
Abstract
Rheumatoid arthritis (RA) is characterized by synovial inflammation, synoviocyte expansion and damage to cartilage and bone. We recently reported that peroxisome proliferator-activated receptor (PPAR)-γ inhibited the proliferation and activation of fibroblast-like synoviocytes (FLS), and was downregulated in RA synovial. In this study we investigated the role of PPAR-γ in RA and the underlying mechanisms. Adjuvant-induced arthritis (AIA) was induced in rats; from D15, AIA rats were orally administered pioglitazone (30 mg·kg−1·d−1) or rosiglitazone (4 mg·kg−1·d−1) for 14 days. Collagen-induced arthritis (CIA) was induced in wild-type and Ppar-γ+/− mice. We showed that the expression of PPAR-γ was significantly reduced, whereas that of TNF-α was markedly increased in human RA FLS. In CIA mice, knockdown of PPAR-γ expression (Ppar-γ+/−) aggravated the ankle inflammation. Similarly, T0070907 (a PPAR-γ antagonist) or si-PPAR-γ promoted the activation and inflammation of TNF-α-induced FLS in vitro. On the contrary, administration of PPAR-γ agonist pioglitazone or rosiglitazone, or injection of ad-Ppar-γ into the ankle of AIA rat in vivo induced overexpression of PPAR-γ, reduced the paw swelling and inflammation, and downregulated activation and inflammation of FLS in RA. Interesting, injection of ad-Ppar-γ into the ankle also reversed the ankle inflammation in Ppar-γ+/− CIA mice. We conducted RNA-sequencing and KEGG pathway analysis, and revealed that PPAR-γ overexpression was closely related to p53 signaling pathway in TNF-α-induced FLS. Co-IP study confirmed that p53 protein was bound to PPAR-γ in RA FLS. Taken together, PPAR-γ alleviates the inflammatory response of TNF-α-induced FLS by binding p53 in RA.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is an inflammatory and systemic autoimmune disease characterized by synovial inflammation, synoviocyte expansion, and damage to cartilage and bone [1]. The average prevalence of RA is estimated at 0.5%–1.0% worldwide and is 2–3 times more common in women than men. During the pathogenesis of RA, the synovial layer will increase from 3 to 20 layers, which is associated with an increase in fibroblast-like synoviocytes (FLS) [2] and the secretion of metalloproteinases. Crosstalk between inflammation and chemokines leads to RA progression by activating FLS [3]. Recent research has shown that FLS promote arthritis severity, and lining FLS can also contribute to bone erosion and cartilage damage in RA [4]. Moreover, TNF-α, which is mainly derived from activated macrophages, is one of the activators of FLS in the early stages of RA [5, 6]. In RA, after FLS activation, the cells become invasive, directly encroaching upon articular cartilage and digesting it by producing matrix metalloproteinases (MMPs) and secreting interleukin (IL)-6 and chemokines [5, 7]. Thus, to treat RA, it is vital to inhibit FLS activation and inflammation.
Recently, we reported that peroxisome proliferator-activated receptor (PPAR)-γ expression was downregulated in RA synovial tissue and inhibited the proliferation and activation of FLS in adjuvant-induced arthritis (AIA) via the Wnt/β-catenin signaling pathway [8]. PPAR-γ activation by 15-deoxy-delta (12,14)-prostaglandin J (2) (15d-PGJ (2)) can induce synoviocyte apoptosis in AIA [9]. PPAR-γ has been widely reported to be an anti-inflammatory target, and a large number of agonists have been designed [10, 11]. Agonists of this receptor suppress Th17 immunity by inhibiting glutaminolysis/2-HG/H3K4me3 and GSH/ROS signaling by GLS1 [12]. Additionally, PPAR-γ mediates the vasculoregenerative program to reverse pulmonary hypertension by binding to p53 [13]. These results indicate that PPAR may be crucial in inflammation; however, the exact mechanism of aberrant PPAR-γ expression in RA and its relationship with inflammation and FLS activation are unclear.
In this study, we stimulated FLS with TNF-α to simulate the inflammatory microenvironment of synovial cells in joints and used a rat AIA model, a collagen-induced arthritis (CIA) and a PPAR-γ+/− CIA mouse model to investigate the critical role and underlying mechanism of PPAR-γ in RA. Our results clearly showed that PPAR-γ plays an essential role by binding to p53 to alleviate the inflammatory response of TNF-α-induced FLS in RA.
Materials and methods
Synovium and blood collection
The synovium of RA patients (n = 8) and osteoarthritis (OA, n = 8) patients undergoing total joint replacement were collected from the Department of Orthopedics of the Second Affiliated Hospital of Anhui Medical University (Hefei, China). The blood of RA patients (n = 22) was collected from the Department of Rheumatology, the First Affiliated Hospital of Anhui Medical University (Hefei, China). Monocytes were separated from the blood of RA patients with a Human Peripheral Blood Mononuclear Cell Isolation Kit (Tianjin Hao Yang Biology, China). During the course of the experiment, all patients gave written, informed consent, and experimental protocols involving human subjects were approved by the Biomedical Ethics Committee of Anhui Medical University (Approval no. 20200970). RA patient information is shown in Supplementary Table S1.
Animals
Ppar-γ+/− mice were designed by GemPharmatech (Nanjing, China) with CRISPR/Cas9. The gRNA sequences of the Ppar-γ were 5′-GTCTGTGGGGATAAAGCATC-3′ and 5′-CTCGGCACTCAATGGCCATG-3′. Sprague–Dawley rats (80–120 g) were obtained from the Experimental Animal Center of Anhui Medical University (Certificate No. 20200498). All animals were cared for according to the guidelines provided by the Center for Developmental Biology of Anhui Medical University. Experiments were conducted in accordance with the regulations of the Experimental Animal Administration and were approved by Anhui Medical University’s subcommittee on animal care (Approval No. 20200963).
AIA model rats were injected with complete Freund’s adjuvant (0.1 mL/100 g body weight; Chondrex, Redmond, WA, USA) under the left hind paw [8, 14,15,16]. On the 14th day, AIA rats were intragastrically administered pioglitazone (30 mg·kg−1·d−1, Tonghua Maoxiang Pharmaceutical Co., Ltd., Tonghua, China) or rosiglitazone (4 mg·kg−1·d−1, Jiangsu Yellow River Pharmaceutical Co., Ltd., Yancheng, China) for 14 days. The adenovirus carrying the rat Ppar-γ gene (ad-Ppar-γ) or negative control gene (ad-NC) (Hanbio, Shanghai, China) was intra-articularly injected into the hind knee (0.1 mL). Secondary (right) hind paw swelling was measured on Days 15, 18, 21, 24, 27 and 30 by a toe volume measuring instrument (Chengdu Taimeng Software Co. Ltd, Chengdu, China).
For the CIA model, 10-to-14-week-old mice (Certificate No. 202133877) were immunized with 0.1 mL of an emulsion (2 mg/mL chicken type II collagen emulsified in 5 mg/mL complete Freund’s adjuvant, Chondrex) at the tail root. After 21 days, the mice were boosted with a 0.1 mL emulsion by subcutaneous injection. The adenovirus carrying the mouse Ppar-γ gene (ad-Ppar-γ) or the negative control gene (ad-NC) (Hanbio, Shanghai, China) was intra-articularly injected into the ankle (0.1 mL) on Day 21. In all models, arthritis was scored in the wrists and ankles as previously described: 0 = normal; 1 = minimal or moderate erythema and mild swelling; 2 = marked erythema and severe swelling, digits not yet involved; and 3 = maximal erythema and swelling, digits involved.
FLS culture
Cells were harvested from RA synovium, separated and cultured in DMEM (HyClone, UT, USA) containing 20% (v/v) FBS (PAN-Biotech GmbH, Adenbach, Germany) with normal penicillin and streptomycin (Beyotime, Shanghai, China) at 37 °C and 5% CO2. FLS were used for experiments between passages 4 and 8.
Histopathology
Samples were fixed in paraformaldehyde for 48 h, and the sectioned synovium was subjected to hematoxylin and eosin (H&E) staining, immunohistochemistry (IHC), and immunofluorescence (IF) analysis and photographed by CaseViewer software (3DHISTECH, Budapest, Hungary).
Enzyme-linked immunosorbent assay (ELISA)
Rat and mouse serum were collected from centrifuged blood, and the human cell supernatant of treated RA FLS was collected at 48 h. Based on the manufacturer’s protocol, ELISA was used to measure the levels of IL-6, IL-1β and TNF-α in mouse serum, TNF-α and IL-6 levels in rat serum and IL-6 and IL-8 levels in human FLS. The kit information can be found in the Supplementary Information.
Immunocytochemical analysis of FLS
PPAR-γ antibodies (Abcam, Cambridge, UK) were used overnight to detect FLS expressing TNF-α. The FLS were covered with DAPI (Beyotime, Shanghai, China) for 10 min and then photographed by an epifluorescence microscope (BX-51; Olympus, Tokyo, Japan).
Gene silencing and overexpression
The PPAR-γ small interfering RNA (PPAR-γ-siRNA, GenePharma, Shanghai, China), overexpression plasmid (PPAR-γ-pcDNA3.1, Hanbio) or P53-siRNA (Hanbio) was transfected into FLS by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The sequences of the sense and antisense oligonucleotides are shown in Supplementary Table S2. After being cultured Opti-MEM for 8 h, the transfected FLS were cultured in complete medium for 48 h at 37 °C.
Real-time quantitative PCR (RT–qPCR)
TRIzol reagent (Invitrogen) was used to extract total RNA, which was reverse transcribed into cDNA with PrimeScript TM RT Master Mix (Takara, Kyoto, Japan). RT–qPCR was performed with a SYBR® Green Premix Pro Taq HS qPCR Kit (AG, Changsha, China). The primers for RT–qPCR were synthesized by Sangon Biotech (Shanghai, China), and the sequences are shown in Supplementary Table S3.
Western blotting (WB)
Total proteins were extracted with RIPA buffer (Beyotime), denatured by boiling, separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, blocked with primary antibodies and incubated overnight. The antibodies were used at a 1:500 dilution. PPAR-γ and tissue inhibitor of metalloproteinase (TIMP)-1 antibodies were provided by Abcam; P53, MMP-3 and MMP-9 antibodies were provided by Cell Signaling Technology (Danvers, MA, USA); and IL-6, IL-1β and β-actin antibodies were provided by Bioworld, Shanghai, China. Immobilon Western Chemiluminescent HRP Substrate (Millipore) was used to incubate the protein bands with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies for 1 h. Then, the blots were photographed with a ChemiDocMP Imaging System (Bio-Rad).
RNA sequencing (RNA-seq)
After being infected with the human PPAR-γ overexpression adenovirus (ad-PPAR-γ, Hanbio), RA FLS were examined by RNA-seq at LC-bio Technologies (Hangzhou) Co., Ltd. (Hangzhou, China).
Paw swelling analysis
After AIA rats were subcutaneously administered complete Freund’s adjuvant in the left hind paw, the amount of swelling in the right hind paw was measured by a toe volume measuring instrument (Chengdu Taimeng Software Co. Ltd) on Days 15, 18, 21, 24, 27 and 30.
Wound-healing assay
After the cells reached 60%–80% confluence in a 24-well plate, the FLS were treated with PPAR-γ-siRNA or PPAR-γ-pcDNA3.1 for 24 h. Then, the cells were scratched with a pipette tip, fixed with methanol, stained with crystal violet for 24 h, and viewed under an Olympus BX-51 microscope.
Coimmunoprecipitation (Co-IP) assay
RA FLS were examined by Co-IP according to the manufacturer’s protocol (Wanlei bio, Shenyang, China). Anti-PPAR-γ and anti-P53 were purchased from Cell Signaling Technology.
Statistical analysis
SPSS v13.0 software was used to analyze the data, which are presented as the mean ± standard deviation. We performed one-way analysis of variance or RMANOVA with a post hoc Dunnett’s test to determine statistical significance. P values < 0.05 were considered statistically significant in all analyses.
Results
PPAR-γ and TNF-α expression showed opposite trends in RA FLS
As a first step in investigating the pivotal role of PPAR-γ in RA, we collected the synovium from RA and OA patients and found inflammatory cell infiltration in the synovium in RA (Fig. 1a). Moreover, PPAR-γ expression was downregulated in RA FLS, as shown by IHC (Fig. 1a), which was confirmed by WB and RT–qPCR (Fig. 1b). PPAR-γ expression was also downregulated in AIA and CIA synovial tissue (Supplementary Fig. S1a). PPAR-γ levels were reduced in FLS stimulated with inflammatory factors such as IL-1β, IL-6, IL-17A, TNF-α, IFN-γ, and lipopolysaccharide (Fig. 1c). Moreover, the expression of PPAR-γ and TNF-α in the RA synovium showed opposite trends, and there was some overlap, as shown in Fig. 1d. PPAR-γ expression was also downregulated in TNF-α-induced RA FLS (Supplementary Fig. S1b). PPAR-γ mRNA levels were negatively correlated with RF, ESR, DAS28-ESR and DAS28-CRP in RA PBMCs (P < 0.05) (Fig. 1e). However, PPAR-γ mRNA levels were negatively correlated with anti-CCP and CRP levels (P > 0.05) (Supplementary Fig. S1c). Surprisingly, PPAR-γ mRNA levels were also negatively correlated with TNF-α, IL-1β, IL-6, IL-8 and IL-17A, in addition to IL-10 (P < 0.05) (Supplementary Fig. S1c).

a Representative images showing PPAR-γ expression in the synovium by H&E staining and IHC (20×). PPAR-γ levels were examined by WB and RT–qPCR (b) after treatment with IL-1β (2 ng/mL), IL-6 (5 ng/mL), IL-17A (10 ng/mL), TNF-α (10 ng/mL), IFN-γ (10 ng/mL), and lipopolysaccharide (LPS; 1 μg/mL) (c). d PPAR-γ and TNF-α expression in the synovium was analyzed by IF analysis (20×). e Correlation analysis of PPAR-γ mRNA and RF, ESR, DAS28-ESR and DAS28-CRP levels in RA PBMCs. The values represent the mean ± SD of 3 different samples. ##P < 0.01 vs. OA group; *P < 0.05, **P < 0.01 vs. RA group.
Knockout of PPAR-γ promoted TNF-α-induced FLS activation and inflammation in RA
Next, we established PPAR-γ-knockout (Ppar-γ+/−) mice via GemPharmatech with CRISPR/Cas9. WB showed that PPAR-γ protein expression was reduced in Ppar+/− mice (Fig. 2a). Joint scoring showed that CIA Ppar+/− mice had exacerbated swelling in the wrists and ankles (Fig. 2b). Moreover, H&E staining suggested that inflammatory cell infiltration and hyperplasia of the synovium were increased in CIA Ppar+/− mouse ankle joints (Fig. 2c). TNF-α expression was also upregulated in the synovium of Ppar+/− mice (Fig. 2d). Moreover, the secretion of IL-1β, IL-6 and TNF-α was increased in CIA Ppar+/− mouse blood.

a PPAR-γ expression in the mouse synovium. b Mouse joint scores. c Representative images showing H&E staining of the synovium (20×). d Representative images showing TNF-α expression by IHC (20×). e IL-1β, IL-6 and TNF-α protein levels in mouse blood were examined by ELISA. The values represent the mean ± SD of 10 different samples. ##P < 0.01 vs. WT group; **P < 0.01 vs. Ppar-γ+/− group; &&P < 0.01 vs. CIA group.
We also downregulated the expression of PPAR-γ by T0070907 or si-PPAR-γ in TNF-α-induced RA FLS (Supplementary Fig. S2a). WB and RT–qPCR showed that IL-1β and IL-6 expression was upregulated by T0070907 or si-PPAR-γ in TNF-α-induced RA FLS (Supplementary Figs. S2a and S2b). Moreover, IL-6 and IL-8 secretion was upregulated by T0070907 or si-PPAR-γ in FLS (Supplementary Fig. S2c). Notably, the expression of the proinflammatory cytokines IL-1β, IL-6 and IL-8, the chemokines C-C motif chemokine ligand (CCL)-2, CCL-3 and CCL-8, and MMPs (MMP-3 and MMP-9) in TNF-α-induced FLS was increased by T0070907 or si-PPAR-γ, as shown by WB and RT–qPCR, except for IL-10 and TIMP-1 (Supplementary Figs. S2b and S2d). Moreover, T0070907 or si-PPAR-γ increased the migration of FLS exposed to TNF-α in RA, as shown by wound-healing assays (Supplementary Fig. S2e). Thus, PPAR-γ knockout promoted TNF-α-induced FLS activation and inflammation in RA.
PPAR-γ overexpression inhibited inflammation and FLS activation in RA
The PPAR-γ agonists pioglitazone and rosiglitazone and the PPAR-γ-pcDNA3.1 overexpression vector were used to overexpress PPAR-γ in RA FLS. WB and RT–qPCR revealed that PPAR-γ expression was increased (Fig. 3a, b). The levels of the proinflammatory cytokines (IL-1β, IL-6 and IL-8) were suppressed (Fig. 3a, b) in TNF-α-induced FLS, whereas IL-10 levels were increased by pioglitazone, rosiglitazone or PPAR-γ-pcDNA3.1. In addition, PPAR-γ overexpression reduced IL-6 and IL-8 secretion (Fig. 3c), as well as the expression of CCL-2, CCL-3, and CCL-8 (Fig. 3b). Moreover, PPAR-γ overexpression downregulated the mRNA and protein expression of MMP-3 and MMP-9, whereas TIMP-1 levels were upregulated (Fig. 3b, d), and the migration of FLS was decreased, as shown by a wound-healing assay (Fig. 3e).

a PPAR-γ, IL-1β and IL-6 protein expression in FLS was analyzed by WB. b PPAR-γ, IL-1β, IL-6, IL-8, IL-10, CCL-2, CCL-3, CCL-8, MMP-3, MMP-9 and TIMP-1 mRNA expression in FLS was analyzed by RT–qPCR. c IL-6 and IL-8 protein levels in RA FLS supernatant were examined by ELISA. d MMP-3, MMP-9 and TIMP-1 protein expression was analyzed by WB. e The migration of FLS was examined by wound-healing assays (20×). The values represent the mean ± SD of 3 different samples. ##P < 0.01 vs. RA group; *P < 0.05, **P < 0.01, @P < 0.05, @@P < 0.01 vs. TNF-α group; &&P < 0.01 vs. pcDNA3.1 group.
To test the effects of PPAR-γ overexpression on synovial tissues in vivo, AIA rats were intragastrically treated with pioglitazone and rosiglitazone and injected with ad-Ppar-γ in the knees (Fig. 4a). PPAR-γ expression was upregulated by pioglitazone, rosiglitazone and ad-Ppar-γ in the synovial lining, as shown by WB, HIC and IF analysis (Fig. 4b, c and Supplementary Fig. S3a). Moreover, IL-1β, IL-6 and TNF-α levels were decreased by PPAR-γ overexpression, as shown by WB and ELISA (Fig. 4b, d). Moreover, treatment reduced inflammation and infiltration in the AIA synovium (Fig. 4c), as well as paw swelling (Fig. 4e).

a The schematic shows the delivery method of pioglitazone, rosiglitazone and ad-Ppar-γ in AIA rats. b IL-1β, IL-6, TNF-α and PPAR-γ protein expression in the synovium of rat knee joints was analyzed by WB. c Representative images of PPAR-γ in the synovium were examined by IHC and H&E staining (20×). d IL-6 and TNF-α protein levels in rat blood examined by ELISA. e Paw swelling. The values represent the mean ± SD of 10 different samples. ##P < 0.01 vs. normal group; *P < 0.05, **P < 0.01, @P < 0.05, @@P < 0.01 vs. AIA group; &&P < 0.01 vs. ad-NC group.
Next, we injected ad-Ppar-γ into the ankles of Ppar-γ+/− mice with CIA (Fig. 5a). After ad-Ppar-γ treatment, the joint scores showed ameliorated swelling in the wrists and ankles of Ppar-γ+/− CIA mice (Fig. 5b). H&E staining showed that the infiltration of inflammatory cells and hyperplasia of the synovium were reduced by ad-Ppar-γ in Ppar-γ+/− CIA ankle joints (Fig. 5c). Moreover, TNF-α expression was also downregulated in Ppar-γ+/− CIA mouse synovium (Fig. 5d). IL-1β, IL-6 and TNF-α expression was reduced by ad-Ppar-γ in Ppar-γ+/− CIA mouse blood (Fig. 5e). In short, by overexpressing PPAR-γ in RA, FLS inflammation and activation were inhibited.

a The schematic shows the delivery of ad-Ppar-γ in Ppar-γ+/− CIA mice. b Joint scores in CIA mice. c Representative images showing H&E staining in the synovium (20×). d Representative images showing TNF-α expression by IHC (20×). e IL-1β, IL-6 and TNF-α protein levels in mouse blood were examined by ELISA. The values represent the mean ± SD of 10 different samples. ##P < 0.01 vs. Ppar-γ+/− CIA group.
PPAR-γ bound to p53 in TNF-α-induced FLS
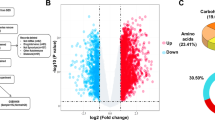
To assess the mechanism of PPAR-γ in TNF-α-induced FLS, we examined the gene expression profiles of FLS with ad-PPAR-γ by RNA-seq. After infection with human ad-PPAR-γ, 101 genes were significantly downregulated, and 170 genes were significantly upregulated in TNF-α-induced FLS (Fig. 6a, b). We also found that many inflammatory factors, chemokines and MMPs were reduced by ad-PPAR-γ, such as IL-21, IL-26, CCL13, CCL16 and MMP28 (Fig. 6c). KEGG pathway analysis showed that PPAR-γ overexpression was closely related to the p53 signaling pathway in FLS (Fig. 6d). Co-IP showed that the p53 protein bound to PPAR-γ in FLS (Fig. 6e). PPAR-γ overexpression with ad-PPAR-γ enhanced p53 expression and reduced p-p53 expression in TNF-α-induced FLS. On the other hand, silencing p53 with p53-siRNA did not change the mRNA level of PPAR-γ; however, silencing p53 increased chemokine (CCL-2, CCL-3 and CCL-8) and proinflammatory cytokine (IL-1β, IL-6 and IL-8) levels (Supplementary Fig. S4a). Thus, PPAR-γ alleviated the inflammatory response of TNF-α-induced FLS by binding to p53 in RA (Supplementary Fig. S4b).

a Volcano plot showing gene expression alterations in TNF-α-induced FLS treated with ad-PPAR-γ. b A heatmap showing representative significantly altered genes (P < 0.05) in TNF-α-induced FLS treated with ad-PPAR-γ. c A histogram showing inflammatory cytokine, chemokine and MMP genes in FLS. d KEGG pathway analysis showed that the p53 signaling pathway participates in PPAR-γ-mediated regulation of TNF-α-induced FLS. e The p53 protein bound to PPAR-γ, as shown by Co-IP. f PPAR-γ, p53 and p-p53 expression in TNF-α-induced FLS was analyzed by WB.
Discussion
In this study, we confirmed that PPAR-γ expression was downregulated in human RA FLS. We also found that knocking out PPAR-γ expression increased ankle inflammation in Ppar-γ+/− CIA mice and promoted RA FLS activation and inflammation in the presence of T0070907 or si-PPAR-γ. Moreover, PPAR-γ overexpression reduced paw swelling and inflammation after intragastric administration of pioglitazone and rosiglitazone and the injection of ad-Ppar-γ in AIA rats in vivo and decreased FLS activation and inflammation in RA. Moreover, mouse ad-Ppar-γ ameliorated ankle inflammation in Ppar-γ+/− CIA mice. Accordingly, the p53 protein bound to PPAR-γ in RA FLS. Thus, PPAR-γ alleviates the inflammatory response of TNF-α-induced FLS by binding to p53 in RA.
There is crosstalk between macrophages and FLS in the RA synovium [17, 18]. TNF-α derived from macrophages can activate FLS, leading to inflammation and FLS exacerbation in the occurrence and development of RA [19]. Activated FLS infiltrate and recruit monocytes and macrophages through the release of proinflammatory cytokines, chemokines, cell adhesion molecules and extracellular matrix proteins in RA [20, 21]. FLS are not targeted with therapeutics; however, inhibiting cytokines and chemokines produced by FLS is a potential treatment strategy for RA. PPAR-γ was shown to preserve OA by promoting demethylation in mice [22]. We previously reported that PPAR-γ overexpression could inhibit FLS proliferation and migration in AIA rats [8]. In the present work, PPAR-γ was downregulated in the synovium, and there was a close relationship between PPAR-γ and TNF-α expression in the RA synovium and blood in vitro and in vivo. Importantly, PPAR-γ mRNA levels were negatively correlated with RF, ESR, DAS28-ESR, DAS28-CRP and TNF-α levels in RA PBMCs (P < 0.05). These results suggested that PPAR-γ might be involved in the inflammatory response in FLS induced by TNF-α.
To clarify the role of PPAR-γ in RA, we investigated FLS activation, inflammation and chemokine production in the presence of TNF-α in RA. During the development of RA, FLS activation is characterized by invasion and migration, and these cells contact cartilage and induce osteoclast formation through activation. Similarly, various cytokines produced by FLS and macrophages form a complex network and exert physiological effects through overlapping and mutual regulation in RA [23, 24]. Chemokine-induced FLS activation has been shown to exacerbate inflammation and bone destruction [19]. In the present study, Ppar-γ+/− mice were established with CRISPR/Cas9. We found that Ppar-γ+/− CIA mice had exacerbated swelling of the wrists and ankles and increased inflammatory cell infiltration and hyperplasia of the synovium. Moreover, TNF-α expression was upregulated in Ppar-γ+/− mouse synovium and blood in vivo. Knocking out PPAR-γ expression with T0070907 or si-PPAR-γ promoted FLS activation (MMP-3 and MMP-9), inflammation (IL-β, IL-6 and IL-8) and chemokine production (CCL-2, CCL-3 and CCL-8) in the presence of TNF-α in RA in vitro. Thus, PPAR-γ knockdown intensified TNF-α-induced FLS activation and the inflammatory response in RA.
PPAR-γ overexpression by pioglitazone, rosiglitazone or PPAR-γ-pcDNA3.1 reduced the activation, inflammation and chemokine expression of TNF-α-induced FLS in RA in vitro. Moreover, inflammatory cell infiltration into the synovium and secondary swelling was reduced by pioglitazone, rosiglitazone and ad-Ppar-γ in AIA rats in vivo, while serum levels of inflammatory cytokines (IL-1β, IL-6 and TNF-α) were decreased. Moreover, after ad-Ppar-γ administration in Ppar-γ+/− mice, swelling of the wrists and ankles, inflammatory cell infiltration and hyperplasia of the synovium were reversed, and inflammatory cytokines were reduced in CIA mice. Therefore, PPAR-γ overexpression relieved TNF-α-induced FLS activation and the inflammatory response in RA.
In this study, we examined the gene expression profiles of FLS with ad-PPAR-γ by RNA-seq and found that 101 genes were significantly downregulated and 170 genes were significantly upregulated in RA. TNF-α is one of the factors that prevents self-injury, and it can also cause the pathogenesis of RA. In this study, RA FLS were activated by TNF-α to simulate the inflammatory environment of synovial tissues and examine the specific mechanism of PPAR-γ. We found that PPAR-γ overexpression was closely related to the p53 signaling pathway in FLS by KEGG pathway analysis. Hennigs et al. showed that PPAR-γ mediated the vasculoregenerative program to reverse pulmonary hypertension via p53 [13]. Moreover, the p53 protein was shown to bind to PPAR-γ by Co-IP, and this binding was reduced by ad-PPAR-γ. Proinflammatory cytokines and chemokines were increased by p53-siRNA in TNF-α-induced FLS. Consistently, p53 regulates macrophage polarization [25] and promotes inflammatory signals by suppressing IL-1β expression [26]. On the other hand, PPAR-γ plays an important role in type 2 diabetes, fat synthesis and inflammation. We hypothesize that patients with clinical complications of RA and diabetes can be treated with NSAIDs combined with PPAR-γ agonists such as pioglitazone to control the disease. However, we lack such conditions. Taken together, our results indicate that PPAR-γ alleviates the inflammatory response of TNF-α-induced RA FLS by binding to p53.
In conclusion, TNF-α, which is one of the activators of FLS, plays a key role in the development of RA. We found that PPAR-γ could inhibit FLS activation and inflammatory factors through the p53 pathway, thus improving the immune response of patients with RA. Based on these findings, therapeutic strategies targeting PPAR-γ may be effective in preventing the development of RA.
References
Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Prim. 2018;4:18001.
de Oliveira PG, Farinon M, Sanchez-Lopez E, Miyamoto S, Guma M. Fibroblast-like synoviocytes glucose metabolism as a therapeutic target in rheumatoid arthritis. Front Immunol. 2019;10:1743.
Muntyanu A, Abji F, Liang K, Pollock RA, Chandran V, Gladman DD. Differential gene and protein expression of chemokines and cytokines in synovial fluid of patients with arthritis. Arthritis Res Ther. 2016;18:296.
Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570:246–51.
Loh C, Park SH, Lee A, Yuan R, Ivashkiv LB, Kalliolias GD. TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: transcriptomic and epigenomic analysis. Ann Rheum Dis. 2019;78:1205–14.
Ross EA, Devitt A, Johnson JR. Macrophages: the good, the bad, and the gluttony. Front Immunol. 2021;12:708186.
Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33.
Li XF, Sun YY, Bao J, Chen X, Li YH, Yang Y, et al. Functional role of PPAR-gamma on the proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Sci Rep. 2017;7:12671.
Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, et al. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106:189–97.
Ju Z, Su M, Hong J, Kim E, Jung JH. Anti-inflammatory effects of an optimized PPAR-gamma agonist via NF-kappaB pathway inhibition. Bioorg Chem. 2020;96:103611.
Liu Y, Wang J, Luo S, Zhan Y, Lu Q. The roles of PPARgamma and its agonists in autoimmune diseases: a comprehensive review. J Autoimmun. 2020;113:102510.
Miao Y, Zheng Y, Geng Y, Yang L, Cao N, Dai Y, et al. The role of GLS1-mediated glutaminolysis/2-HG/H3K4me3 and GSH/ROS signals in Th17 responses counteracted by PPARgamma agonists. Theranostics. 2021;11:4531–48.
Hennigs JK, Cao A, Li CG, Shi M, Mienert J, Miyagawa K, et al. PPARgamma-p53-mediated vasculoregenerative program to reverse pulmonary hypertension. Circ Res. 2021;128:401–18.
Li XF, Chen X, Bao J, Xu L, Zhang L, Huang C, et al. PTEN negatively regulates the expression of pro-inflammatory cytokines and chemokines of fibroblast-like synoviocytes in adjuvant-induced arthritis. Artif Cells Nanomed Biotechnol. 2019;47:3687–96.
Li XF, Song B, Xu QQ, Yin SQ, Chen X, Xu L, et al. Over-expression of PTEN suppresses the proliferation and migration of fibroblast-like synoviocytes in adjuvant-induced arthritis. Cell Physiol Biochem. 2019;52:1446–62.
Li XF, Wu S, Yan Q, Wu YY, Chen H, Yin SQ, et al. PTEN methylation promotes inflammation and activation of fibroblast-like synoviocytes in rheumatoid arthritis. Front Pharmacol. 2021;12:700373.
Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16:316–33.
Wu YY, Li XF, Wu S, Niu XN, Yin SQ, Huang C, et al. Role of the S100 protein family in rheumatoid arthritis. Arthritis Res Ther. 2022;24:35.
Zhang Y, Aldridge J, Vasileiadis GK, Edebo H, Ekwall AH, Lundell AC, et al. Recombinant adiponectin induces the production of pro-inflammatory chemokines and cytokines in circulating mononuclear cells and fibroblast-like synoviocytes from non-inflamed subjects. Front Immunol. 2020;11:569883.
Cao C, Wu F, Niu X, Hu X, Cheng J, Zhang Y, et al. Cadherin-11 cooperates with inflammatory factors to promote the migration and invasion of fibroblast-like synoviocytes in pigmented villonodular synovitis. Theranostics. 2020;10:10573–88.
Frey O, Huckel M, Gajda M, Petrow PK, Brauer R. Induction of chronic destructive arthritis in SCID mice by arthritogenic fibroblast-like synoviocytes derived from mice with antigen-induced arthritis. Arthritis Res Ther. 2018;20:261.
Zhu X, Chen F, Lu K, Wei A, Jiang Q, Cao W. PPARgamma preservation via promoter demethylation alleviates osteoarthritis in mice. Ann Rheum Dis. 2019;78:1420–9.
Umar S, Palasiewicz K, Volin MV, Zanotti B, Al-Awqati M, Sweiss N, et al. IRAK4 inhibitor mitigates joint inflammation by rebalancing metabolism malfunction in RA macrophages and fibroblasts. Life Sci. 2021;287:120114.
Choi BY, Choi Y, Park JS, Kang LJ, Baek SH, Park JS, et al. Inhibition of Notch1 induces population and suppressive activity of regulatory T cell in inflammatory arthritis. Theranostics. 2018;8:4795–804.
Mukhopadhyay S, Antalis TM, Nguyen KP, Hoofnagle MH, Sarkar R. Myeloid p53 regulates macrophage polarization and venous thrombus resolution by inflammatory vascular remodeling in mice. Blood. 2017;129:3245–55.
Ubertini V, Norelli G, D’Arcangelo D, Gurtner A, Cesareo E, Baldari S, et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene. 2015;34:2493–504.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 82002269), the Anhui Provincial Science and Technology Major Project (No. 8212929035), the China Postdoctoral Science Foundation (No. 2020M671839), and the Postdoctoral Science Foundation from Anhui Medical University (No. BSH201902).
Author information
Authors and Affiliations
Contributions
XFL performed all experiments, analyzed the data, and wrote the manuscript. SQY and HL helped to collect the RA and OA synovial tissue samples. YLY, SW, YYW, XC, and BS helped to isolate FLS and assisted with WB, RT–qPCR analysis. HW and JL conceived the study and revised the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Xf., Yin, Sq., Li, H. et al. PPAR-γ alleviates the inflammatory response in TNF-α-induced fibroblast-like synoviocytes by binding to p53 in rheumatoid arthritis. Acta Pharmacol Sin 44, 454–464 (2023). https://doi.org/10.1038/s41401-022-00957-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00957-9
Keywords
This article is cited by
-
Daturataturin A Ameliorates Psoriasis by Regulating PPAR Pathway
Biochemical Genetics (2024)
-
Bavachinin Ameliorates Rheumatoid Arthritis Inflammation via PPARG/PI3K/AKT Signaling Pathway
Inflammation (2023)
