
Abstract
Type 2 bradykinin receptor (B2R) is an essential G protein-coupled receptor (GPCR) that regulates the cardiovascular system as a vasodepressor. Dysfunction of B2R is also closely related to cancers and hereditary angioedema (HAE). Although several B2R agonists and antagonists have been developed, icatibant is the only B2R antagonist clinically used for treating HAE. The recently determined structures of B2R have provided molecular insights into the functions and regulation of B2R, which shed light on structure-based drug design for the treatment of B2R-related diseases. In this review, we summarize the structure and function of B2R in relation to drug discovery and discuss future research directions to elucidate the remaining unknown functions of B2R dimerization.
Similar content being viewed by others

Introduction
The kallikrein–kinin system (KKS) plays essential roles in maintaining the homeostasis of the cardiovascular system [1]. The KKS consists of kallikreins, kininogens, kinins, kininases, and kinin receptors [2]. There are 15 tissue kallikrein genes, KLK1–KLK15, and one plasma kallikrein gene in the human genome [3]. Kallikreins belong to the serine protease family and are widely distributed in the kidneys, pancreas, salivary glands, and plasma [4]. Two splice variants of kininogens are expressed in the liver: high-molecular-weight kininogen (HK, MW: 72 kDa) and low-molecular-weight kininogen (LK, MW: 48 kDa) [5]. Kinins are short-lived peptides originating from kininogens, and the nonapeptide bradykinin (RPPGFSPFR) is liberated from HK with the help of plasma kallikrein, while the decapeptide kallidin (KRPPGFSPFR) is liberated from LK by tissue kallikrein [6] (Fig. 1). The C-terminal arginines of bradykinin and kallidin can be cleaved by carboxypeptidases N and M into des-Arg9-bradykinin and des-Arg10-kallidin [6]. Kinins function as vasodilators by interacting with kinin receptors on the cell membrane [7]. Kinin receptors belong to the G protein-coupled receptor (GPCR) superfamily, with two subtypes, type 1 and type 2 bradykinin receptors (B1R and B2R) [8]. Although B1R and B2R show 32% sequence identity, they exhibit very different expression and ligand selectivity [9]. B2R is ubiquitously expressed in physiological and pathological conditions, while B1R is expressed rarely in normal tissues [10]. In particular, B1R is supposed to function as an essential mediator in oxidative stress and inflammation [11]. Bradykinin shows higher affinity for B2R than for B1R [12]. In contrast, kinins lacking C-terminal arginines, such as des-Arg9-bradykinin and des-Arg10-kallidin, preferentially bind B1R over B2R [12]. The mechanisms of kinin selectivity on receptors lie in the interactions between the C-termini of kinins and receptors [9]. After activating the receptors, kinins are degraded by kininases, such as angiotensin-converting enzymes (ACEs) and neutral endopeptidase (NEP), to terminate their functions [13].

In endothelial cells, upon bradykinin binding, GDP of the Gαq subunit is replaced by GTP, accompanying with the dissociation of Gαq and Gβγ subunits. Gαq activates PLC and calcium mobilization, and the increasing level of Ca2+ enhances the activities of eNOS and PLA2, which promote the release of NO and prostaglandins, respectively, and lead to vasodilation. Angiotensin II could either bind to AT1R to elevate the blood pressure, induce pro-inflammatory and pro-fibrosis activities, or bind AT2R to reduce the blood pressure, perform anti-inflammation and anti-fibrosis activities (PCP prolyl carboxypeptidase, APA aminopeptidases A, PK plasma kallikrein, TK tissue kallikrein, DAG diacyl glycerol, PKC protein kinase C, MAPK mitogen-activated protein kinase).
In the human genome, the gene encoding B2R (BDKRB2) is located on chromosome 14q32, and the B2R protein contains 391 residues [14]. The N-terminus of B2R contains Met1 to Asn57, while the C-terminus ranges from Gln352 to Gln391 [15]. There are three putative glycosylation sites at Asn30, Asn39, and Asn207 and a palmitoylation site at Cys351 [16]. In addition, several serines and threonines are predicted to be phosphorylated by G protein-coupled receptor kinases (GRKs) [17].
B2R functions
B2R is activated upon the binding of endogenous agonists on the extracellular side, and in combination with the conformational transitions, G proteins can occupy the intracellular cleft surrounded by TMs2-7 [18, 19]. B2R mainly mediates the Gq signaling pathway, but in some cases, it also couples to Gi, Gs, and G12/13 [20,21,22]. In endothelial cells, Gq proteins couple to B2R upon receptor activation and then disassociate into the Gαq subunit and Gβγ heterodimers [3] (Fig. 1). Gαq induces the activation of phospholipase C (PLC), the cleavage of phosphatidylinositol (4,5) bisphosphate (PIP2), and the liberation of Ca2+ from endoplasmic reticulum (ER) [23, 24]. Increasing the level of intracellular Ca2+ promotes the activation of calcineurin and endothelial nitric oxide synthase (eNOS) and finally increases the concentration of nitric oxide (NO) in vessels [25]. Ca2+ also enhances the phosphorylation of phospholipase A2 (PLA2) and triggers the release of prostaglandins [3] (Fig. 1).
Both NO and prostaglandins are vasodilators and are effective in lowering blood pressure [3, 26]. In addition to regulating the circulation, activated B2R also induces a transient increase in endothelial permeability and the exudation of protein-rich fluid into the interstitium [27], which leads to inflammation. Additionally, B2R localizes to the sensory ganglia, dorsal horn, and peripheral nociceptors, and bradykinin activates B2R in the nervous system and evokes pain [28]. Dysfunction of B2R leads to cardiovascular diseases, such as hypertension, ventricular hypertrophy, and myocardosis [29, 30]. Studies on kallikrein-deficient mice show that the posterior wall and septum of the heart become thinner and that the heart tends to dilate, resulting in reduced left ventricular mass [31]. In addition, B2R is closely related to the development of renal diseases [32], respiratory diseases [33], neurological diseases [34], cancers [35, 36], and hereditary angioedema (HAE) [37]. In vitro and in vivo experiments have shown that peptide and nonpeptide B2R antagonists stimulate apoptosis in cancers through the activation of the MAP kinase pathway and the blockage of intracellular calcium mobilization activity, which activates the caspase pathway and leads to apoptosis [38]. HAE is an autosomal dominant inherited disease characterized by increased activation of the KKS [39]. HAE leads to edema in the limbs, fauces, gastrointestinal tract, and respiratory passage and results in asphyxiation [40].
The Gq signaling of B2R is terminated once GRKs bind to the receptor and phosphorylate specific serines and threonines at the C-terminus, such as Ser366, Thr369, Ser373, and Ser375 [17, 41]. Then, β-arrestins (β-arrestin1 and β-arrestin2) recognize the phosphorylation sites and promote the internalization of B2R with the help of clathrin and adaptor protein 2 (AP2) [42]. In endocytic vesicles, agonists and β-arrestin dissociate from B2R, and then receptors are recycled to the plasma membrane or degraded in lysosomes [43]. Based on the results of a B2R recycling experiment, B2R internalizes into endosomes with β-arrestin2 and is rapidly recycled to the plasma membrane [43]. Regarding the interactions between B2R and β-arrestins, the endocytosis level of B2R is mainly correlated with the β-arrestin2 level, while β-arrestin1 shows a compensatory behavior [44]. Previous results suggest that β-arrestin2 binds B2R with a deficient C-terminus and induces receptor internalization, although more weakly than for wild-type B2R, and the K342P mutation in helix 8 of B2R decreases signaling through β-arrestin2, indicating that helix 8 is important for B2R-β-arrestin2 coupling [44]. However, the results from β-arrestin1 show that it requires both helix 8 and the C-terminus [44]. Thus, β-arrestin1 mediates the phosphorylation-dependent internalization of B2R, while β-arrestin2 mediates both pathways [44].
The biological functions of B2R are determined not only by the actions of kinins but also by interactions with other GPCRs, such as angiotensin II type 1 receptor (AT1R), apelin receptor (APJ), and κ-opioid receptor (κ-OR) (Fig. 2). Dimerization influences the agonist affinity, pharmacology, downstream signaling pathways, and trafficking of GPCRs [45]. AT1R, functioning as a vasopressor, is a key component in the renin–angiotensin system, while B2R acts as a vasodepressor [46] (Fig. 1). AT1R-B2R heterodimers reinforce Ca2+ signaling of AT1R in preeclampsia and block the β-arrestin-dependent internalization of B2R [47]. APJ is widely expressed in the cardiovascular system. APJ-B2R heterodimers expressed in human umbilical vein endothelial cells (HUVECs) enhance the phosphorylation of eNOS and extracellular signal regulated kinases1/2 (ERK1/2), thus facilitating cell proliferation [48]. κ-OR is distributed in the nervous system, which is activated by dynorphin (Dyn) and participates in analgesia, fluid homeostasis, and anti-pruritic activity. κ-OR-B2R heterodimers enhance cell proliferation through Gs/cAMP/PKA pathways when Dyn A (1–13) binds κ-OR in the heterodimers [49].

AT1R-B2R heterodimer enhances the Ca2+-induced hypertension in preeclampsia and blocks the β-arrestin-mediated B2R internalization. APJ-B2R and κ-OR-B2R heterodimers promote cell proliferation through PLC-ERK1/2-eNOS and cAMP-PKA signaling pathways, respectively.
B2R structures
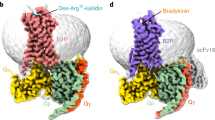
Several structures of B2R-Gq complexes have been recently solved using single-particle cryo-electron microscopy (cryo-EM) [18, 19] (Fig. 3a). B2R shares common structural features with other class A GPCRs, such as canonical seven-transmembrane helices (7TMs), a disulfide bond between TM3 and extracellular loop 2 (ECL2), and helix 8 lying parallel to the plasma membrane (Fig. 3b, c). ECL2 of B2R adopts a β-sheet conformation similar to that of AT1R, angiotensin II type 2 receptor (AT2R), and neurotensin receptor 1 (NTSR1) [50,51,52] (Fig. 3b). The N/C-termini of B2R form short α-helices at P4-I7 and S366-S389, as predicted by Jpred [53], SOPMA [54], and Alphafold [55], which may facilitate the binding of peptide agonists and coupling of intracellular signaling proteins. In addition, the other disulfide bond between C47N-term and C3047.25 (Ballesteros-Weinstein numbering [56]) locks the orthosteric binding pocket, and three structured cholesterols are observed at the intracellular clefts between TM2–TM4, TM3–TM4, and TM6–TM7 and probably regulate the B2R conformation in the allosteric mode [18] (Fig. 3b, c).

a Overall structures of bradykinin-bound B2R-Gq complex (PDB ID: 7F6H, B2R, green; bradykinin, violet; Gα, salmon; Gβ, cyan; Gγ, yellow) and kallidin-bound B2R-Gq complex (PDB ID: 7F6I, B2R, violet; kallidin, green). b ECL2 of B2R exhibits a β-sheet, with two disulfide bonds formed between C130-C211 and C47-C304. c At the intracellular side, ICL2 adopts a short α-helical conformation and three cholesterols (CHOL 1–3) lie in clefts between TM2–TM4, TM3–TM4, and TM6–TM7. d Hydrogen bonds (dash lines) formed between ECL1 and ECL2. e Anion trap formed by aspartic acids and glutamates locates at the entrance of ligand-binding pocket.
The funnel-like orthosteric binding pocket of B2R is covered by ECL2, which forms hydrogen bonds with D122, W123, and E127 in ECL1, functioning as a lid to decelerate the dissociation of peptide agonists [18] (Fig. 3d). Negatively charged residues, such as D203, E204, E221, D293, E307, and D311, are located at the entrance of the pocket, forming an anion trap to lock the positively charged peptide agonists in the pocket (Fig. 3e). Bradykinin and kallidin in the pocket adopt S-shaped conformations, with the N-termini extending to the extracellular side, while the C-termini insert into the bottom of the pocket. Pro3 of bradykinin (P3B) or Pro4 of kallidin (P4K) forms hydrogen bonds with I213 of B2R, while G4B or G5K forms an additional hydrogen bond with R1964.64 (Fig. 4a). These two interactions anchor peptide agonists in the pocket, while intramolecular hydrogen bonds among G4B/G5K, S6B/S7K, and R9B/R10K stabilize the conformations of the peptide agonists [18]. Extensive polar, ionic, and hydrophobic interactions also contribute to agonist binding to B2R.

a Binding poses of bradykinin (violet) and kallidin (green) in the pocket, key hydrogen bonds are formed between P3B/P4K-I213ECL2 and G4B/G5K-R1964.64. Hydrogen bonds are labeled as blue (bradykinin) and red (kallidin) dash lines (bradykinin-bound B2R, PDB ID: 7F6H, green; kallidin-bound B2R, PDB ID: 7F6I, violet). b, c Key hydrogen bonds (b) and hydrophobic interactions (c) involved in Gq coupling (Gαq, salmon). d Conformations of toggle switch and PIF motif in kallidin-bound B2R structure.
The structural superposition of des-Arg10-kallidin-bound B1R and bradykinin-bound B2R shows similar conformations (Fig. 5a), while the interactions with Phe9 of des-Arg10-kallidin (F9DK) and F8B/ R9B determine the peptide agonist selectivity between B1R and B2R. F9DK is negatively charged and forms electrostatic interactions with K1183.33 and R2025.38 of B1R (Fig. 5b). However, F8B is electroneutral and unable to contact the equivalent S1383.33 and T2245.38 of B2R (Fig. 5c). Furthermore, it is difficult to accommodate R9B in the narrow gap surrounded by R2025.38, Y2666.51, and E2736.58 of B1R, and steric hindrance occurs between R9B and R2025.38 of B1R [18, 19] (Fig. 5d). In contrast, no steric hindrance is observed between R9B and T2245.38, F2866.51, or D2936.58 of B2R (Fig. 5e). Thus, des-Arg10-kallidin prefers B1R, and bradykinin prefers B2R.

a Structural comparison between B2R-bradykinin complex (PDB ID: 7F6H) and B1R-des-Arg10-kallidin complex (PDB ID: 7EIB). b–e Ionic interactions between B1R and F9DK as well as steric hindrance between ligand pocket of B1R and R9B determine the preference of des-Arg10-kallidin for B1R and bradykinin for B2R. (B1R, yellow-orange; B2R, green; des-Arg10-kallidin, cyan; bradykinin, violet).
At the B2R-Gq interface, the α5-helix of Gαq contributes the most contacts with B2R. Y356G.H5.23 (CGN numbering [57]) of Gαq forms hydrogen bonds with V1513.46, D1543.49, and R1553.50 of B2R (Fig. 4b). In addition, several pairs of polar interactions, namely, R167ICL2-R37G.hns1.02, N2545.68-Q350G.H5.17, and R2676.32-L358G.H5.25/V359G.H5.26, enhance the coupling between B2R and Gαq. The intracellular loop 2 (ICL2) of B2R forms a short helix, positioning M163ICL2 in the hydrophobic surface surrounded by F341, K345, I348, and S198 of Gαq (Fig. 4c).
In the inactive-state structures of muscarinic M1 acetylcholine receptor (M1R) [58] and histamine H1 receptor (H1R) [59], the conserved DRY motif at the intracellular tip of TM3 forms hydrogen bonds with the residues in TM6 and ICL2, stabilizing the closed conformation of the intracellular cavity. Additionally, N7.49 of the conserved NPxxY motif forms hydrogen bonds with aspartic acids in TM2. In the active state B2R structure, F8B/F9K interacts with the conserved toggle switch W2836.48, inducing the outward movement of F2796.44 in the PIF motif and intracellular tip of TM6, which breaks the hydrogen bonds between the DRY motif and TM6, as well as the NPxxY motif and TM2 (Fig. 4d). Then, the α5-helix of Gαq inserts into the intracellular cleft and couples to B2R by forming hydrogen bonds between DRY motif-Y356G.H5.23 and NPxxY motif-N357G.H5.24 for B2R activation (Fig. 4b).
B2R agonists and antagonists
B2R is an essential drug target in maintaining the homeostasis of the cardiovascular system and relieving symptoms of edema and pain [60]. Due to the pro-inflammatory activity and widespread distribution of B2R in the central nervous system (CNS), the potent B2R-selective agonist labradimil (Arg-Pro-Hyp-Gly-Thi-Ser-Pro-Tyr(Me)-psi(CH2NH)-Arg) is able to temporarily increase the permeability of the blood brain barrier (BBB) in the RG2 rat model of glioma, facilitating the entry of chemotherapeutics into the CNS to kill tumors in the brain [61]. The mechanisms underlying this phenomenon include the modification of vasculature characteristics around tumors, which prevents drug delivery to the tumor interstitium, as well as the release of nitric oxide and prostaglandin E2 to change the vascular physiology and morphology [62, 63].
The development of B2R antagonists began in the 1960s. The first generation of B2R antagonists are bradykinin analogs that replace several residues with D-Phe, Hyp, or Thi, such as NPC-567 (D-Arg-Arg-Pro-Hyp-Gly-Phe-Ser-D-Phe-Phe-Arg) and NPC-349 (D-Arg-Arg-Pro-Hyp-Gly-Thi-Ser-D-Phe-Thi-Arg) [64] (Table 1). However, these agents show low affinity for B2R and are sensitive to peptidases, which hinder their clinical use. According to the results of solid-state NMR spectroscopy and molecular modeling [65, 66], the C-terminus of bradykinin may form a β-turn when binding to B2R. Several unnatural amino acids mimic the β-turn to strengthen the binding of antagonists and improve the subtype selectivity between B1R and B2R. The second generation of antagonists includes icatibant and NPC17731. Icatibant (D-Arg-Arg-Pro-Hyp-Gly-Thi-Ser-D-Tic-Oic-Arg) is a highly potent B2R antagonist for treating HAE [67] that was approved in 2011. Patients show poor compliance, however, as icatibant is administered by hypodermic injection. Thus, the exploitation of the third generation of B2R antagonists is imperative. Selective nonpeptide antagonists of B2R appeared in the 1990s, including Win64338 [68], FR173657 [69], bradyzide [70], anatibant [71], fasitibant [72], and JSM10292 [73] (Fig. 6). In addition to the convenience provided by oral administration, these nonpeptide antagonists are more resistant to metabolism and show fewer off-target effects [74, 75]. B-9430 (D-Arg-Arg-Pro-Hyp-Gly-Igl-Ser-D-Igl-Oic-Arg) is a derivative of bradykinin and binds to both human B1R and B2R with pIC50 values of 7.9 and 9.6, respectively [76]. In contrast, anatibant preferentially binds B2R over B1R [71]. Antagonists of B2R are promising therapeutics for relieving symptoms of inflammation, pain, and diabetes. In addition to icatibant, many other antagonists have been in clinical trials. Deltibant is a peptide antagonist for the treatment of pain, stroke, and severe brain injury [77]. Anatibant is helpful in the treatment of traumatic brain injury, while fasitibant is used for osteoarthritis. More information about these antagonists is summarized below.

Chemical structures of WIN64338, FR 173657, Bradyzide, Anatibant, Fasitibant, and JSM10292.
Icatibant
Icatibant, also known as HOE-140, is a bradykinin analog with high affinity (Ki value of 0.064 nM) for human B2R [78]. In a phase 2 clinical study, 27 patients took intravenous prednisolone plus clemastine as the standard therapy protocol or subcutaneous icatibant to analyze the efficiency of icatibant in ACE-inhibitor-induced angioedema by observing the median time of the resolution of edema [79]. The results indicate that the administration of icatibant shortened the median time by up to 19 h compared with the standard therapy. B2R is also an essential mediator of chronic bronchial asthma, and objective pulmonary function tests (PFTs) were carried out for patients with chronic asthma to evaluate the effects of icatibant [33]. After 4 weeks of treatment with icatibant, a 10% improvement in PFTs was achieved compared with the placebo group.
Deltibant
As a B2R antagonist, deltibant has been confirmed to alleviate the depressor response of bradykinin in rats and rabbits. In addition, a single dose of deltibant could reverse the profound hypotensive response and low survival rate caused by LPS in piglet models of endotoxic shock [80]. To investigate the clinical effects of deltibant on traumatic brain injury (TBI), 11 of 20 candidate patients took deltibant, while the others received placebo [81]. After treatment with deltibant, patients faced less risk of brain swelling and cerebral edema.
Anatibant
Anatibant, previously known as LF 16-0687, displays a sub-nanomolar affinity to human B2R with a Ki value of 0.67 nM [71]. Anatibant is a selective B2R antagonist that hinders downstream Gq signaling and the production of inositol 1,4,5-triphosphate (IP3). In vivo experiments performed in anesthetized rats showed that anatibant antagonizes bradykinin-induced edema [71]. In clinical trials, 25 patients suffering severe traumatic brain injury were selected to investigate the pharmacokinetics, safety, tolerability, and pharmacological effects of the anatibant [82]. TBI patients and healthy volunteers both tolerated the subcutaneous injection of 22.5 mg of anatibant, which reached therapeutic concentrations in the plasma within 2 h. BK1-5 is a metabolite of bradykinin, whose levels in plasma and cerebrospinal fluid increase excessively after trauma, which suggests that the active state B2R may participate in the progression of TBI. However, the competitive binding of anatibant at B2R may impede the activation of B2R and function as a therapeutic choice for TBI patients.
Fasitibant
Fasitibant, a nonpeptide B2R antagonist, shares similar chemical scaffolds with anatibant. The affinity of fasitibant to human B2R was measured through [3H]bradykinin competition assays with a pKi value of 10.3 [83]. In addition, fasitibant is more effective than anatibant in blocking G protein signals. In vivo studies were performed in rats pretreated with monosodium iodoacetate (MIA) as a knee joint osteoarthritis model and used to evaluate the effects of icatibant and fasitibant in relieving the sense of pain [84]. MIA treatment induces the upregulation of bradykinin activities and the production of prostaglandin E2. Compared to the analgesic effect of icatibant, fasitibant is more potent and long-lasting in eliminating the physiological effects of activated B2R.
The search for new scaffolds targeting B2R could be accelerated by structure-based drug design and virtual screening. According to the successful cases of μ-OR [85] and M1R [86], commercially available lead-like compounds could be docked into the B2R structure. Compounds with the potential to interact with the key residues of B2R could be further optimized with diverse chemical moieties. After evaluation of the affinity, subtype selectivity, and pharmacological effects of compounds, the in vivo performance of the promising candidates could be further analyzed in animal or clinical trials.
In addition, allosteric modulators are expected to increase on-target selectivity and decrease side effects caused by off-target effects at other receptors [87]. Based on the B2R structures, there are putative sites for allosteric modulator binding, although the B2R structures bound to the allosteric modulators are needed to elucidate the allosteric mechanism.
Future directions
B2R is a key regulator in the KKS, and dysfunction of B2R leads to cardiovascular diseases, neurological diseases, hereditary angioedema and cancers [60]; thus, B2R has been an ideal target for drug discovery for several decades, but only icatibant is approved for clinical use [68, 71, 72, 78, 88]. It is quite challenging and time-consuming to explore new chemical entities without structural information on the target. The stereochemical requirements for B2R antagonist binding have been revealed by molecular dynamics simulations, molecular modeling, and solid-state NMR spectroscopy; however, the detailed receptor‒ligand interactions remained elusive [89, 90]. With the recently determined B1R and B2R structures, the molecular mechanisms of ligand binding, subtype selectivity, receptor activation, and G protein coupling have been elucidated and are expected to accelerate the structure-based design of novel B2R agonists. Nevertheless, the active state B2R structures seem not to be ideal for antagonist design, especially for nonpeptide antagonists. Peptide antagonists may adopt different binding poses with different interactions to hinder the conformational changes necessary for B2R activation. The key pharmacophores for nonpeptide antagonists are still elusive based on the current agonist-bound structures and need to be identified from the B2R-antagonist structures.
The binding sites for allosteric modulators have been revealed in several GPCR structures. In the P2Y1R structure, the antagonist BPTU binds to the interface between P2Y1R and the lipid bilayer through hydrophobic interactions [91]. It blocks the movements of TM2 and TM3 required for P2Y1R activation. The CCR9-selective antagonist vercirnon occupies the intracellular cavity that binds G proteins, preventing CCR9 activation and G protein coupling [92]. The extracellular domain (ECD) is quite suitable as an allosteric regulatory site for class C GPCRs [87]. Structures have revealed that positive and negative allosteric modulators can be accommodated in the extracellular vestibule or the bottom of the orthosteric binding site [93,94,95,96,97,98,99,100]. However, no obvious vestibule or large ECD exists in the B2R structures, no lipidic ligand targeting B2R has been reported previously, and the orthosteric binding pocket of B2R is relatively shallow [18]. The intracellular cavity might be a potential binding site for negative allosteric modulators to block the downstream release of NO and prostaglandins.
GPCR heterodimers exhibit different physiological roles from monomers. B2R is associated with many GPCRs, such as AT1R, κ-OR, APJ, and dopamine D2R, that regulate blood pressure, cell proliferation, and neutrophil adhesion to endothelial cells [46, 48, 49, 101, 102]. However, no structure of B2R heterodimers has been determined. Several heterodimeric structures of class C GPCRs have been revealed, such as metabotropic glutamate receptors (mGluRs) [103] and GABAB receptors [104]. In both structures, the transmembrane helices provide hydrophobic interactions for dimerization, while G proteins couple to only one monomer. In the GABAB structure, the positive allosteric modulator BHFF resides within the crevice between monomers.
Increasing evidence has indicated that B2R is related to the development of the COVID-19 pandemic and is a potential drug target for related disorders. SARS-CoV-2 uses angiotensin-converting enzyme 2 (ACE2) for its infection, which is widely distributed in lung alveolar cells and degrades des-Arg9-bradykinin [105]. The loss of ACE2 during infection leads to the accumulation of des-Arg9-bradykinin and bradykinin and the upregulation of B2R activities in the lungs as well as pulmonary angioedema [106]. Eighty-nine percent of COVID-19 patients administered icatibant showed a 3 L/min reduction in oxygen supplementation after 24 h, while the proportion was 17% in the control group [107]. The clinical application of icatibant and the development of new B2R antagonists are thus needed to treat pulmonary angioedema and suppress thromboinflammation [108].
References
Rhaleb NE, Yang XP, Carretero OA. The Kallikrein-Kinin system as a regulator of cardiovascular and renal function. Compr Physiol. 2011;1:971–93.
Kayashima Y, Smithies O, Kakoki M. The kallikrein-kinin system and oxidative stress. Curr Opin Nephrol Hypertens. 2012;21:92–6.
Madeddu P, Emanueli C, El-Dahr S. Mechanisms of disease: the tissue kallikrein-kinin system in hypertension and vascular remodeling. Nat Clin Pract Nephr. 2007;3:208–21.
Hillmeister P, Persson PB. The Kallikrein-Kinin system. Acta Physiol. 2012;206:215–9.
Schmaier AH, Schutsky D, Farber A, Silver LD, Bradford HN, Colman RW. Determination of the bifunctional properties of high-molecular-weight Kininogen by studies with monoclonal-antibodies directed to each of its chains. J Biol Chem. 1987;262:1405–11.
Marceau F, Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nat Rev Drug Discov. 2004;3:845–52.
Cruden NLM, Lang NN, MacGillivray TJ, Uren NG, Fox KAA, Newby DE. Vasomotor and fibrinolytic responses to kinin receptor agonists in the atherosclerotic human lower limb. Heart Vessels. 2012;27:179–85.
Sriramula S. Kinin B1 receptor: a target for neuroinflammation in hypertension. Pharmacol Res. 2020;155:104715.
Marceau F, Bachelard H, Bouthillier J, Fortin JP, Morissette G, Bawolak MT, et al. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int Immunopharmacol. 2020;82:106305.
da Costa PLN, Sirois P, Tannock IF, Chammas R. The role of kinin receptors in cancer and therapeutic opportunities. Cancer Lett. 2014;345:27–38.
Parekh RU, Robidoux J, Sriramula S. Kinin B1 receptor blockade prevents angiotensin II-induced neuroinflammation and oxidative stress in primary hypothalamic neurons. Cell Mol Neurobiol. 2020;40:845–57.
Lau J, Rousseau J, Kwon D, Benard F, Lin KS. A systematic review of molecular imaging agents targeting bradykinin B1 and B2 receptors. Pharmaceuticals. 2020;13:199.
Blais C, Marceau F, Rouleau JL, Adam A. The kallikrein-kininogen-kinin system: lessons from the quantification of endogenous kinins. Peptides. 2000;21:1903–40.
Powell SJ, Slynn G, Thomas C, Hopkins B, Briggs I, Graham A. Human bradykinin B2-receptor—nucleotide-sequence analysis and assignment to chromosome-14. Genomics. 1993;15:435–8.
Kooistra AJ, Mordalski S, Pandy-Szekeres G, Esguerra M, Mamyrbekov A, Munk C, et al. GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021;49:D335–43.
Renaux A, Consortium U. UniProt: the universal protein knowledgebase (vol 45, pg D158, 2017). Nucleic Acids Res. 2018;46:2699.
Blaukat A, Pizard A, Breit A, Wernstedt C, Alhenc-Gelas F, Muller-Esterl W, et al. Determination of bradykinin B-2 receptor in vivo phosphorylation sites and their role in receptor function. J Biol Chem. 2001;276:40431–40.
Shen JK, Zhang DQ, Fu Y, Chen AQ, Yang XL, Zhang HT. Cryo-EM structures of human bradykinin receptor-G(q) proteins complexes. Nat Commun. 2022;13:714.
Yin YL, Ye CY, Zhou FL, Wang J, Yang DH, Yin WC, et al. Molecular basis for kinin selectivity and activation of the human bradykinin receptors. Nat Struct Mol Biol. 2021;28:755–61.
Gohla A, Offermanns S, Wilkie TM, Schultz G. Differential involvement of G alpha(12) and G alpha(13) in receptor-mediated stress fiber formation. J Biol Chem. 1999;274:17901–7.
Liebmann C, Graness A, Ludwig B, Adomeit A, Boehmer A, Boehmer FD, et al. Dual bradykinin B-2 receptor signalling in A431 human epidermoid carcinoma cells: activation of protein kinase C is counteracted by a G(s)-mediated stimulation of the cyclic AMP pathway. Biochem J. 1996;313:109–18.
de Weerd WF, Leeb-Lundberg LM. Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J Biol Chem. 1997;272:17858–66.
Liao JK, Homcy CJ. The G-Proteins of the G-Alpha(I) and G-Alpha(Q) family coupled the bradykinin receptor to the release of endothelium-derived relaxing factor. J Clin Invest. 1993;92:2168–72.
Gutowski S, Smrcka A, Nowak L, Wu DG, Simon M, Sternweis PC. Antibodies to the Alpha-Q-subfamily of guanine nucleotide-binding regulatory protein-alpha subunits attenuate activation of phosphatidylinositol 4,5-bisphosphate hydrolysis by hormones. J Biol Chem. 1991;266:20519–24.
Michel T, Vanhoutte PM. Cellular signaling and NO production. Pflug Arch Eur J Phy. 2010;459:807–16.
Mcgiff JC, Quilley J. Prostaglandins, Kinins and the regulation of blood-pressure. Clin Exp Hypertens. 1980;2:729–40.
Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vasc Pharmacol. 2002;39:187–99.
Paterson KJ, Zambreanu L, Bennett DLH, McMahon SB. Characterisation and mechanisms of bradykinin-evoked pain in man using iontophoresis. Pain. 2013;154:782–92.
Tang M, He FP, Ma L, Liu P, Wang JW, Zhu XC. Bradykinin receptors in Ischemic Injury. Curr Neurovasc Res. 2018;15:359–66.
Osorio JC, Cheema FH, Martens TP, Mahmut N, Kinnear C, Gonzalez AMD, et al. Simvastatin reverses cardiac hypertrophy caused by disruption of the bradykinin 2 receptor. Can J Physiol Pharmacol. 2008;86:633–42.
Meneton P, Bloch-Faure M, Hagege AA, Ruetten H, Huang W, Bergaya S, et al. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc Natl Acad Sci USA. 2001;98:2634–9.
Naicker S, Naidoo S, Ramsaroop R, Moodley D, Bhoola K. Tissue kallikrein and kinins in renal disease. Immunopharmacology. 1999;44:183–92.
Akbary AM, Wirth KJ, Scholkens BA. Efficacy and tolerability of Icatibant (Hoe 140) in patients with moderately severe chronic bronchial asthma. Immunopharmacology. 1996;33:238–42.
Huang HM, Lin TA, Sun GY, Gibson GE. Increased inositol 1,4,5-trisphosphate accumulation correlates with an up-regulation of bradykinin receptors in Alzheimers-disease. J Neurochem. 1995;64:761–6.
Wang GJ, Ye YW, Zhang XF, Song JM. Bradykinin stimulates IL-6 production and cell invasion in colorectal cancer cells. Oncol Rep. 2014;32:1709–14.
Wang GJ, Sun JF, Liu GH, Fu Y, Zhang XF. Bradykinin promotes cell proliferation, migration, invasion, and tumor growth of gastric cancer through ERK signaling pathway. J Cell Biochem. 2017;118:4444–53.
Nussberger J, Cugno M, Cicardi M, Agostoni A. Local bradykinin generation in hereditary angioedema. J Allergy Clin Immun. 1999;104:1321–2.
Stewart JM. Bradykinin antagonists as anti-cancer agents. Curr Pharm Des. 2003;9:2036–42.
Schneider L, Lumry W, Vegh A, Williams AH, Schmalbach T. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immun. 2007;120:416–22.
Farkas H, Reshef A, Aberer W, Caballero T, McCarthy L, Hao J, et al. Treatment effect and safety of icatibant in pediatric patients with hereditary angioedema. J Allergy Clin Immunol Pract. 2017;5:1671–8.
Gurevich EV, Tesmer JJG, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69.
Khoury E, Nikolajev L, Simaan M, Namkung Y, Laporte SA. Differential regulation of endosomal GPCR/beta-arrestin complexes and trafficking by MAPK. J Biol Chem. 2014;289:23302–17.
Simaan M, Bedard-Goulet S, Fessart D, Gratton JP, Laporte SA. Dissociation of beta-arrestin from internalized bradykinin B2 receptor is necessary for receptor recycling and resensitization. Cell Signal. 2005;17:1074–83.
Feierler J, Wirth M, Welte B, Schussler S, Jochum M, Faussner A. Helix 8 plays a crucial role in bradykinin B-2 receptor trafficking and signaling. J Biol Chem. 2011;286:43282–93.
Wang WJ, Qiao YH, Li ZJ. New insights into modes of GPCR activation. Trends Pharmacol Sci. 2018;39:367–86.
Quitterer U, AbdAlla S. Vasopressor meets vasodepressor: the AT1-B2 receptor heterodimer. Biochem Pharmacol. 2014;88:284–90.
Mikusic NLR, Silva MG, Pineda AM, Gironacci MM. Angiotensin receptors heterodimerization and trafficking: how much do they influence their biological function? Front Pharmacol. 2020;11:1179.
Ji BY, Shang LY, Wang CM, Wan L, Cheng BH, Chen J. Roles for heterodimerization of APJ and B2R in promoting cell proliferation via ERK1/2-eNOS signaling pathway. Cell Signal. 2020;73:109671.
Ji BY, Liu HQ, Zhang RM, Jiang YL, Wang CM, Li S, et al. Novel signaling of dynorphin at K-opioid receptor/bradykinin B2 receptor heterodimers. Cell Signal. 2017;31:66–78.
Wingler LM, McMahon C, Staus DP, Lefkowitz RJ, Kruse AC. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell. 2019;176:479–90.
Zhang HT, Han GW, Batyuk A, Shchenko AI, White KL, Patel N, et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature. 2017;544:327–32.
White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–13.
Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. JPred: a consensus secondary structure prediction server. Bioinformatics. 1998;14:892–3.
Geourjon C, Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. 1995;11:681–4.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9.
Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428.
Flock T, Ravarani CNJ, Sun DW, Venkatakrishnan AJ, Kayikci M, Tate CG, et al. Universal allosteric mechanism for G alpha activation by GPCRs. Nature. 2015;524:173–9.
Thal DM, Sun BF, Feng D, Nawaratne V, Leach K, Felder CC, et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature. 2016;531:335–40.
Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, et al. Structure of the human histamine H-1 receptor complex with doxepin. Nature. 2011;475:65–70.
Leeb-Lundberg LMF, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77.
Bartus RT, Elliott P, Hayward N, Dean R, McEwen EL, Fisher SK. Permeability of the blood brain barrier by the bradykinin agonist, RMP-7: Evidence for a sensitive, auto-regulated, receptor-mediated system. Immunopharmacology. 1996;33:270–8.
Emerich DF, Dean RL, Snodgrass P, Lafreniere D, Agostino M, Wiens T, et al. Bradykinin modulation of tumor vasculature: II. Activation of nitric oxide and phospholipase A(2)/prostaglandin signaling pathways synergistically modifies vascular physiology and morphology to enhance delivery of chemotherapeutic agents to tumors. J Pharmacol Exp Ther. 2001;296:632–41.
Emerich DF, Snodgrass P, Dean RL, Lafreniere D, Agostino M, Wiens T, et al. Bradykinin modulation of tumor vasculature: I. Activation of B-2 receptors increases delivery of chemotherapeutic agents into solid peripheral tumors, enhancing their efficacy. J Pharmacol Exp Ther. 2001;296:623–31.
Steranka LR, Farmer SG, Burch RM. Antagonists of B2-bradykinin receptors. Faseb J. 1989;3:2019–25.
Kyle DJ, Blake PR, Smithwick D, Green LM, Martin JA, Sinsko JA, et al. Nmr and computational evidence that high-affinity bradykinin receptor antagonists adopt C-terminal beta-turns. J Med Chem. 1993;36:1450–60.
Lopez JJ, Shukla AK, Reinhart C, Schwalbe H, Michel H, Glaubitz C. The structure of the neuropeptide bradykinin bound to the human G-protein coupled receptor bradykinin B2 as determined by solid-state NMR spectroscopy. Angew Chem Int Ed. 2008;47:1668–71.
Hock FJ, Wirth K, Albus U, Linz W, Gerhards HJ, Wiemer G, et al. Hoe-140 a new potent and long-acting bradykinin-antagonist—invitro studies. Br J Pharmacol. 1991;102:769–73.
Sawutz DG, Salvino JM, Dolle RE, Casiano F, Ward SJ, Houck WT, et al. The Nonpeptide Win-64338 Is a Bradykinin B-2 Receptor Antagonist. Proc Natl Acad Sci USA. 1994;91:4693–7.
Asano M, Inamura N, Hatori C, Sawai H, Fujiwara T, Katayama A, et al. The identification of an orally active, nonpeptide bradykinin B-2 receptor antagonist, FR173657. Br J Pharmacol. 1997;120:617–24.
Burgess GM, Perkins MN, Rang HP, Campbell EA, Brown MC, McIntyre P, et al. Bradyzide, a potent non-peptide B-2 bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. Br J Pharmacol. 2000;129:77–86.
Pruneau D, Paquet JL, Luccarini JM, Defrene E, Fouchet C, Franck RM, et al. Pharmacological profile of LF 16-0687, a new potent non-peptide bradykinin B-2 receptor antagonist. Immunopharmacology. 1999;43:187–94.
Valenti C, Giuliani S, Cialdai C, Tramontana M, Maggi CA. Fasitibant chloride, a kinin B2 receptor antagonist, and dexamethasone interact to inhibit carrageenan-induced inflammatory arthritis in rats. Br J Pharmacol. 2012;166:1403–10.
Dziadulewicz EK, Brown MC, Dunstan AR, Lee W, Said NB, Garratt PJ. The design of non-peptide human Bradykinin B2 receptor antagonists employing the benzodiazepine peptidomimetic scaffold. Bioorg Med Chem Lett. 1999;9:463–8.
Rassias G, Leonardi S, Rigopoulou D, Vachlioti E, Afratis K, Piperigkou Z, et al. Potent antiproliferative activity of bradykinin B2 receptor selective agonist FR-190997 and analogue structures thereof: a paradox resolved? Eur J Med Chem. 2021;210:112948.
Sawutz DG, Salvino JM, Dolle RE, Seoane PR, Farmer SG. Pharmacology and structure activity relationships of the nonpeptide bradykinin receptor antagonist Win-64338. Can J Physiol Pharmacol. 1995;73:805–11.
Stewart JM, Gera L, Hanson W, Zuzack JS, Burkard M, McCullough R, et al. A new generation of bradykinin antagonists. Immunopharmacology. 1996;33:51–60.
Marmarou A, Nichols J, Burgess J, Newell D, Troha J, Burnham D, et al. Effects of the bradykinin antagonist Bradycor (TM) (deltibant, CP-1027) in severe traumatic brain injury: results of a multi-center, randomized, placebo-controlled trial. J Neurotraum. 1999;16:431–44.
Amblard M, Daffix I, Bedos P, Berge G, Pruneau D, Paquet JL, et al. Design and synthesis of potent bradykinin agonists containing a benzothiazepine moiety. J Med Chem. 1999;42:4185–92.
Bas M, Greve J, Stelter K, Havel M, Strassen U, Rotter N, et al. A Randomized Trial of Icatibant in ACE-Inhibitor-Induced Angioedema. N Engl J Med. 2015;372:418–25.
Siebeck M, Spannagl E, Schorr M, Stumpf B, Fritz H, Whalley ET, et al. Effect of combined B-1 and B-2 kinin receptor blockade in porcine endotoxin shock. Immunopharmacology. 1996;33:81–4.
Narotam PK, Rodell TC, Nadvi SS, Bhoola KD, Troha JM, Parbhoosingh R, et al. Traumatic brain contusions: A clinical role for the kinin antagonist CP-0127. Acta Neurochir. 1998;140:793–803.
Marmarou A, Guy M, Murphey L, Roy F, Layani L, Combal JP, et al. A single dose, three-arm, placebo-controlled, phase I study of the bradykinin B-2 receptor antagonist anatibant (LF16-0687Ms) in patients with severe traumatic brain injury. J Neurotraum. 2005;22:1444–55.
Fattori D, Rossi C, Fincham CI, Caciagli V, Catrambone F, D’Andrea P, et al. Design and synthesis of novel sulfonamide-containing bradykinin hB2 receptor antagonists. 2. Synthesis and structure-activity relationships of alpha,alpha-cycloalkylglycine sulfonamides. J Med Chem. 2007;50:550–65.
Cialdai C, Giuliani S, Valenti C, Tramontana M, Maggi CA. Effect of Intra-articular 4-(S)-Amino-5-(4-{4-[2,4-dichloro-3-(2,4-dimethyl-8-quinolyloxymethyl)phenylsulfonamido]-tetrahydro-2H-4-pyranylcarbonyl} piperazino)-5-oxopentyl](trimethyl)ammonium chloride hydrochloride (MEN16132), a Kinin B-2 receptor antagonist, on nociceptive response in monosodium iodoacetate-induced experimental osteoarthritis in rats. J Pharmacol Exp Ther. 2009;331:1025–32.
Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–90.
Brown AJH, Bradley SJ, Marshall FH, Bown GA, Bennett KA, Brown J, et al. From structure to clinic: Design of a muscarinic M1 receptor agonist with potential to treatment of Alzheimer’s disease. Cell. 2021;184:5886–901.
Thal DM, Glukhova A, Sexton PM, Christopoulos A. Structural insights into G-protein-coupled receptor allostery. Nature. 2018;559:45–53.
Valenti C, Cialdai C, Giuliani S, Tramontana M, Quartara L, Maggi CA. MEN 16132, a kinin B-2 receptor antagonist, prevents the endogenous bradykinin effects in guinea-pig airways. Eur J Pharmacol. 2008;579:350–6.
Joedicke L, Mao JF, Kuenze G, Reinhart C, Kalavacherla T, Jonker HRA, et al. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. Nat Chem Biol. 2018;14:284–90.
Lupala CS, Gomez-Gutierrez P, Perez JJ. New insights into the stereochemical requirements of the bradykinin B-2 receptor antagonists binding. J Comput Aid Mol Des. 2016;30:85–101.
Zhang DD, Gao ZG, Zhang KH, Kiselev E, Crane S, Wang J, et al. Two disparate ligand-binding sites in the human P2Y(1) receptor. Nature. 2015;520:317–21.
Oswald C, Rappas M, Kean J, Dore AS, Errey JC, Bennett K, et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature. 2016;540:462–5.
Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature. 2017;545:112–5.
Kruse AC, Ring AM, Manglik A, Hu JX, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–6.
Wu HX, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64.
Dore AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–62.
Christopher JA, Aves SJ, Bennett KA, Dore AS, Errey JC, Jazayeri A, et al. Fragment and structure-based drug discovery for a class C GPCR: discovery of the mGlu(5) negative allosteric modulator HTL14242 (3-Chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). J Med Chem. 2015;58:6653–64.
Christopher JA, Orgovan Z, Congreve M, Dore AS, Errey JC, Marshall FH, et al. Structure-based optimization strategies for G protein-coupled receptor (GPCR) allosteric modulators: a case study from analyses of new metabotropic glutamate receptor 5 (mGIu(5)) X-ray structures. J Med Chem. 2019;62:207–22.
Wang C, Wu HX, Evron T, Vardy E, Han GW, Huang XP, et al. Structural basis for smoothened receptor modulation and chemoresistance to anticancer drugs. Nat Commun. 2014;5:4355.
Hollenstein K, Kean J, Bortolato A, Cheng RKY, Dore AS, Jazayeri A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–43.
Niewiarowska-Sendo A, Polit A, Piwowar M, Tworzydlo M, Kozik A, Guevara-Lora I. Bradykinin B2 and dopamine D2 receptors form a functional dimer. Biochim Biophys Acta Mol Cell Res. 2017;1864:1855–66.
Niewiarowska-Sendo A, Labedz-Maslowska A, Kozik A, Guevara-Lora I. Bradykinin B2 receptor and dopamine D2 receptor cooperatively contribute to the regulation of neutrophil adhesion to endothelial cells. Acta Biochim Pol. 2018;65:367–75.
Lin SL, Han S, Cai XQ, Tan QX, Zhou KX, Wang DJ, et al. Structures of G(i)-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature. 2021;594:583–8.
Shen CS, Mao CY, Xu CJ, Jin N, Zhang HB, Shen DD, et al. Structural basis of GABA(B) receptor-G(i) protein coupling. Nature. 2021;594:594–8.
Mansour E, Palma AC, Ulaf RG, Ribeiro LC, Bernardes AF, Nunes TA, et al. Safety and outcomes associated with the pharmacological inhibition of the Kinin-Kallikrein system in severe COVID-19. Viruses-Basel. 2021;13:309.
Nagashima S, Dutra AA, Arantes MP, Zeni RC, Klein CK, de Oliveira FC, et al. COVID-19 and lung mast cells: the Kallikrein-Kinin activation pathway. Int J Mol Sci. 2022;23:1714.
van de Veerdonk FL, Kouijzer IJE, de Nooijer AH, van der Hoeven HG, Maas C, Netea MG, et al. Outcomes associated with use of a Kinin B2 receptor antagonist among patients with COVID-19. Jama Netw Open. 2020;3:e2017708.
van de Veerdonk FL, Giamarellos-Bourboulis E, Pickkers P, Derde L, Leavis H, van Crevel R, et al. A guide to immunotherapy for COVID-19. Nat Med. 2022;28:39–50.
Hess JF, Borkowski JA, Macneil T, Stonesifer GY, Fraher J, Strader CD, et al. Differential pharmacology of cloned human and mouse B2 bradykinin receptors. Mol Pharmacol. 1994;45:1–8.
Leeb T, Mathis SA, LeebLundberg LMF. The sixth transmembrane domains of the human B1 and B2 bradykinin receptors are structurally compatible and involved in discriminating between subtype-selective agonists. J Biol Chem. 1997;272:311–7.
Aramori I, Zenkoh J, Morikawa N, O’Donnell N, Asano M, Nakamura K, et al. Novel subtype-selective nonpeptide bradykinin receptor antagonists FR167344 and FR173657. Mol Pharmacol. 1997;51:171–6.
Gibson C, Schnatbaum K, Pfeifer JR, Locardi E, Paschke M, Reimer U, et al. Novel small molecule bradykinin B2 receptor antagonists. J Med Chem. 2009;52:4370–9.
Acknowledgements
HTZ is supported by National Key R&D Program of China (2018YFA0508100), National Natural Science Foundation of China (81722044, 91753115, 21778049, 81861148018), and National Science and Technology Major Project of China (2018ZX09711002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shen, Jk., Zhang, Ht. Function and structure of bradykinin receptor 2 for drug discovery. Acta Pharmacol Sin 44, 489–498 (2023). https://doi.org/10.1038/s41401-022-00982-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00982-8
