020;256:117913.
Majidi Z, Emamgholipour S, Omidifar A, Rahmani Fard S, Poustchi H, Shanaki M. The circulating levels of CTRP1 and CTRP5 are associated with obesity indices and carotid intima-media thickness (cIMT) value in patients with type 2 diabetes: a preliminary study. Diabetol Metab Syndr. 2021;13:14.
Yang Y, Liu S, Zhang RY, Luo H, Chen L, He WF, et al. Association between C1q/TNF-related protein-1 levels in human plasma and epicardial adipose tissues and congestive heart failure. Cell Physiol Biochem. 2017;42:2130–43.
Li L, Aslam M, Siegler BH, Niemann B, Rohrbach S. Comparative analysis of CTRP-mediated effects on cardiomyocyte glucose metabolism: cross talk between AMPK and Akt signaling pathway. Cells. 2021;10:905.
Wan Y, Wang J, Yang B, Huang C, Tang X, Yi H, et al. Effects and mechanisms of CTRP3 overexpression in secondary brain injury following intracerebral hemorrhage in rats. Exp Ther Med. 2022;23:35.
Yavuz YC, Altınkaynak K, Sevinc C, Ozbek Sebin S, Baydar I. The cartonectin levels at different stages of chronic kidney disease and related factors. Ren Fail. 2019;41:42–46.
Li Q, Wang L, Tan W, Peng Z, Luo Y, Zhang Y, et al. Identification of C1qTNF-related protein 4 as a potential cytokine that stimulates the STAT3 and NF-κB pathways and promotes cell survival in human cancer cells. Cancer Lett. 2011;308:203–14.
Mandal MN, Vasireddy V, Reddy GB, Wang X, Moroi SE, Pattnaik BR, et al. CTRP5 is a membrane-associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Invest Ophthalmol Vis Sci. 2006;47:5505–13.
Liu J, Yan X, Wang Z, Zhang N, Lin A, Li Z. Adipocyte factor CTRP6 inhibits homocysteine-induced proliferation, migration, and dedifferentiation of vascular smooth muscle cells through PPARγ/NLRP3. Biochem Cell Biol. 2021;99:596–605.
Murayama MA, Kakuta S, Inoue A, Umeda N, Yonezawa T, Maruhashi T, et al. CTRP6 is an endogenous complement regulator that can effectively treat induced arthritis. Nat Commun. 2015;6:8483.
Qu LH, Hong X, Zhang Y, Cong X, Xiang RL, Mei M, et al. C1q/tumor necrosis factor-related protein-6 attenuates TNF-α-induced apoptosis in salivary acinar cells via AMPK/SIRT1-modulated miR-34a-5p expression. J Cell Physiol. 2021;236:5785–800.
Appari M, Breitbart A, Brandes F, Szaroszyk M, Froese N, Korf-Klingebiel M, et al. C1q-TNF-related protein-9 promotes cardiac hypertrophy and failure. Circ Res. 2017;120:66–77.
Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Ge G, Spooner E, Hug C, et al. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 2009;23:241–58.
Wei Z, Seldin MM, Natarajan N, Djemal DC, Peterson JM, Wong GW. C1q/tumor necrosis factor-related protein 11 (CTRP11), a novel adipose stroma-derived regulator of adipogenesis. J Biol Chem. 2013;288:10214–29.
Omidifar A, Toolabi K, Rahimipour A, Emamgholipour S, Shanaki M. The gene expression of CTRP12 but not CTRP13 is upregulated in both visceral and subcutaneous adipose tissue of obese subjects. Diabetes Metab Syndr. 2019;13:2593–99.
Tan BK, Lewandowski KC, O’Hare JP, Randeva HS. Insulin regulates the novel adipokine adipolin/CTRP12: in vivo and ex vivo effects. J Endocrinol. 2014;221:111–9.
Wei Z, Peterson JM, Wong GW. Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): activation OF AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J Biol Chem. 2011;286:15652–65.
Zhang Q, Niu X, Tian L, Liu J, Niu R, Quan J, et al. CTRP13 attenuates the expression of LN and CAV-1 Induced by high glucose via CaMKKβ/AMPK pathway in rLSECs. Aging. 2020;12:11485–99.
Recinella L, Orlando G, Ferrante C, Chiavaroli A, Brunetti L, Leone S. Adipokines: new potential therapeutic target for obesity and metabolic, rheumatic, and cardiovascular diseases. Front Physiol. 2020;11:578966.
Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5’ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1667–71.
Tsao TS, Murrey HE, Hug C, Lee DH, Lodish HF. Oligomerization state-dependent activation of NF-kappa B signaling pathway by adipocyte complement-related protein of 30 kDa (Acrp30). J Biol Chem. 2002;277:29359–62.
Tsao TS, Tomas E, Murrey HE, Hug C, Lee DH, Ruderman NB, et al. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. Different oligomers activate different signal transduction pathways. J Biol Chem. 2003;278:50810–7.
Peterson JM, Wei Z, Wong GW. C1q/TNF-related protein-3 (CTRP3), a novel adipokine that regulates hepatic glucose output. J Biol Chem. 2010;285:39691–701.
Weigert J, Neumeier M, Schäffler A, Fleck M, Schölmerich J, Schütz C, et al. The adiponectin paralog CORS-26 has anti-inflammatory properties and is produced by human monocytic cells. FEBS Lett. 2005;579:5565–70.
Peterson JM, Wei Z, Wong GW. CTRP8 and CTRP9B are novel proteins that hetero-oligomerize with C1q/TNF family members. Biochem Biophys Res Commun. 2009;388:360–5.
Kim KY, Kim HY, Kim JH, Lee CH, Kim DH, Lee YH, et al. Tumor necrosis factor-alpha and interleukin-1beta increases CTRP1 expression in adipose tissue. FEBS Lett. 2006;580:3953–60.
Li Y, Wright GL, Peterson JM. C1q/TNF-related protein 3 (CTRP3) function and regulation. Compr Physiol. 2017;7:863–78.
Petersen PS, Wolf RM, Lei X, Peterson JM, Wong GW. Immunomodulatory roles of CTRP3 in endotoxemia and metabolic stress. Physiol Rep. 2016;4:e12735.
Kopp A, Bala M, Buechler C, Falk W, Gross P, Neumeier M, et al. C1q/TNF-related protein-3 represents a novel and endogenous lipopolysaccharide antagonist of the adipose tissue. Endocrinology. 2010;151:5267–78.
Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–27.
Naïmi M, Gautier N, Chaussade C, Valverde AM, Accili D, Van Obberghen E. Nuclear forkhead box O1 controls and integrates key signaling pathways in hepatocytes. Endocrinology. 2007;148:2424–34.
Yoo HJ, Hwang SY, Hong HC, Choi HY, Yang SJ, Choi DS, et al. Implication of progranulin and C1q/TNF-related protein-3 (CTRP3) on inflammation and atherosclerosis in subjects with or without metabolic syndrome. PLoS One. 2013;8:e55744.
Lei X, Seldin MM, Little HC, Choy N, Klonisch T, Wong GW. C1q/TNF-related protein 6 (CTRP6) links obesity to adipose tissue inflammation and insulin resistance. J Biol Chem. 2017;292:14836–50.
Huang Z, Zhao D, Wang Y, Li X, Li J, Han J, et al. C1q/TNF-related protein 9 decreases cardiomyocyte hypoxia/reoxygenation-induced inflammation by inhibiting the TLR4/MyD88/NF-κB signaling pathway. Exp Ther Med. 2021;22:1139.
Li Y, Geng X, Wang H, Cheng G, Xu S. CTRP9 ameliorates pulmonary arterial hypertension through attenuating inflammation and improving endothelial cell survival and function. J Cardiovasc Pharmacol. 2016;67:394–401.
Zheng S, Ren J, Gong S, Qiao F, He J. CTRP9 protects against MIA-induced inflammation and knee cartilage damage by deactivating the MAPK/NF-κB pathway in rats with osteoarthritis. Open Life Sci. 2020;15:971–80.
Lei S, Chen J, Song C, Li J, Zuo A, Xu D, et al. CTRP9 alleviates foam cells apoptosis by enhancing cholesterol efflux. Mol Cell Endocrinol. 2021;522:111138.
Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019–31.
Wang Y, Zhang H. Regulation of autophagy by mTOR signaling pathway. Adv Exp Med Biol. 2019;1206:67–83.
Wang H, Liu Q, Zhang X. C1q/tumor necrosis factor-related protein-1 attenuates microglia autophagy and inflammatory response by regulating the Akt/mTOR pathway. Life Sci. 2020;256:117992.
Chen L, Qin L, Liu X, Meng X. CTRP3 alleviates Ox-LDL-induced inflammatory response and endothelial dysfunction in mouse aortic endothelial cells by activating the PI3K/Akt/eNOS pathway. Inflammation. 2019;42:1350–59.
Si Y, Fan W, Sun L. A review of the relationship between CTRP family and coronary artery disease. Curr Atheroscler Rep. 2020;22:22.
Xiang H, Xue W, Li Y, Zheng J, Ding C, Dou M, et al. C1q/TNF-related protein 6 (CTRP6) attenuates renal ischaemia-reperfusion injury through the activation of PI3K/Akt signalling pathway. Clin Exp Pharmacol Physiol. 2020;47:1030–40.
Zhao Q, Zhang CL, Xiang RL, Wu LL, Li L. CTRP15 derived from cardiac myocytes attenuates TGFβ1-induced fibrotic response in cardiac fibroblasts. Cardiovasc Drugs Ther. 2020;34:591–604.
Paskeh MDA, Asadi A, Mirzaei S, Hashemi M, Entezari M, Raesi R, et al. Targeting AMPK signaling in ischemic/reperfusion injury: From molecular mechanism to pharmacological interventions. Cell Signal. 2022;94:110323.
Townley R, Shapiro L. Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science. 2007;315:1726–9.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62.
Peterson JM, Aja S, Wei Z, Wong GW. CTRP1 protein enhances fatty acid oxidation via AMP-activated protein kinase (AMPK) activation and acetyl-CoA carboxylase (ACC) inhibition. J Biol Chem. 2012;287:1576–87.
Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J Biol Chem. 1994;269:22162–8.
Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–12.
McGarry JD, Leatherman GF, Foster DW. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J Biol Chem. 1978;253:4128–36.
Park SY, Choi JH, Ryu HS, Pak YK, Park KS, Lee HK, et al. C1q tumor necrosis factor alpha-related protein isoform 5 is increased in mitochondrial DNA-depleted myocytes and activates AMP-activated protein kinase. J Biol Chem. 2009;284:27780–89.
Liu Q, Zhang H, Lin J, Zhang R, Chen S, Liu W, et al. C1q/TNF-related protein 9 inhibits the cholesterol-induced Vascular smooth muscle cell phenotype switch and cell dysfunction by activating AMP-dependent kinase. J Cell Mol Med. 2017;21:2823–36.
Zhang H, Gong X, Ni S, Wang Y, Zhu L, Ji N. C1q/TNF-related protein-9 attenuates atherosclerosis through AMPK-NLRP3 inflammasome singling pathway. Int Immunopharmacol. 2019;77:105934.
Liu B, Zhang R, Wei S, Yuan Q, Xue M, Hao P, et al. ALDH2 protects against alcoholic cardiomyopathy through a mechanism involving the p38 MAPK/CREB pathway and local renin-angiotensin system inhibition in cardiomyocytes. Int J Cardiol. 2018;257:150–59.
Wei WY, Ma ZG, Xu SC, Zhang N, Tang QZ. Pioglitazone protected against cardiac hypertrophy via inhibiting AKT/GSK3β and MAPK signaling pathways. PPAR Res. 2016;2016:9174190.
Li L, Fan D, Wang C, Wang JY, Cui XB, Wu D, et al. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc Res. 2011;91:80–9.
Zhang B, Zhang P, Tan Y, Feng P, Zhang Z, Liang H, et al. C1q-TNF-related protein-3 attenuates pressure overload-induced cardiac hypertrophy by suppressing the p38/CREB pathway and p38-induced ER stress. Cell Death Dis. 2019;10:520.
Akiyama H, Otani M, Sato S, Toyosawa S, Furukawa S, Wakisaka S, et al. A novel adipokine C1q/TNF-related protein 1 (CTRP1) regulates chondrocyte proliferation and maturation through the ERK1/2 signaling pathway. Mol Cell Endocrinol. 2013;369:63–71.
Liao J, Barthel A, Nakatani K, Roth RA. Activation of protein kinase B/Akt is sufficient to repress the glucocorticoid and cAMP induction of phosphoenolpyruvate carboxykinase gene. J Biol Chem. 1998;273:27320–4.
Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–36.
Schmid A, Kopp A, Hanses F, Karrasch T, Schäffler A. C1q/TNF-related protein-3 (CTRP-3) attenuates lipopolysaccharide (LPS)-induced systemic inflammation and adipose tissue Erk-1/-2 phosphorylation in mice in vivo. Biochem Biophys Res Commun. 2014;452:8–13.
Cartier A, Hla T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science. 2019;366:eaar5551.
Tsai HC, Han MH. Sphingosine-1-phosphate (S1P) and S1P signaling pathway: therapeutic targets in autoimmunity and inflammation. Drugs. 2016;76:1067–79.
Yuasa D, Ohashi K, Shibata R, Mizutani N, Kataoka Y, Kambara T, et al. C1q/TNF-related protein-1 functions to protect against acute ischemic injury in the heart. FASEB J. 2016;30:1065–75.
Damirin A, Tomura H, Komachi M, Tobo M, Sato K, Mogi C, et al. Sphingosine 1-phosphate receptors mediate the lipid-induced cAMP accumulation through cyclooxygenase-2/prostaglandin I2 pathway in human coronary artery smooth muscle cells. Mol Pharm. 2005;67:1177–85.
Hofmann C, Chen N, Obermeier F, Paul G, Büchler C, Kopp A, et al. C1q/TNF-related protein-3 (CTRP-3) is secreted by visceral adipose tissue and exerts antiinflammatory and antifibrotic effects in primary human colonic fibroblasts. Inflamm Bowel Dis. 2011;17:2462–71.
Zhang R, Zhong L, Zhou J, Peng Y. Complement-C1q TNF-related protein 3 alleviates mesangial cell activation and inflammatory response stimulated by secretory IgA. Am J Nephrol. 2016;43:460–8.
Chu LY, Ramakrishnan DP, Silverstein RL. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood. 2013;122:1822–32.
Roberts DD, Miller TW, Rogers NM, Yao M, Isenberg JS. The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biol. 2012;31:162–9.
Liu K, Wang J, Gao X, Ren W. C1q/TNF-related protein 9 inhibits coxsackievirus B3-induced injury in cardiomyocytes through NF-κB and TGF-β1/Smad2/3 by modulating THBS1. Mediators Inflamm. 2020;2020:2540687.
Wu XM, Zhang N, Li JS, Yang ZH, Huang XL, Yang XF. Purinergic receptors mediate endothelial dysfunction and participate in atherosclerosis. Purinergic Signal. 2022. https://doi.org/10.1007/s11302-021-09839-x. Online ahead of print.
Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120:713–35.
Price DT, Loscalzo J. Cellular adhesion molecules and atherogenesis. Am J Med. 1999;107:85–97.
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604.
Liao XH, Wang N, Zhao DW, Zheng DL, Zheng L, Xing WJ, et al. STAT3 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem. 2015;290:19641–52.
Fang S, Xu Y, Zhang Y, Tian J, Li J, Li Z, et al. Irgm1 promotes M1 but not M2 macrophage polarization in atherosclerosis pathogenesis and development. Atherosclerosis. 2016;251:282–90.
Nakamura R, Sene A, Santeford A, Gdoura A, Kubota S, Zapata N, et al. IL10-driven STAT3 signalling in senescent macrophages promotes pathological eye angiogenesis. Nat Commun. 2015;6:7847.
Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64:2028–41.
Lu L, Zhang RY, Wang XQ, Liu ZH, Shen Y, Ding FH, et al. C1q/TNF-related protein-1: an adipokine marking and promoting atherosclerosis. Eur Heart J. 2016;37:1762–71.
Shen Y, Li C, Zhang RY, Zhang Q, Shen WF, Ding FH, et al. Association of increased serum CTRP5 levels with in-stent restenosis after coronary drug-eluting stent implantation: CTRP5 promoting inflammation, migration and proliferation in vascular smooth muscle cells. Int J Cardiol. 2017;228:129–36.
Ogawa H, Ohashi K, Ito M, Shibata R, Kanemura N, Yuasa D, et al. Adipolin/CTRP12 protects against pathological vascular remodelling through suppression of smooth muscle cell growth and macrophage inflammatory response. Cardiovasc Res. 2020;116:237–49.
Choi KM, Hwang SY, Hong HC, Choi HY, Yoo HJ, Youn BS, et al. Implications of C1q/TNF-related protein-3 (CTRP-3) and progranulin in patients with acute coronary syndrome and stable angina pectoris. Cardiovasc Diabetol. 2014;13:14.
Zhang L, Liu Q, Zhang H, Wang XD, Chen SY, Yang Y, et al. C1q/TNF-related protein 9 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. J Cardiovasc Pharmacol. 2018;72:167–75.
Jung TW, Hong HC, Hwang HJ, Yoo HJ, Baik SH, Choi KM. C1q/TNF-related protein 9 (CTRP9) attenuates hepatic steatosis via the autophagy-mediated inhibition of endoplasmic reticulum stress. Mol Cell Endocrinol. 2015;417:131–40.
Peterson JM, Wei Z, Seldin MM, Byerly MS, Aja S, Wong GW. CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction. Am J Physiol Regul Integr Comp Physiol. 2013;305:R522–33.
Sanchez-Lopez E, Zhong Z, Stubelius A, Sweeney SR, Booshehri LM, Antonucci L, et al. Choline uptake and metabolism modulate macrophage IL-1β and IL-18 production. Cell Metab. 2019;29:1350–62. e7
Mohamadinarab M, Ahmadi R, Gholamrezayi A, Rahvar F, Naghdalipour M, Setayesh L, et al. Serum levels of C1q/TNF-related protein-3 in inflammatory bowel disease patients and its inverse association with inflammatory cytokines and insulin resistance. IUBMB Life. 2020;72:1698–704.
Raghavan S, Singh NK, Gali S, Mani AM, Rao GN. Protein kinase Cθ via activating transcription factor 2-mediated CD36 expression and foam cell formation of Ly6C(hi) cells contributes to atherosclerosis. Circulation. 2018;138:2395–412.
Wang C, Xu W, Liang M, Huang D, Huang K. CTRP13 inhibits atherosclerosis via autophagy-lysosome-dependent degradation of CD36. FASEB J. 2019;33:2290–300.
Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21.
Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–67.
Davis FM, Tsoi LC, Melvin WJ, denDekker A, Wasikowski R, Joshi AD, et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J Exp Med. 2021;218:e20201839.
Song H, Yang Y, Sun Y, Wei G, Zheng H, Chen Y, et al. Circular RNA Cdyl promotes abdominal aortic aneurysm formation by inducing M1 macrophage polarization and M1-type inflammation. Mol Ther. 2022;30:915–31.
Xu W, Chao Y, Liang M, Huang K, Wang C. CTRP13 mitigates abdominal aortic aneurysm formation via NAMPT1. Mol Ther. 2021;29:324–37.
Jiang W, Li W, Hu X, Hu R, Li B, Lan L. CTRP1 prevents sepsis-induced cardiomyopathy via Sirt1-dependent pathways. Free Radic Biol Med. 2020;152:810–20.
Yi W, Sun Y, Yuan Y, Lau WB, Zheng Q, Wang X, et al. C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation. 2012;125:3159–69.
Jin AP, Zhang QR, Yang CL, Ye S, Cheng HJ, Zheng YY. Up-regulation of CTRP12 ameliorates hypoxia/re-oxygenation-induced cardiomyocyte injury by inhibiting apoptosis, oxidative stress, and inflammation via the enhancement of Nrf2 signaling. Hum Exp Toxicol. 2021;40:2087–98.
Jiang W, Song J, Zhang S, Ye Y, Wang J, Zhang Y. CTRP13 protects H9c2 cells against hypoxia/reoxygenation (H/R)-induced injury via regulating the AMPK/Nrf2/ARE signaling pathway. Cell Transpl. 2021;30:9636897211033275.
Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–45.
Zhu H, Ding Y, Zhang Y, Ding X, Zhao J, Ouyang W, et al. CTRP3 induces an intermediate switch of CD14(++)CD16(+) monocyte subset with anti-inflammatory phenotype. Exp Ther Med. 2020;19:2243–51.
Liu M, Yin L, Li W, Hu J, Wang H, Ye B, et al. C1q/TNF-related protein-9 promotes macrophage polarization and improves cardiac dysfunction after myocardial infarction. J Cell Physiol. 2019;234:18731–47.
Iacobellis G, Willens HJ. Echocardiographic epicardial fat: a review of research and clinical applications. J Am Soc Echocardiogr. 2009;22:1311–9. quiz 417-8
Babapour B, Doustkami H, Avesta L, Moradi A, Saadat S, Piralaei K, et al. Correlation of serum adipolin with epicardial fat thickness and severity of coronary artery diseases in acute myocardial infarction and stable angina pectoris patients. Med Princ Pr. 2021;30:52–61.
Wu L, Gao L, Zhang D, Yao R, Huang Z, Du B, et al. C1QTNF1 attenuates angiotensin II-induced cardiac hypertrophy via activation of the AMPKa pathway. Free Radic Biol Med. 2018;121:215–30.
Judge A, Dodd MS. Metabolism. Essays Biochem. 2020;64:607–47.
Moradi N, Fadaei R, Khamseh ME, Nobakht A, Rezaei MJ, Aliakbary F, et al. Serum levels of CTRP3 in diabetic nephropathy and its relationship with insulin resistance and kidney function. PLoS One. 2019;14:e0215617.
Jeon JH, Kim KY, Kim JH, Baek A, Cho H, Lee YH, et al. A novel adipokine CTRP1 stimulates aldosterone production. FASEB J. 2008;22:1502–11.
Li Q, Wu J, Xi W, Chen X, Wang W, Zhang T, et al. Ctrp4, a new adipokine, promotes the differentiation of osteoblasts. Biochem Biophys Res Commun. 2019;512:224–29.
Bai B, Ban B, Liu Z, Zhang MM, Tan BK, Chen J. Circulating C1q complement/TNF-related protein (CTRP) 1, CTRP9, CTRP12 and CTRP13 concentrations in Type 2 diabetes mellitus: In vivo regulation by glucose. PLoS One. 2017;12:e0172271.
Guo B, Zhuang T, Xu F, Lin X, Li F, Shan SK, et al. New Insights Into Implications of CTRP3 in Obesity, Metabolic Dysfunction, and Cardiovascular Diseases: Potential of Therapeutic Interventions. Front Physiol. 2020;11:570270.
Li X, Jiang L, Yang M, Wu YW, Sun JZ, Sun SX. CTRP3 improves the insulin sensitivity of 3T3-L1 adipocytes by inhibiting inflammation and ameliorating insulin signalling transduction. Endokrynol Pol. 2014;65:252–8.
Wei Z, Peterson JM, Lei X, Cebotaru L, Wolfgang MJ, Baldeviano GC, et al. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem. 2012;287:10301–15.
Xu E, Yin C, Yi X, Liu Y. Knockdown of CTRP6 inhibits high glucose-induced oxidative stress, inflammation and extracellular matrix accumulation in mesangial cells through regulating the Akt/NF-κB pathway. Clin Exp Pharmacol Physiol. 2020;47:1203–11.
Hu TY, Li LM, Pan YZ. CTRP3 inhibits high glucose-induced human glomerular mesangial cell dysfunction. J Cell Biochem. 2019;120:5729–36.
Yan Z, Zhao J, Gan L, Zhang Y, Guo R, Cao X, et al. CTRP3 is a novel biomarker for diabetic retinopathy and inhibits HGHL-induced VCAM-1 expression in an AMPK-dependent manner. PLoS One. 2017;12:e0178253.
Zhang J, He J. CTRP3 inhibits high glucose-induced oxidative stress and apoptosis in retinal pigment epithelial cells. Artif Cells Nanomed Biotechnol. 2019;47:3758–64.
Li W, Ma N, Liu MX, Ye BJ, Li YJ, Hu HY, et al. C1q/TNF-related protein-9 attenuates retinal inflammation and protects blood-retinal barrier in db/db mice. Eur J Pharmacol. 2019;853:289–98.
Ajoolabady A, Liu S, Klionsky DJ, Lip GYH, Tuomilehto J, Kavalakatt S, et al. ER stress in obesity pathogenesis and management. Trends Pharmacol Sci. 2022;43:97–109.
Han S, Park JS, Lee S, Jeong AL, Oh KS, Ka HI, et al. CTRP1 protects against diet-induced hyperglycemia by enhancing glycolysis and fatty acid oxidation. J Nutr Biochem. 2016;27:43–52.
Ye JJ, Bian X, Lim J, Medzhitov R. Adiponectin and related C1q/TNF-related proteins bind selectively to anionic phospholipids and sphingolipids. Proc Natl Acad Sci USA. 2020;117:17381–88.
Akiyama H, Furukawa S, Wakisaka S, Maeda T. Elevated expression of CTRP3/cartducin contributes to promotion of osteosarcoma cell proliferation. Oncol Rep. 2009;21:1477–81.
Santos SS, Brunialti MK, Rigato O, Machado FR, Silva E, Salomao R. Generation of nitric oxide and reactive oxygen species by neutrophils and monocytes from septic patients and association with outcomes. Shock. 2012;38:18–23.
Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274:330–53.
Pierrakos C, Velissaris D, Bisdorff M, Marshall JC, Vincent JL. Biomarkers of sepsis: time for a reappraisal. Crit Care. 2020;24:287.
Yagmur E, Otto S, Koek GH, Weiskirchen R, Trautwein C, Koch A, et al. Decreased CTRP3 plasma concentrations are associated with sepsis and predict mortality in critically Ill patients. Diagnostics. 2019;9:63.
Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascón GA, et al. The endothelium in sepsis. Shock. 2016;45:259–70.
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95.
Cao L, Tan W, Chen W, Huang H, He M, Li Q, et al. CTRP4 acts as an anti-inflammatory factor in macrophages and protects against endotoxic shock. Eur J Immunol. 2021;51:380–92.
Cheng L, Li B, Chen X, Su J, Wang H, Yu S, et al. CTRP9 induces mitochondrial biogenesis and protects high glucose-induced endothelial oxidative damage via AdipoR1 -SIRT1- PGC-1α activation. Biochem Biophys Res Commun. 2016;477:685–91.
Liu J, Meng Z, Gan L, Guo R, Gao J, Liu C, et al. C1q/TNF-related protein 5 contributes to diabetic vascular endothelium dysfunction through promoting Nox-1 signaling. Redox Biol. 2020;34:101476.
Schmid A, Schäffler A, Karrasch T. CTRP-3 regulates NOD1-mediated inflammation and NOD1 expression in adipocytes and adipose tissue. Inflammation. 2021;44:2260–69.
Ho J, Chan H, Liang Y, Liu X, Zhang L, Li Q, et al. Cathelicidin preserves intestinal barrier function in polymicrobial sepsis. Crit Care. 2020;24:47.
Kumagai Y, Murakami T, Kuwahara A, Iba T, Reich J, Nagaoka I. Antimicrobial peptide LL-37 ameliorates a murine sepsis model via the induction of microvesicle release from neutrophils. Innate Immun. 2020;26:565–79.
Nagaoka I, Tamura H, Reich J. Therapeutic potential of cathelicidin peptide LL-37, an antimicrobial agent, in a murine sepsis model. Int J Mol Sci. 2020;21:5973.
Karrasch T, Höpfinger A, Schäffler A, Schmid A. The adipokine C1q/TNF-related protein-3 (CTRP-3) inhibits Toll-like receptor (TLR)-induced expression of Cathelicidin antimicrobial peptide (CAMP) in adipocytes. Cytokine. 2021;148:155663.
Kirketerp-Møller N, Bayarri-Olmos R, Krogfelt KA, Garred P. C1q/TNF-related protein 6 is a pattern recognition molecule that recruits collectin-11 from the complement system to ligands. J Immunol. 2020;204:1598–606.
Wei WY, Ma ZG, Zhang N, Xu SC, Yuan YP, Zeng XF, et al. Overexpression of CTRP3 protects against sepsis-induced myocardial dysfunction in mice. Mol Cell Endocrinol. 2018;476:27–36.
Zhou MQ, Jin E, Wu J, Ren F, Yang YZ, Duan DD. CTRP12 ameliorated lipopolysaccharide-induced cardiomyocyte injury. Chem Pharm Bull. 2020;68:133–39.
Zhao L, Chen S, Sherchan P, Ding Y, Zhao W, Guo Z, et al. Recombinant CTRP9 administration attenuates neuroinflammation via activating adiponectin receptor 1 after intracerebral hemorrhage in mice. J Neuroinflammation. 2018;15:215.
Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol. 2014;122:173–89.
Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–9.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33.
Murayama MA, Chi HH, Matsuoka M, Ono T, Iwakura Y. The CTRP3-AdipoR2 axis regulates the development of experimental autoimmune encephalomyelitis by suppressing Th17 cell differentiation. Front Immunol. 2021;12:607346.
Shabani P, Emamgholipour S, Doosti M. CTRP1 in liver disease. Adv Clin Chem. 2017;79:1–23.
Wolf RM, Lei X, Yang ZC, Nyandjo M, Tan SY, Wong GW. CTRP3 deficiency reduces liver size and alters IL-6 and TGFβ levels in obese mice. Am J Physiol Endocrinol Metab. 2016;310:E332–45.
Zhang J, Xu J, Lin X, Tang F, Tan L. CTRP3 ameliorates fructose-induced metabolic associated fatty liver disease via inhibition of xanthine oxidase-associated oxidative stress. Tissue Cell. 2021;72:101595.
Gracey E, Burssens A, Cambré I, Schett G, Lories R, McInnes IB, et al. Tendon and ligament mechanical loading in the pathogenesis of inflammatory arthritis. Nat Rev Rheumatol. 2020;16:193–207.
Cho Y, Kim HS, Kang D, Kim H, Lee N, Yun J, et al. CTRP3 exacerbates tendinopathy by dysregulating tendon stem cell differentiation and altering extracellular matrix composition. Sci Adv. 2021;7:eabg6069.
Qian M, Yang Q, Li J, Zhao B, Zhang Y, Zhao Y. C1q/TNF-related protein-9 alleviates airway inflammation in asthma. Int Immunopharmacol. 2020;81:106238.
Millar NL, Silbernagel KG, Thorborg K, Kirwan PD, Galatz LM, Abrams GD, et al. Tendinopathy. Nat Rev Dis Prim. 2021;7:1.
Seldin MM, Tan SY, Wong GW. Metabolic function of the CTRP family of hormones. Rev Endocr Metab Disord. 2014;15:111–23.
Li Y, Ozment T, Wright GL, Peterson JM. Identification of putative receptors for the novel adipokine CTRP3 using ligand-receptor capture technology. PLoS One. 2016;11:e0164593.
Liu ZH. The molecular mechanisms by which RAGE new ligand CTRP1 and HMGB2 promote the progression of coronary artery disease. Chinese Doctoral Dissertations Full-text Database. 2016;5.
Klonisch T, Glogowska A, Thanasupawat T, Burg M, Krcek J, Pitz M, et al. Structural commonality of C1q TNF-related proteins and their potential to activate relaxin/insulin-like family peptide receptor 1 signalling pathways in cancer cells. Br J Pharm. 2017;174:1025–33.
Glogowska A, Kunanuvat U, Stetefeld J, Patel TR, Thanasupawat T, Krcek J, et al. C1q-tumour necrosis factor-related protein 8 (CTRP8) is a novel interaction partner of relaxin receptor RXFP1 in human brain cancer cells. J Pathol. 2013;231:466–79.
Sarver DC, Stewart AN, Rodriguez S, Little HC, Aja S, Wong GW. Loss of CTRP4 alters adiposity and food intake behaviors in obese mice. Am J Physiol Endocrinol Metab. 2020;319:E1084–e100.
Di Florio DN, Sin J, Coronado MJ, Atwal PS, Fairweather D. Sex differences in inflammation, redox biology, mitochondria and autoimmunity. Redox Biol. 2020;31:101482.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81871607 and 82070422), Youth Science and Technology Rising Star Project of Shaanxi Province (2020KJXX-036), High-end Foreign Expert Introduction Program of National Science and Technology (G2022040014L), Innovation Capability Strong Foundation Plan of Xi’an City (Medical Research Project, 21YXYJ0037), and Major Research Projects of Xi’an Science and Technology Plan [201805104YX12SF38(2)].
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, H., Zhang-Sun, Zy., Xue, Cx. et al. CTRP family in diseases associated with inflammation and metabolism: molecular mechanisms and clinical implication. Acta Pharmacol Sin 44, 710–725 (2023). https://doi.org/10.1038/s41401-022-00991-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00991-7
Keywords
This article is cited by
-
CTRP5 Attenuates Doxorubicin-Induced Cardiotoxicity Via Inhibiting TLR4/NLRP3 Signaling
Cardiovascular Drugs and Therapy (2023)
