
Abstract
Tax1 banding protein 1 (Tax1bp1) was originally identified as an NF-κB regulatory protein that participated in inflammatory, antiviral and innate immune processes. Tax1bp1 also functions as an autophagy receptor that plays a role in autophagy. Our previous study shows that Tax1bp1 protects against cardiomyopathy in STZ-induced diabetic mice. In this study we investigated the role of Tax1bp1 in heart failure. Pressure overload-induced heart failure model was established in mice by aortic banding (AB) surgery, and angiotensin II (Ang II)-induced heart failure model was established by infusion of Ang II through osmotic minipump for 4 weeks. We showed that the expression levels of Tax1bp1 in the heart were markedly increased 2 and 4 weeks after AB surgery. Knockdown of Tax1bp1 in mouse hearts significantly ameliorated both AB- and Ang II infusion-induced heart failure parameters. On the contrary, AB-induced heart failure was aggravated in cardiac-specific Tax1bp1 transgenic mice. Similar results were observed in neonatal rat cardiomyocytes (NRCMs) under Ang II insult. We demonstrated that the pro-heart failure effect of Tax1bp1 resulted from its interaction with the E3 ligase ITCH to promote the transcription factor P73 ubiquitination and degradation, causing enhanced BCL2 interacting protein 3 (BNIP3)-mediated cardiomyocyte apoptosis. Knockdown ITCH or BNIP3 in NRCMs significantly reduced Ang II-induced apoptosis in vitro. Similarly, BNIP3 knockdown attenuated heart failure in cardiac-specific Tax1bp1 transgenic mice. In the left ventricles of heart failure patients, Tax1bp1 expression level was significantly increased; Tax1bp1 gene expression was negatively correlated with left ventricular ejection fraction in heart failure patients. Collectively, the Tax1bp1 increase in heart failure enhances ITCH-P73-BNIP3-mediated cardiomyocyte apoptosis and induced cardiac injury. Tax1bp1 may serve as a potent therapeutic target for the treatment of heart failure.
• Cardiac Tax1bp1 transgene mice were more vulnerable to cardiac dysfunction under stress.
• Cardiac Tax1bp1 transgene mice were more vulnerable to cardiac dysfunction under stress.
• Knockout of Tax1bp1 in mouse hearts ameliorated heart failure induced by pressure overload.
• Tax1bp1 interacts with the E3 ligase Itch to promote P73 ubiquitination and degradation, causing enhanced BNIP3-mediated apoptosis.
• Tax1bp1 may become a target of new therapeutic methods for treating heart failure.

Similar content being viewed by others


Introduction
Heart failure (HF) is a syndrome caused by structural or functional cardiac abnormalities that lead to elevated intracardiac pressures or reduced cardiac output at rest or during stress [1]. An estimated 64.3 million people are living with heart failure worldwide, and the absolute numbers have been increasing due to aging and improved survival after acute heart disease [2]. Cardiac injury occurring in any cardiovascular disease can cause myocyte cell loss and myocardial strain increase. The increased strain in the remaining myocytes collaborates with neurohormonal activation, causing eccentric hypertrophy. Then, cardiac fibrosis presses on left ventricular dilatation and often results in functional mitral regurgitation, leading to worse outcomes [3, 4]. Treatment of heart failure is mainly based on diuretics to relieve symptoms and neurohormonal antagonists to improve outcomes. However, the 5- and 10-year survival rates of patients with HF remain low at 57% and 35%, respectively [5]. Thus, new interventional procedures to counteract myocyte loss and suppress left ventricular remodeling are urgently needed.
Loss of cardiac myocytes is a central feature of heart disease to heart failure. The Bcl-2 gene family contains a group of proteins that both promote and repress cell death [6]. BNIP3 is a subfamily member of the Bcl-2 family of proteins that antagonize the activity of prosurvival proteins and promote apoptosis [6]. BNIP3 can cause opening of the mitochondrial permeability transition pore (mPTP) and trigger cytochrome c release, DNA fragmentation and early loss of plasma membrane integrity [7, 8]. In HF, the role of BNIP3 in cardiac myocyte cell death has been confirmed [9]. Thus, targeting BNIP3 is a novel concept in HF therapy [10].
Tax1bp1 was originally identified as an NF-κB regulatory protein that participates in inflammatory, antiviral and innate immune processes [11,12,13]. Tax1bp1 also functions as an autophagy receptor that plays a role in autophagy [14]. Our previous study found that Tax1bp1 could protect against cardiac dysfunction and remodeling by promoting autophagy in diabetic cardiomyopathy [15]. Studies have found that Tax1bp1 recruits E3 ligases that participate in the protein quality control process [16] as well as the cell death process [17]. These results indicate that Tax1bp1 acts as a promising functional protein in heart failure.
In this study, we provide evidence that Tax1bp1 specifically prompts cardiomyocyte apoptosis initiated by pressure overload or neurohormonal activation. Cardiac Tax1bp1 transgene mice were more vulnerable to cardiac dysfunction under stress. Tax1bp1 interacts with the E3 ligase Itch to promote P73 ubiquitination and degradation, causing enhanced BNIP3-mediated apoptosis. These findings provide new insight into the functional role of Tax1bp1 in cardiovascular disease, and Tax1bp1 acts as a proapoptotic protein.
Materials and methods
Animal
All animal operations and procedures obeyed to the Animals (Scientific Procedures) Act 1986, of the UK Parliament, Directive 2010/63/EU of the European Parliament; the Guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication, revised 2011) and were approved by the Animal Use Committees of Renmin Hospital of Wuhan University (WHU-RM-20180812). Cardiac-specific Tax1bp1 transgene mouse Tax1bp1 flox was generated by Cyagen (Shanghai, China). Mice carrying the α-Mhc-MerCreMer transgene (C57BL/6J background) were purchased from the Jackson Laboratory (stock no. 005650). Briefly, full-length Tax1bp1 cDNA was cloned downstream of the cardiac MEM-α-Mhc promoter. The linearized cardiac-specific plasmid was microinjected into fertilized mouse embryos to produce cardiac-specific mice and was identified by PCR analysis. Transgene PCR primers: forward primer, 5′-CTGGTTATTGTGCTGTCTCATCAT-3′; reverse primer, 5′-ACCTCTACAAATGTGGTATGGC-3′. Tax1bp1 loxp mice were bred with Cre-α-Mhc transgenic mice to generate Tax1bp1 loxp/MEM-Creα-Mhc mice. For the cardiac-specific transgene of Tax1bp1, tamoxifen (20 mg·kg−1·d−1, T-5648, Sigma, USA) was injected into 6-week-old male Tax1bp1 loxp/MEM-Cre mice for 5 consecutive days. The wild type littermates were used as control. To induce cardiac knockdown of Tax1bp1, C57BL/6J mice (purchased from the Chinese Academy of Medical Sciences, Beijing, China) were subjected to retro-orbital venous plexus injections of either adeno-associated virus (AAV9)-shTax1bp1 or AAV9-ScRNA to knockdown Tax1bp1. To induce BNIP3 knockdown in mouse hearts, C57BL/6J mice were subjected to retro-orbital venous plexus injections of AAV9-shBNIP3. The assignment of mice to different groups was randomized via a random number table.
Animal model
8- to 10-week-old male mice were subjected to aortic banding as described in our previous study [18]. Briefly, the mice were anaesthetized with an ip injection of 3% sodium pentobarbital at a dose of 40 mg/kg. Mice were subjected to ventilation, and then the chest was opened at the second intercostal space. We exposed the thoracic aorta and tied it with a 27-gauge needle with a 7–0 silk suture. Then, needle was removed after ligation. Successful aortic banding was confirmed by Doppler analysis. Ligation was not operated in sham-group mice. To treat post-operative pain, temegesic (0.1 mg/kg) was applied once daily for 6 days post-surgery.
An Ang II infusion-induced heart failure model was also performed in mice. Briefly, after anesthetization with pentobarbital sodium, each mouse was implanted with an osmotic minipump (Alzet model 2004, Alza Corp, USA) with Ang II (dissolved in 0.9% NaCl, set at a rate of 1.4 mg·kg−1·d−1) for 4 weeks. For the control group, mice were infused with the same volume of saline. 4 weeks after treatment the mice were sacrificed by cervical dislocation.
Adeno-associated virus vector
Recombinant AAV9s, including AAV9-shTax1bp1 and AAV9-shBNIP3, were constructed by Vigene Bioscience Company (Jinan, China). Briefly, plasmids with the cytomegalovirus (CMV) promoter drive shTax1bp1, and shBNIP3 was used to generate viral particles. HEK 293T cells were transfected with AAV9 capsids. Then, the cells were collected, and each AAV9 was purified with FPLC. Quantitative real-time PCR was used to determine the titers of the AAV vectors (viral genomes/mL). Each mice received a total of 60–80 μL AAV9-shTax1bp1, AAV9-shBNIP3 or AAV9-shRNA (5.0 × 1013 – 6.5 × 1013 VG/mL) injection from the retro-orbital venous plexus 2 weeks before aortic banding or the Ang II infusion model.
Echocardiographic and hemodynamic data
Echocardiographic and hemodynamic data were collected via a MyLab 30CV ultrasound system (Biosound Esaote, Genoa, Italy) and a microtip catheter transducer (SPR-839; Millar Instruments, Houston, TX, USA) as described in our previous study [18].
Histological analysis
Heart samples from each mice were stained with H&E and PSR to assess cell surface area and heart tissue fibrosis. For H&E staining, more than 200 cells were quantified in each group with Image-Pro Plus, version 6.0. For PSR staining, the left ventricular (LV) collagen volume was calculated by dividing the total section via Image-Pro Plus, version 6.0. TUNEL staining was also used to assess apoptosis with a terminal deoxynucleotide transferase-mediated dUTP nick end-labeling (TUNEL) reagent (S7111, Millipore Corporation, Burlington, MA, USA). In addition, cardiomyocytes were stained with α-actin, and nucleus were stained with DAPI and quantified as described in our previous study [19].
Neonatal rat cardiomyocyte (NRCM) isolation and culture
1- to 3-day-old neonatal Sprague–Dawley rats were used to harvest NRCMs. Briefly, hearts were removed from rats and cut into 1–3 mm3 pieces. Then, 0.125% trypsin–EDTA (2520-072; Gibco, USA) was used to digest heart tissue 15 min×5 times. Digestion was collected and stopped in Dulbecco’s modified Eagle’s medium/F-12 (C11330500BT, Gibco, USA) containing 20% fetal bovine serum (FBS, 10099-141, Gibco, USA) culture medium. Then, cells were filtered after centrifugation. Collected cells were incubated for 90 min to remove noncardiac myocytes (mainly adhering to plastic). Finally, cells were plated at a density of 5 × 105/well. NRCMs were transfected with adenovirus (Ad-) Tax1bp1 (MOI = 100) or Tax1bp1 siRNA for 8 h to overexpress or knockdown Tax1bp1. Cells were also transfected with BNIP3 or ITCH siRNA for 8 h to knockdown BNIP3 or ITCH. For induction in the vitro model, cells were stimulated with Ang II (1μmol/L) for 48 h. Cells were stained with α-actin to identify the cell surface area. More than 50 cells were quantified in each group with Image-Pro Plus, version 6.0. Cells were stained with TUNEL reagent as well as DAPI to quantify TUNEL-positive cells.
Adult mouse cardiomyocyte, fibroblast and endothelial cell isolation
The Langendorff method was used to isolate adult mouse cardiomyocytes 4 weeks after the mice received aortic banding surgery according to our previous study [20]. Briefly, after heparin injection, the hearts were cannulated through the aorta and mounted on a modified Langendorff perfusion system. The ventricular tissue was disaggregated with forceps and gently pipetted. The cell suspension was filtered, and the isolated cells were collected and plated on laminin-coated 35-mm culture dishes. Cells were subjected to Western blot analysis. For fibroblast and endothelial cell isolation, the hearts were cut into 1-mm3 tissue pieces, and 0.125% trypsin and collagenase were used to digest the left ventricles for 15 min at 34 °C five times. The digestion fluid was collected and centrifuged. The cells were resuspended, filtered and then seeded onto 100-mm plates for 90 min. After removing the cardiomyocytes, the cardiac fibroblasts were collected for Western blot analysis. For endothelial cells (ECs), CD31 beads were used to bind ECs. ECs were then washed and cultured in dishes precoated with 2% gelatin (Sigma, USA) in endothelial basal medium with 10% FBS.
Real-time PCR and western blotting
Real-time PCR and Western blotting were performed according to our previous study [19]. The primers used in this study are listed in Table S1. The antibodies used are listed in Table S2.
Coimmunoprecipitation assays
Cultured NRCMs were cotransfected with psicoR-HA-Tax1bp1 and psicoR-Flag-ITCH. Immunoprecipitation buffer was used to collect cell lysates. Each sample was incubated with 10 μL of protein A/G-agarose beads and 1 μg of antibody overnight at 4 °C. Then, the immunoprecipitation buffer was incubated with the indicated primary antibodies with immunoblotting.
Luciferase assay
The synthesized promoter regions (−1638 and −186 bp from the transcription start site) of the BNIP3 gene were subcloned into the luciferase reporter vector (Promega, USA). Luciferase reporter constructs were packed with an adenoviral system and then cotransfected into NRCMs with a control plasmid with either Ad-P73 or P73 siRNA (either Ad-Tax1bp1 or Tax1bp1 siRNA). The Dual Luciferase Reporter Assay Kit (Promega,USA) was used to determine the luciferase activity.
Ubiquitination assays
An in vivo ubiquitination assay was performed with 293T cells, transiently transfected with plasmids expressing Myc-ubiquitin, Flag-Tax1bp1, and HA-P73 for p73 ubiquitination or ITCH siRNA. After 24 h, the cells were treated with 10 mM MG132 (Sigma, USA) to block protein degradation for 6 h. Cells were harvested, lysed and immunoprecipitated as described above. Polyubiquitinated proteins were detected with the anti-Myc antibody. Flag-tagged point mutants Tax1bp1(Y741A and Y768A), were generated with the QuikChange site-directed mutagenesis kit (Stratagene, USA). 293T cells were transfected with plasmids expressing Myc-ubiquitin, Flag-Tax1bp1, HA-P73, and Flag-mutants Taxbp1-PPXY (Y741A and Y768A) to detect the active region of Tax1bp1 to ITCH.
GST pull-down assay
In vitro GST pulldown assay was performed as described previously [21]. The GST-fused full-length and mutants Tax1bp1(Y741A and Y768A) and Tax1bp1 (ZF: F764A) were generated using pGEX-4T1 (Amersham Biosciences, SE).
Human heart samples
Samples of failing human hearts were collected from the LVs of HF patients undergoing heart transplants. Control samples were obtained from the LVs of normal heart donors. Clinical and echocardiographic data (including tissue Doppler imaging) were obtained at the time of admission. The samples were obtained after informed consent and with approval of the local Ethical Committee (Renmin Hospital of Wuhan University Human Research Ethics Committee, Wuhan, China) and the ethic number is WDRY2021-KS058. The investigation conforms with the principles outlined in the Declaration of Helsinki.
Statistical analysis
SPSS 23.3 was used for data analysis. All measurements were obtained while blinded using a random number. The assignment of mice to different groups was randomized via a random number table. The group sizes of the in vivo experiments were estimated based on power analysis of HW/BW with an α error of 5% and a power of 80%, consistent with a previously published article [18]. Variance similar between the groups was statistically compared. One-way ANOVA was used to compare the data between groups followed by a post hoc Tukey test. Student's unpaired t-test was used to compare the data between two groups. Categorical variables were compared using the chi-square test. Between-variable correlations were assessed by Spearman’s test. Statistical significance was confirmed as a P-value less than 0.05.
Results
Tax1bp1 was upregulated during the pathology of cardiac remodeling
We assessed Tax1bp1 protein levels in heart tissue and cardiomyocytes. Mice were subjected to aortic banding (AB) to establish a pressure overload-induced heart failure model. Tax1bp1 levels were increased in mouse hearts 2 and 4 weeks after AB surgery (Fig. 1a, b). NRCMs were stimulated with angiotensin II (Ang II) to establish an in vitro HF model. We detected Tax1bp1 protein levels in NRCMs under Ang II stimulation. In this in vitro experiment, we found that the Tax1bp1 level was sharply increased after 48 h of Ang II stimulation (Fig. 1c). Coimmunofluorescence staining of Tax1bp1 and wheat germ agglutinin (WGA), which binds to glycoproteins of cardiomyocyte membranes (routinely used for the staining of cardiac sarcolemma), was used to detect the cellular location of the Tax1bp1 protein. In heart tissue, Tax1bp1 was expressed in the cytoplasm of cardiomyocytes and was upregulated after 4 weeks of pressure overload (Fig. 1b). In NRCMs, Tax1bp1 protein was also present in the cytoplasm and increased during Ang II stimulation (Fig. 1d). We also isolated cardiomyocytes, fibroblasts and endothelial cells from mouse hearts 1, 2, and 4 weeks after AB. Tax1bp1 expression levels were upregulated mainly in cardiomyocytes 1, 2, and 4 weeks after AB (Fig. 1e–g).

a Tax1bp1 protein levels in mouse hearts undergoing aortic banding surgery (n = 6 for each group). b Tax1bp1 and wheat germ agglutinin (WGA) staining in mouse hearts (n = 5, Scale bars: 50 μm). c Tax1bp1 protein levels in cardiomyocytes under Ang II insult (n = 6). d Tax1bp1 staining in cardiomyocytes (n = 5, Scale bars: 50 μm). Tax1bp1 and α-actinin protein levels in cardiomyocytes (e), Tax1bp1 and α-SMA protein levels in fibroblasts (f), and Tax1bp1 and CD31 protein levels in endothelial cells (g) isolated from mouse hearts 1, 2, and 4 weeks after AB (n = 6). **P < 0.01. One-way ANOVA followed by a post hoc Tukey test was used to compare the data in Fig. 1a, e–g Student’s unpaired t test was used to compare the data in Fig. 1b–d.
Tax1bp1 knockdown in vivo ameliorated cardiac remodeling in mice under AB
In the next step, we injected mice with AAV9-shTax1bp1 to downregulate Tax1bp1 in the heart tissue. Then, the mice were subjected to AB surgery 2 weeks after AAV9 injection. The Tax1bp1 protein was upregulated during the HF process. We detected Tax1bp1 protein levels 1, 2, 4 and 6 weeks after AB surgery in mice receiving AAV9-shTax1bp1 injection. As a result, Tax1bp1 levels were sharply reduced at 4 weeks and 6 weeks in mouse heart tissue receiving AAV9-shTax1bp1 injection (Supplementary Fig. 1a). HF performance was also ameliorated 4 weeks after AB in mice receiving AAV9-shTax1bp1 injection (Fig. 2a–e). We observed that heart weight gain and lung weight gain were diminished in mice receiving AAV9-shTax1bp1 injection compared with the ScRNA group after AB surgery (Fig. 2a, Supplementary Fig. 1b). Additionally, the cell hypertrophic response (Fig. 2b, Supplementary Fig. 1c) and fibrosis response (Fig. 2c, Supplementary Fig. 1d) were considerably reduced in AB surgery mice receiving AAV9-shTax1bp1 injection compared with control mice (ScRNA). Cardiac function in mice receiving AAV9-shTax1bp1 injection was also improved 4 weeks after AB (Fig. 2d, Supplementary Fig. 1e). The parameters detected by pressure loop analysis were also improved in AAV9-shTax1bp1 group mice 4 weeks after AB. As the end diastolic pressure dropped, cardiac output increased, dp/dtmax increased and dp/dtmin dropped (Fig. 2e, Supplementary Fig. 1f). Of note, changes in Tax1bp1 knockdown did not affect the structure and function of mouse hearts under normal conditions (Fig. 2b–e, Supplementary Fig. 1b–g).

a Heart weight (HW)/body weight (BW) and HW/tibia length (TL) ratios in mice subjected to AAV9-shTax1BP1 injection and AB surgery (n = 12). b H&E staining and WGA staining and cross-section area (CSA) quantification (n = 6, Scale bars: 50 μm). c PSR staining and left ventricular (LV) collagen volume (n = 6, Scale bars: 100 μm). d Echocardiography results in mice subjected to AAV9-shTax1BP1 injection and AB surgery (n = 10). LVEF left ventricular ejection fraction, LVFS left ventricular fractional shortening. e Hemodynamic results. dp/dtmax, maximal rate of pressure development; dp/dtmin, maximal rate of pressure decay. f α-actin staining in NRCMs transfected with Tax1bp1 siRNA under Ang II insult (n = 6, Scale bars: 50 μm). **P<0.01. One-way ANOVA followed by a post hoc Tukey test was used to compare the data in Fig. 2a–f.
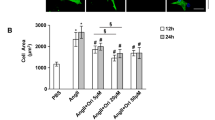
To determine the role of Tax1bp1 in cardiomyocytes under Ang II insult, NRCMs were transfected with Tax1bp1 siRNA to decrease Tax1bp1 expression (Supplementary Fig. 2a). The hypertrophic response to Ang II was first examined. As a result, the cell surface area and mRNA level of hypertrophic genes were decreased in Tax1bp1-silenced cells (Fig. 2f, Supplementary Fig. 2b).
Tax1bp1 exacerbated HF in mice under AB
We used cardiac-specific Tax1bp1 transgenic mice to overexpress Tax1bp1 in heart tissue (Supplementary Fig. 3a). Tax1bp1 transgene mice revealed an aggressive HF phenotype 4 weeks after AB surgery compared to the wild-type (WT) littermates. We observed heavily increased heart weight (Fig. 3a) and lung weight (Supplementary Fig. 3b), enhanced cross-sectional area (Fig. 3b), interstitial and perivascular fibrosis (Fig. 3c), and aggregated hypertrophic and fibrosis marker gene transcription (Supplementary Fig. 3c, d) in Ta1xbp1 transgenic mice subjected to AB compared to WT mice. Cardiac function also worsened in Ta1xbp1 transgenic mice 4 weeks after AB surgery (Fig. 3d, e; Supplementary Fig. 3e–g).

a Heart weight (HW)/body weight (BW) and HW/tibia length (TL) ratios in Tax1bp1 transgenic mice 4 weeks after AB (n = 12). b H&E staining and cross-sectional area (CSA) quantification (n = 6, Scale bars: 50 μm). c PSR staining and left ventricular (LV) collagen volume (n = 6, Scale bars: 100 μm). d Echocardiography results in Tax1bp1 transgenic mice subjected to AB surgery (n = 10). LVEF, left ventricular ejection fraction, LVFS left ventricular fractional shortening. e Hemodynamic results. dp/dtmax, maximal rate of pressure development; dp/dtmin, maximal rate of pressure decay. f α-actin staining in NRCMs transfected with Ad-Tax1bp1 under Ang II insult (n = 6, Scale bars: 50 μm). **P < 0.01. One-way ANOVA followed by a post hoc Tukey test was used to compare the data in Fig. 3a–f.
To determine the role of Tax1bp1 in cardiomyocytes under Ang II insult, NRCMs were transfected with Ad-Tax1bp1 to elevate the expression of Tax1bp1 (Supplementary Fig. 2c). The hypertrophic response to Ang II was first examined. As a result, the cell surface area and mRNA level of hypertrophic genes were sharply increased in Tax1bp1-overexpressing cells (Fig. 3f, Supplementary Fig. 2d).
Tax1bp1 knockdown improved Ang II infusion-induced cardiac remodeling
Whether Tax1bp1 silencing could hamper the progression of HF induced by neuroendocrine overactivation is unclear. Mice were subjected to AAV9-shTax1bp1 knockdown (Supplementary Fig. 4a), and Ang II infusion was used to induce HF. Four weeks after Ang II infusion, mice exhibited structural remodeling (hypertrophy and fibrosis) (Supplementary Fig. 4b, d), increased heart weight and lung weight (Supplementary Fig. 4c), and cardiac dysfunction (Supplementary Fig. 4e). However, HF signals in mice with Tax1bp1 knockdown were decreased compared with those in control mice after Ang II infusion (Supplementary Fig. 4b–e).
Tax1bp1 affected cell death
Given that cardiomyocyte loss is the main cause of HF in heart disease, we detected cell apoptosis as the classical form of cell death during HF. We stained myocytes with both TUNEL and α-actin in mouse hearts 4 weeks after AB surgery. Decreased myocyte apoptosis was observed in mice receiving AAV9-shTax1bp1 injection compared to mice receiving AAV9-ScRNA (Fig. 4a). A converse result was also observed in Tax1bp1 transgene mice 4 weeks after AB compared to the WT littermates (Fig. 4b). The changes in apoptotic markers, such as Bax, Bcl-2, and cleaved (c-) caspase 3, were also ameliorated in mice receiving AAV9-shTax1bp1 injection but deteriorated in Tax1bp1 TG mice (Fig. 4c and d). In vitro NRCMs exposed to Ang II stimuli were also stained using a TUNEL kit. Myocytes with Tax1bp1 silencing exhibited less apoptosis, whereas myocytes overexpressing Tax1bp1 exhibited more apoptosis than the corresponding controls (Fig. 4e).

a TUNEL and α-actin staining in mice subjected to AAV9-shTax1BP1 injection and AB surgery (n = 6, Scale bars: 50 μm). b TUNEL and α-actin staining in Tax1bp1 transgenic mice subjected to AB surgery (n = 6, Scale bars: 50 μm). c Bax, Bcl-2, and c-caspase 3 in hearts from mice subjected to AAV9-shTax1BP1 injection and AB surgery (n = 4). d Bax, Bcl-2, and c-caspase 3 in Tax1bp1 transgenic mice (n = 4). e TUNEL staining in NRCMs transfected with Ad-Tax1bp1 or Tax1bp1 siRNA (n = 6, Scale bars: 50 μm). **P < 0.01. Student’s unpaired t test was used to compare the data in Fig. 4a, b, e. One-way ANOVA followed by a post hoc Tukey test was used to compare the data in Fig. 4c, d.
ITCH-P73-BNIP3 was the target of Tax1bp1
We searched for possible proteins that link Tax1bp1 and apoptosis [11]. BNIP3, a protein regulating both autophagy and apoptosis, was elevated by Tax1bp1 overexpression in myocytes under Ang II insult (Fig. 5a). We also assessed the upstream target of BNIP3 and found that Tax1bp1 did not affect the protein level of ITCH but reduced the P73 level (Fig. 5a, b). In myocytes under Ang II insult with Tax1bp1 silencing, the BNIP3 level decreased with unchanged ITCH and increased P73 (Fig. 5c, d). The same results were observed in mouse heart tissues under AB surgery in both Tax1bp1 transgenic mice and AAV9-shTax1bp1-injected mice (Supplementary Fig. 5a–d). Previous studies have reported that Tax1bp1 plays an important role in autophagy [14, 15]. We also detected the autophagy-related protein level, as shown in Supplementary Fig. 5e and f. The P62 level was reduced, and the LC3II level was increased in mouse hearts after AB surgery, indicating that autophagy was increased in HF hearts, while the Tax1bp1 transgene further reduced the P62 level and enhanced the LC3II level. Consistently, Tax1bp1 knockdown increased P62 and reduced LC3II levels in HF hearts. These data suggested that Tax1bp1 further enhanced the increased autophagy during HF.

a and b BNIP3, P73, and ITCH protein levels in NRCMs transfected with Ad-Tax1bp1 under Ang II insult (n = 6). c and d BNIP3, P73, and ITCH protein levels in NRCMs transfected with Tax1bp1 siRNA under Ang II insult (n = 6). **P < 0.01. NS, no significance. One-way ANOVA followed by a post hoc Tukey test was used to compare the data in Fig. 5a–d.
To further verify the rationale of Tax1bp1 in autophagy during HF. NRCMs were transfected with Atg5 siRNA to inhibit autophagy, which was validated in our previous study [15]. As shown in Supplementary Fig. 5g and h, Atg5 knockdown counteracted Tax1bp1-induced cell apoptosis under Ang II stimulation.
Tax1bp1 interacted with ITCH, affecting P73 ubiquitination
To clarify the relationship of these targeting proteins, co-IP was performed between Tax1bp1 and ITCH, a ubiquitin ligase. As shown in Fig. 6a and b, the Tax1bp1 and ITCH proteins could interact with each other. This interaction was increased under Ang II and AB surgery stimuli (Fig. 6c). We also detected the interaction between Tax1bp1 and P73 but did not find a positive result with either endogenous or exogenous co-IP assays (Supplementary Fig. 6a, b). A previous study confirmed that ITCH mediates the ubiquitination degradation of P73 [22]. P73 is a transcription factor that regulates the expression of genes involved in cell proliferation and apoptosis. We then determined the regulatory effect of P73 on BNIP3. As shown in Fig. 6d and e, P73 downregulated the transcriptional activity of the BNIP3 promoter, whereas P73 silencing upregulated the transcriptional activity of the BNIP3 reporter (Fig. 6e). We also confirmed the regulatory function of Tax1bp1 on BNIP3. Conversely, Tax1bp1 overexpression upregulated the transcriptional activity of the BNIP3 reporter, whereas Tax1bp1 silencing reduced the transcription of BNIP3 (Fig. 6e). We detected the mRNA level of BNIP3 using hypoxia as a positive control. A consistent result was observed at the BNIP3 mRNA level (Fig. 6e). An in vivo ubiquitination assay was performed to assess whether P73 can serve as a substrate for the ubiquitin ligase activity of ITCH. 293 T cells were transfected with HA-p73, Myc-ubiquitin, and Flag-Tax1bp1 or ITCH siRNA and treated with the proteasome inhibitor MG132 prior to harvesting. As shown in Fig. 6f, Tax1bp1 significantly increased P73 ubiquitination, but siRNA-mediated knockdown of ITCH significantly reduced P73 ubiquitination. A previous study [11] found that Tax1bp1 interacts with ITCH via its two PPXY (P, proline; X, any amino acid; Y, tyrosine) motifs. We generated double mutants of the two PPXY motifs (Y741A and Y768A) in the context of the Tax1bp1 isoform and transfected them together with HA-P73. P73 ubiquitination was clearly reduced when the two PPXY motifs in Tax1bp1 were mutated (Fig. 6g). Together, these findings support the idea that by interacting with ITCH, Tax1bp1 increases the degradation of P73. To confirm this notion, we transfected myocytes with BNIP3 siRNA or ITCH siRNA (Supplementary Fig. 6c, d) and found that Tax1bp1-mediated enhanced apoptosis under Ang II insult was blocked by either BNIP3 knockdown or ITCH knockdown (Supplementary Fig. 6e). It is unclear whether mutation of the Tax1bp1 PPXY motif should only disrupt protein–protein interactions or whether it will also render the zinc finger domains incapable of binding ubiquitin. As shown in Supplementary Fig. 6f, GST-pull down results revealed that double mutants of the two PPXY motifs (Y741A and Y768A) abolished ubiquitin binding of Tax1bp1. The same result was observed when we mutated the conserved Phe residues to the Ala Zn finger (ZF: F764A) (Supplementary Fig. 6f), and the ubiquitin binding of Tax1bp1 was also abolished. In addition, to explore the effect of the Tax1bp1 mutant on myocyte apoptosis, we performed TUNEL staining. As shown in Supplementary Fig. 6g, mutation of the Tax1bp1 PPXY motif could not aggravate myocyte apoptosis like Tax1bp1 under Ang II stimulation.

a Endogenous Co-IP assay in NRCMs. b Exogenous Co-IP assay in NRCMs cotransfected with psicoR-HA-Tax1bp1 and psicoR-Flag-ITCH. c Co-IP assay in NRCMs stimulated with Ang II and Co-IP assay in protein from heart tissue in mice subjected to AB surgery. d P73 expression levels in NRCMs transfected with either Ad-P73 or P73 siRNA (n = 6). e Upper: Luciferase assay in NRCMs cotransfected with Ad-P73, P73 siRNA or Ad-Tax1bp1, Tax1bp1 siRNA; lower panel: mRNA level of BNIP3 in the indicated condition (n = 6). f 293 T cells were transfected with HA-p73, Myc-ubiquitin, and Flag-Tax1bp1 or ITCH siRNA and treated with the proteasome inhibitor MG132 prior to harvesting. Ubiquitination assays and Western blotting to detect P73 ubiquitination. g 293 T cells were transfected with plasmids expressing Myc-ubiquitin, Flag-Tax1bp1, HA-P73, and Tax1bp1 mutants (Y741A and Y768A). Ubiquitination assays and Western blotting to detect P73 ubiquitination. **P < 0.01. Student’s unpaired t test was used to compare the data in Fig. 6d and e.
BNIP3 knockdown in vivo abolished the role of Tax1bp1 in mice under AB
To further confirm that Tax1bp1 accelerates the HF process by BNIP3-mediated apoptosis, Tax1bp1 transgenic mice were subjected to AAV9-shBNIP3 injection to knockdown BNIP3 (Supplementary Fig. 7a). Four weeks after AB surgery, HF signs were improved in mice receiving AAV9-shBNIP3 injection compared with control mice (mice receiving AAV9-NC injection) (Supplementary Fig. 7b–g). These ameliorated HF signs were evidenced by diminished heart and lung weight, decreased myocyte cross-sectional area, decreased apoptosis and fibrosis volume and improved cardiac systolic and diastolic function (Supplementary Fig. 7b–g). These data demonstrate that BNIP3 is the target of Tax1bp1 in the progression of HF.
Association between Tax1bp1 level and HF
Heart tissue from heart failure patients with heart transplantation was harvested. Tax1bp1 protein levels were increased in the LV tissue of HF patients compared with those in control patients (Fig. 7a). Consistently, the BNIP3 level was upregulated, and P73 was reduced in the LV tissue of HF patients (Fig. 7b). Tax1bp1 protein levels in heart tissue were positively related to BNIP3 in HF patients (R2 = 0.749, P < 0.001) (Fig. 7c). The clinical characteristics are listed in Table S3. We also explored the association of Tax1bp1 levels with cardiac function in all control and HF patients. As a result, the Tax1bp1 mRNA level was negatively related to LVEF (R2 = 0.775, P < 0.001). Positive correlation was found with LVEDD (R2 = 0.661, P < 0.001) (Fig. 7d).

a Tax1bp1 protein levels in patients with heart failure (n = 8 in each group). b BNIP3 and P73 protein levels in patients with heart failure (n = 8 in each group). **P < 0.01. c Correlation model for Tax1bp1 and BNIP3 protein levels in patients with heart failure (n = 8 in each group). d Correlation model for Tax1bp1 mRNA level and LVEF and LVEDD in patients with heart failure (n = 8 in the control group, n = 8 in the HF group). Student’s unpaired t test was used to compare data in Fig. 7a, b; Spearman’s test was used in Fig. 7c, d.
Discussion
HF is common in adults, accounting for substantial morbidity and mortality worldwide [2]. Despite the efficacy of many therapies, its prevalence is still increasing [3]. A better understanding of the mechanism and the identification of more efficient therapies are urgently needed. Tax1bp1, a protein that initially acts as an NF-κB negative regulating factor, is now found to participate in many pathological processes [23,24,25]. In this study, we first uncovered a deteriorating role of Tax1bp1 in HF. Using cardiomyocyte-specific transgenic mice, we found that Tax1bp1 accelerated pressure overload-induced HF. When we subjected mice to Tax1bp1 knockdown in the heart, the HF process induced either by pressure overload or Ang II infusion was suppressed. This functional role of Tax1bp1 in HF affects cardiomyocyte apoptosis. We determined that Tax1bp1 interacted with ITCH and prompted P73 degradation. This activity subsequently enhanced the transcription of BNIP3, and cells with more BNIP3 were prone to apoptosis. When we silenced BNIP3 in Tax1bp1 transgenic mice, the deleterious effects of Tax1bp1 decreased.
Tax1bp1 is a ubiquitin-binding adaptor protein [16]. Tax1bp1 plays a key role in the negative regulation of NF-κB and IRF3 signaling in the innate immune system and antiviral pathway. In addition, Tax1bp1 was also reported as an autophagy receptor that modulated selective autophagic degradation [26], xenophagy [14], and macroautophagy [15, 24]. In our previously published study, we found that Tax1bp1 acted as a positive regulator that protected against diabetic cardiomyopathy via autophagy [15]. Iha et al. also reported that mice genetically knocked out for Tax1bp1 were born normal but developed age-dependent inflammatory cardiac valvulitis [27]. However, in this study, we observed the opposite result. In the process of HF (both induced by the pressure overload model and Ang II induction model), Tax1bp1 functions as a negative factor that facilitates HF pathology. These contradictory results may account for the different pathological processes between diabetic cardiomyopathy and pressure overload/Ang II-induced HF. In these two studies, under physical conditions, we did not observe any changes in cardiac structure or function in mice with knockdown of Tax1bp1 or transgenic Tax1bp1 [15]. This finding suggests that Tax1bp1 function varies in the heart under different stresses. Because cardiomyocyte inflammation is the main feature of diabetic cardiomyopathy [28], Tax1bp1, as an NF-κB P65 negative regulator, inhibits NF-κB P65 in diabetic cardiomyopathy. However, in this study, we did not observe an inhibitory effect of Tax1bp1 on NF-κB P65 in cardiomyocytes under Ang II insult or in heart tissues after AB surgery (Supplementary Fig. VIII), which was consistent with our previous study [15]. This inconsistency may account for different cell types or disease pathologies. In addition, Tax1bp1 increased autophagy in a diabetic cardiomyopathy model, as reported by Xiao Y [15]. Consistently, we also found that cardiomyocyte autophagy was increased in the HF model induced by Ang II and pressure overload. Tax1bp1-increased autophagy may further deteriorate cell injury and cause cell death. Currently, it is agreed upon that autophagy promotes or inhibits cell death depending on the internal and external environment. During heart failure, excessive autophagy will degrade proteins and organelles with normal function and inhibit the survival of cardiomyocytes, leading to deterioration of heart failure [29]. Thus, the increased autophagy in the HF model may further enhance cell death, promoting cardiac dysfunction.
There are nine types of cell death: death receptor-mediated apoptosis, death receptor-mediated necrosis (necroptosis), mitochondrial-mediated apoptosis, mitochondrial-mediated necrosis, autophagy-dependent cell death, ferroptosis, pyroptosis, parthanatos, and immunogenic cell death [30]. In HF, myocyte apoptosis is one of the main types of cell death. Mitochondrial-mediated apoptosis and mitochondrial outer membrane permeabilization (MOMP) lead to various mitochondrial apoptogens in the cytosol, promoting caspase activation [30]. In our study, Tax1bp1 enhanced cardiomyocyte apoptosis under Ang II insult. These results are inconsistent with Choi’s study showing that Tax1bp1 restricted virus-induced apoptosis in mouse fibroblasts [11].
BNIP3 is a BH3-only factor that can heterodimerize with antiapoptotic factors such as Bcl-2 and Bcl-Xl in the heart, hampering Bax and Bak activation [8]. We assessed BNIP3 levels in cardiomyocytes overexpressing or silencing Tax1bp1 and found that Tax1bp1 positively regulated BNIP3 protein levels under Ang II insult. The canonical function of proapoptotic BNIP3 is to initiate outer mitochondrial membrane pore formation and permit mitochondrial cytochrome c release, leading to programmed cardiomyocyte death [7]. This BNIP3 protein change regulated by Tax1bp1 is consistent with our observed increased apoptosis phenotype induced by Tax1bp1 overexpression in the heart and cardiomyocytes. Notably, under baseline conditions, Tax1bp1 did not alter BNIP3 protein levels in cardiomyocytes. We then explored the possible link between Tax1bp1 and BNIP3. A previous study reported that P73, a transcription factor belonging to the P53 family, could directly bind to the promoter region of BNIP3 and repress its transcription in cancer cells [31]. We found that the P73 protein level was downregulated by Tax1bp1 in cardiomyocytes, and our luciferase reporter assay further confirmed the transcriptional regulation of BNIP3 by P73 in cardiomyocytes. Thus, P73 may be a crucial molecule that mediates the positive regulation of Tax1bp1 to BNIP3 in cardiomyocytes under stress. As a ubiquitin-binding adaptor protein, Tax1bp1 may affect P73 through ubiquitination degradation. Many studies have confirmed that P73, as a member of the tumor suppressor p53 family, is tightly regulated by distinct ubiquitin-protein isopeptide (E3) ligases, including ITCH [22, 32, 33]. In our study, in cardiomyocytes, Tax1bp1 bound to ITCH, and under Ang II insult, Tax1bp1-ITCH binding increased. In the presence of HF, Tax1bp1 binding with ITCH mediates the degradation of P73, which subsequently reduces the transcriptional regression of BNIP3, thus inducing cardiomyocyte apoptosis. When we knocked down either BNIP3 or ITCH, the proapoptotic effect of Tax1bp1 on cardiomyocytes was blocked. This pathway was also confirmed in an in vivo study. When we knocked down BNIP3 levels in mouse hearts, the deleterious effect of the Tax1bp1 transgene was abolished during HF progression. However, Choi previously reported that Tax1bp1 restrained virus-induced murine embryonic fibroblast apoptosis by facilitating ITCH-mediated degradation [11]. This contradictory result may account for different cell types and different stresses that Tax1bp1-ITCH binding may target on diverse proteins.
Physiological autophagy plays an important role in maintaining cardiac homeostasis, but excessive autophagy aggravates heart-related diseases [34]. In end-stage heart failure, extensive autophagy has been detected in patient heart tissue [35]. This increased autophagy proceeds with extensive cytoplasmic destruction and nuclear disintegration leading to cardiomyocyte necrosis or at least apoptosis [35]. In this study, we also found that the autophagy level was increased after AB and that Tax1bp1 could further increase the autophagy level, which may account for the increased cardiomyocyte apoptosis. Indeed, BNIP3 also positively regulates autophagy, which may lead to more cell death [36]. Thus, the proapoptotic effects of Tax1bp1 during HF may occur via BNIP3-mediated apoptosis and autophagy, leading to increased cardiomyocyte death.
As a clinical transformation, we also demonstrated that Tax1bp1 was remarkably altered in LV tissue of HF patients and was negatively associated with LV function, such as LVEF and LVEDD, a sensitive indicator for patient prognosis and cardiovascular events.
Taken together, this evidence suggests that Tax1bp1 accelerates HF progression and impairs cardiac function. This effect was mediated by ITCH-mediated degradation of P73, leading to the activation of BNIP3-associated apoptosis. Thus, targeting increased Tax1bp1 during the HF process may be a promising therapy.
Our current study, for the first time, revealed that Tax1bp1 was upregulated in the LV of patients with HF and in the LV of pressure overload mice. Tax1bp1 deteriorated pressure overload as well as Ang II-induced HF via activation of ITCH-P73-BNIP3-mediated cardiomyocyte apoptosis. Tax1bp1 knockout was sufficient to inhibit cardiomyocyte apoptosis and HF.
Limitations
The demographics, medications and echocardiographic data in HF and non-HF patients in Table S3 showed that no difference was observed in sex or age between the two groups, but the comorbidity rate and cardiac function showed differences. However, due to the limitation of sample size, it is difficult to conduct multivariate regression analysis. Therefore, it should be noted that this may bias our clinical observations.
Conclusion
Heart failure is one of the major leading causes of death worldwide, and the absolute numbers have been increasing due to aging and improved survival after acute heart disease. Knockdown of Tax1bp1 in cardiomyocytes attenuates cardiomyocyte apoptosis and ventricular dysfunction in mice by limiting ITCH-P73-BNIP3 activation.
Future studies are warranted to determine the potential therapeutic benefit of Tax1bp1 silencing in cardiomyocytes for the prevention of cardiac remodeling and heart failure following pressure overload and neuroendocrine activation.
Data availability
The datasets used and/or analyzed in this study are available from the corresponding author upon reasonable request.
References
Di Palo KE, Barone NJ. Hypertension and heart failure: prevention, targets, and treatment. Heart Fail Clin. 2020;16:99–106.
Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Failure. 2020;22:1342–56.
Braunwald E. Heart failure. Jacc Heart Fail. 2013;1:1–20.
Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C, et al. Mechanisms contributing to cardiac remodelling. Clin Sci (Lond). 2017;131:2319–45.
Jones NR, Roalfe AK, Adoki I, Hobbs FDR, Taylor CJ. Survival of patients with chronic heart failure in the community: a systematic review and meta-analysis. Eur J Heart Fail. 2019;21:1306–25.
Vasagiri N, Kutala VK. Structure, function, and epigenetic regulation of BNIP3: a pathophysiological relevance. Mol Biol Rep. 2014;41:7705–14.
Webster KA, Graham RM, Bishopric NH. BNip3 and signal-specific programmed death in the heart. J Mol Cell Cardiol. 2005;38:35–45.
Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–46.
Dhingra A, Jayas R, Afshar P, Guberman M, Maddaford G, Gerstein J, et al. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic Biol Med. 2017;112:411–22.
Fordjour PA, Wang L, Gao H, Li L, Wang Y, Nyagblordzro M, et al. Targeting BNIP3 in inflammation-mediated heart failure: a novel concept in heart failure therapy. Heart Fail Rev. 2016;21:489–97.
Choi YB, Shembade N, Parvatiyar K, Balachandran S, Harhaj EW. TAX1BP1 restrains virus-induced apoptosis by facilitating Itch-mediated degradation of the mitochondrial adaptor MAVS. Mol Cell Biol. 2016;37:e00422–16.
Matsushita N, Suzuki M, Ikebe E, Nagashima S, Inatome R, Asano K, et al. Regulation of B cell differentiation by the ubiquitin-binding protein TAX1BP1. Sci Rep. 2016;6:31266.
Nakano S, Ikebe E, Tsukamoto Y, Wang Y, Matsumoto T, Mitsui T, et al. Commensal microbiota contributes to chronic endocarditis in TAX1BP1 deficient mice. PLoS One. 2013;8:e73205.
Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, et al. The autophagy receptor TAX1BP1 and the molecular motor Myosin VI are required for clearance of salmonella typhimurium by autophagy. PLoS Pathog. 2015;11:e1005174.
Xiao Y, Wu QQ, Duan MX, Liu C, Yuan Y, Yang Z, et al. TAX1BP1 overexpression attenuates cardiac dysfunction and remodeling in STZ-induced diabetic cardiomyopathy in mice by regulating autophagy. Biochim Biophys Acta. 2018;1864:1728–43.
Whang MI, Tavares RM, Benjamin DI, Kattah MG, Advincula R, Nomura DK, et al. The ubiquitin binding protein TAX1BP1 mediates autophagasome induction and the metabolic transition of activated T Cells. Immunity. 2017;46:405–20.
De Valck D, Jin DY, Heyninck K, Van de Craen M, Contreras R, Fiers W, et al. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene. 1999;18:4182–90.
Wu QQ, Xiao Y, Duan MX, Yuan Y, Jiang XH, Yang Z, et al. Aucubin protects against pressure overload-induced cardiac remodelling via the beta3 -adrenoceptor-neuronal NOS cascades. Br J Pharmacol. 2018;175:1548–66.
Wu QQ, Xu M, Yuan Y, Li FF, Yang Z, Liu Y, et al. Cathepsin B deficiency attenuates cardiac remodeling in response to pressure overload via TNF-alpha/ASK1/JNK pathway. Am J Physiol Heart Circ Physiol. 2015;308:H1143–54.
Wu QQ, Liu C, Cai Z, Xie Q, Hu T, Duan M, et al. High-mobility group AT-hook 1 promotes cardiac dysfunction in diabetic cardiomyopathy via autophagy inhibition. Cell Death Dis. 2020;11:160.
Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–4.
Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007;14:743–51.
Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 2010;285:14999–5009.
Sarraf SA, Shah HV, Kanfer G, Pickrell AM, Holtzclaw LA, Ward ME, et al. Loss of TAX1BP1-directed autophagy results in protein aggregate accumulation in the brain. Mol Cell. 2020;80:779–95 e10.
Verstrepen L, Verhelst K, Carpentier I, Beyaert R. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem Sci. 2011;36:347–54.
Yang Q, Liu TT, Lin H, Zhang M, Wei J, Luo WW, et al. TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 2017;13:e1006600.
Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, Smith CD, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 2008;27:629–41.
Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. 2018;61:21–8.
Du J, Liu Y, Fu J. Autophagy and heart failure. Adv Exp Med Biol. 2020;1207:223–7.
Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1765–817.
Petrova V, Mancini M, Agostini M, Knight RA, Annicchiarico-Petruzzelli M, Barlev NA, et al. TAp73 transcriptionally represses BNIP3 expression. Cell Cycle. 2015;14:2484–93.
Levy D, Reuven N, Shaul Y. A regulatory circuit controlling Itch-mediated p73 degradation by Runx. J Biol Chem. 2008;283:27462–8.
Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–48.
Corsetti G, Pasini E, Romano C, Chen-Scarabelli C, Scarabelli TM, Flati V, et al. How can malnutrition affect autophagy in chronic heart failure? Focus and Perspectives. Int J Mol Sci. 2021; 22.
Corsetti G, Chen-Scarabelli C, Romano C, Pasini E, Dioguardi FS, Onorati F, et al. Autophagy and oncosis/necroptosis are enhanced in cardiomyocytes from heart failure patients. Med Sci Monit Basic Res. 2019;25:33–44.
Gao A, Jiang J, Xie F, Chen L. Bnip3 in mitophagy: novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin Chim Acta. 2020;506:72–83.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 82170382 and 81700353), Hubei Province’s Outstanding Medical Academic Leader Program, National Key R&D Program of China(2018YFC1311300)and the Fundamental Research Funds of the Central Universities (2042017kf0060).
Author information
Authors and Affiliations
Contributions
QQW and QZT: conceived and designed the experiments; YY, QY, TTH, and YW: performed the experiments; QWX and JHZ: analyzed the data; QQW: wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Wu, Qq., Yao, Q., Hu, Tt. et al. Tax1 banding protein 1 exacerbates heart failure in mice by activating ITCH-P73-BNIP3-mediated cardiomyocyte apoptosis. Acta Pharmacol Sin 43, 2562–2572 (2022). https://doi.org/10.1038/s41401-022-00950-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00950-2
