
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder with cognitive impairment that currently is uncurable. Previous study shows that trilobatin (TLB), a naturally occurring food additive, exerts neuroprotective effect in experimental models of AD. In the present study we investigated the molecular mechanisms underlying the beneficial effect of TLB on experimental models of AD in vivo and in vitro. APP/PS1 transgenic mice were administered TLB (4, 8 mg· kg−1 ·d−1, i.g.) for 3 months; rats were subjected to ICV injection of Aβ25-35, followed by administration of TLB (2.5, 5, 10 mg· kg−1 ·d−1, i.g.) for 14 days. We showed that TLB administration significantly and dose-dependently ameliorated the cognitive deficits in the two AD animal models, assessed in open field test, novel object recognition test, Y-maze test and Morris water maze test. Furthermore, TLB administration dose-dependently inhibited microglia and astrocyte activation in the hippocampus of APP/PS1 transgenic mice accompanied by decreased expression of high-mobility group box 1 (HMGB1), TLR4 and NF-κB. In Aβ25-25-treated BV2 cells, TLB (12.5−50 μM) concentration-dependently increased the cell viability through inhibiting HMGB1/TLR4/NF-κB signaling pathway. HMGB1 overexpression abrogated the beneficial effects of TLB on BV2 cells after Aβ25-35 insults. Molecular docking and surface plasmon resonance assay revealed that TLB directly bound to HMGB1 with a KD value of 8.541×10−4 M. Furthermore, we demonstrated that TLB inhibited Aβ25-35-induced acetylation of HMGB1 through activating SIRT3/SOD2 signaling pathway, thereby restoring redox homeostasis and suppressing neuroinflammation. These results, for the first time, unravel a new property of TLB: rescuing cognitive impairment of AD via targeting HMGB1 and activating SIRT3/SOD2 signaling pathway.

Similar content being viewed by others


Introduction
Alzheimer’s disease (AD), a progressive neurodegenerative disorder and the most fatal common type of dementia, is characterized by progressive cognitive impairment, multiple behavioral and neuropsychiatric disorders [1, 2]. The neuropathological features of AD are intracellular formation of hyperphosphorylated tau proteins in neurofibrillary tangles and the abnormal deposit of extracellular amyloid-β (Aβ) protein in senile plaques in the brain [3]. Emerging evidence suggests that deposition of Aβ is a critical initiating step in the pathogenesis of AD, which triggers a series of detrimental events including neuroinflammation, oxidative stress, and neuronal loss [4, 5]. And deposition of Aβ drives neuroinflammation by activating microglia and astrocyte [6]. Adjacent to the amyloid core, microglial cells are frequently found, whereas astrocytes usually surround the plaques [7]. Activated microglia and astrocyte are elicited by release of pro-inflammatory cytokines, then induce neurotoxicity and accelerate the pathological process of AD [8]. Although the mechanism by which Aβ triggers the course of neuroinflammatory possesses is sophisticated, there is compelling evidence that high-mobility group box 1 (HMGB1) protein, a promoter and activator of neuroinflammatory responses, is involved in the pathogenesis of AD [9, 10]. HMGB1 acts as a typical damage-associated molecular patterns and a pro-inflammatory mediator via direct binding to the advanced glycosylation end product-specific receptor or the receptor toll-like receptor (TLR) 4 [11]. Upon stimulation of Aβ, HMGB1 translocates from the nucleus to the cytosol, and then extracellular HMGB1 binds with TLR4 to activate the nuclear factor (NF)-κB signaling, especially p65 activation, leading to release of inflammatory cytokines [12]. Of note, translocation of HMGB1 from the nucleus to cytosol is affected by post-translational modifications, including acetylation, which are mediated through the equilibrium of histone deacetylases (HDACs) and histone acetyltransferases. Sirtuins are an evolutionarily conserved family of nicotinamide adenine dinucleotide-dependent lysine deacylase, and also classified as class III HDACs due to their capacity to eliminate acetyl from lysine residues on histones proteins [13]. Among the SIRT family (SIRT1-7), SIRT3 is a primary mitochondrial lysine deacetylase with the highest deacetylase activity, and regulates mitochondrial function, reactive oxygen species (ROS) levels and neuroinflammation [14]. However, whether SIRT3 can affect the HMGB1 transcription and the relationship between them during the process of neuroinflammation in AD remains still unclear.
Unfortunately, the exploitation and approval of novel drugs against AD is a struggling and expensively task with a high rate of failure in clinical endpoints [15]. Learning from these failures, unexpected adverse drug effects and tolerability are the critical issues, and the exploitation track should escape from unpredictable derailment because of toxicities not expected by preclinical study [16]. Thus, more safe and effective drugs or strategies are desperately required. Notably, food materials appointed as “generally recognized as safe” (GRAS), similar to drug repurposing such as metformin or aspirin [17, 18], are deemed as a credible resource of safe chemical substances for drug discovery; in particular, it is currently of great significance to detect potential secondary benefits of food additive [19]. TLB is a naturally occurring sweetener found in abundance in leaves of Lithocarpus polystachyus Rehd., which is also named “sweet tea” in the folk, and has already been approved as new food materials in China since 2017 [20]. Most excitingly, Flavor and Extract Manufactures Association of the United States (FEMA) designated TLB as a GRAS (FEMA No. 4674) since 2011; subsequently, the World Health Organization and Food and Agriculture Organization concurs with the FEMA designation and further approved TLB for use as a food additive (JECFA No. 2171) since 2012 [21]. Emerging evidence indicates that TLB exerts pleiotropic pharmacological activities including anti-tumor activity, anti-HIV-1 activity, anti-inflammatory effect, etc. [22,23,24]. Of note, previous studies have revealed that TLB effectively attenuated cerebral ischemia-reperfusion injury due to its antioxidant properties and anti-inflammatory activities through activation of SIRT3 activity and inhibition of TLR4 signaling pathway [25]. Recently, preliminary experiments suggested that TLB supplementation efficiently prolonged lifespan of Caenorhabditis elegans in an anti-oxidative stress-dependent fashion, reduced Aβ25-35-induced HT22 cell apoptosis and alleviated cognitive deficits in 3×FAD AD animal model [26,27,28]. However, to date, there is no comprehensive description about the detailed mechanism of TLB on AD, and whether TLB can penetrate the BBB and the reciprocity between HMGB1 and SIRT3 is involved in the beneficial effects of TLB on AD still remains a mystery. Thus, two animal models: (1) APPswe/PS1dE9 (APP/PS1) double transgenic AD mice, an AD-like pathology animal model and (2) Aβ25-35-induced AD rats, an acute toxicity of amyloid animal model, and Aβ25-25-induced injury in BV2 cells in vitro were used to elucidate the precise mechanism of TLB on AD.
Materials and methods
Animal
APP/PS1 mice and littermate wild-type (WT) mice were purchased from Cavins Laboratory Animal Co. Ltd of Changzhou (Changzhou, China) [specific pathogen free (SPF), certificate No. SCXK 2016-0010)]. Adult male Sprague-Dawley rats weighing 280 ± 20 g were obtained from the Experimental Animal Center of Daping Hospital (SPF, certificate No. SCXK 2014-0011). The animals were raised 5 to 6 per cage with a 12 h light-dark cycles and an appropriate temperature (23 ± 1 °C) and humidity (55% ± 5%) and allowed to drink water ad libitum. All animal experimental operations were approved by the Experimental Animal Ethics Committee of the Zunyi Medical University and all the animal care and experimental procedures were performed in accordance with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978). Randomization was used to allocate animals to various experimental groups and data analysis was performed by an investigator, who was blinded to the treatment.
Experimental designs
Six-month-old male APP/PS1 mice and age-matched WT mice were randomly divided into four groups: WT group, APP/PS1 group, APP/PS1 + TLB (4 mg/kg) group, APP/PS1 + TLB (8 mg/kg) group. TLB was administered intragastrically with the dose of 4, 8 mg/kg (twice a day) for 3 months. The age-matched WT group and the APP/PS1 group were treated with volume-matched saline.
The rats were randomly divided into six groups: sham group, sham + TLB (10 mg/kg) group, Aβ25–35 group, Aβ25–35 + TLB (2.5 mg/kg) group, Aβ25–35 + TLB (5 mg/kg) group, Aβ25–35 + TLB (10 mg/kg) group. An animal model of AD was built using intracerebroventricular (ICV) injection of Aβ25–35, which is considered as a toxic fragment of full-length Aβ that could be properly utilized to stimulate oxidative stress, microglial activation and neuroinflammation [29]. In brief, the Aβ25–35 was aggregated through incubation in distilled water (1 mg/kg) at 37 °C for 4 days and diluted to the final concentration with saline directly prior to the experiment. The rats were anaesthetized by intraperitoneal (i.p.) injection of sodium pentobarbital (30 mg/kg), and small burr holes were drilled on bilateral sides of the skull (1.0 mm posterior to bregma and 1.5 mm lateral to the midline) to allow ICV injection of Aβ25–35 at the depth of 3.5 mm. The injection lasted 5 min and the needle was left in place for 5 min after the injection. Sham group underwent the same operation as mentioned above except injection of Aβ25–35. TLB was administered intragastrically at the dose of 2.5, 5, 10 mg/kg (twice a day) from the 2nd day after the neurosurgery for 14 days. The sham group and the Aβ25–35 group were treated with volume-matched saline.
Morris water maze (MWM)
The MWM test was performed to evaluate the spatial learning and memory ability of the mice or rats as described in our previous study [30]. Briefly, the test was performed using the water maze with a circular pool with diameter 120 cm, height 50 cm, a water temperature of 23 ± 1 °C, and the pool area was divided into four equal quadrants with a platform (diameter: 10 cm) submerged 1 cm under the water surface. After a 5-day navigation experiment to find the platform that submerged 1 cm under the water surface, on the 6th day, a probe trial was conducted to determine memory retention. The escape latency, time spent in the target quadrant, number of target crossings and swimming speed, were measured by the analysis-management system.
Y-maze
The Y-maze was conducted to test spatial recognition memory of mice or rats as described in our previous study [25]. In brief, the Y-maze was performed with an apparatus with identical arms (30 cm × 10 cm × 20 cm). Mice or rats were put at the beginning arm and allowed to explore other arm at liberty for 8 min. Subsequently, mice or rats were got back to the same beginning arm and permitted to explore all three arms at liberty for 5 min, and then the number entries into an arm were recorded. Spontaneous alternation (%) = [(number of actual alternations)/(number of total arm entries − 2)] × 100, was utilized as an index to determine spatial recognition memory.
New object recognition (NOR)
The NOR was conducted to explore the recognition memory of AD model as described in our previous study [31]. Briefly, mice or rats were allowed to acquaint an experimental apparatus (50 cm × 50 cm × 50 cm) with two same objects. In acquisition phase, the mice or rats were allowed to explore the two objects for 10 min at liberty. In familiarization phase, the mice or rats were put into the same experimental apparatus with two objects for 10 min. Then, the mice or rats were taken out of experimental apparatus for a 10 min intertrial interval, then one of the familiar objects was replaced by a novel object that the mice or rats did not habituate before. The nose and/or forepaws of mice or rats directed to the objects within 2 cm or touched were identified as exploratory behavior. The percentage of time spent exploring the novel object relative to the total time spent in both objects was calculated by discrimination index. After the behavioral tests, the mice or rats were anaesthetized by i.p. injection of sodium pentobarbital (50 mg/kg or 30 mg/kg), then the mice or rats were sacrificed and the brains were removed immediately for the following experiments.
RNA sequencing (RNA-seq) and data processing
Total RNA was extracted from hippocampal tissues of mice in WT, APP/PS1, and APP/PS1 + TLB (8 mg/kg) groups using TRIzol buffer and then quantified using an Agilent 2100 (Agilent Technologies Co. Ltd., Palo Alto, CA, USA) analysis meter. Purification of poly-A containing RNA molecules, RNA fragmentation, strand-specific random primed cDNA library preparation, and single-read sequencing on Illumina Nova seq 6000 were conducted by the LC-Bio Co., LTD. (Hangzhou, China). RSEM was used to quantify the genes expression level, and the differentially expressed genes (DEGs) were selected with fold change (FC) greater than 1.5 and P value <0.05 by R package-Ballgown in the present study. Thereafter, Venn diagram was generated by Venny 2.1 online tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html), enrichment analyses were based on the KEGG pathway (https://www.genome.jp/kegg/pathway.html), protein/protein interactions network analysis was based on STRING 11.0 (https://string-db.org/) and presented by Cytoscape 3.6.0 software.
Thioflavin S staining
Thioflavin S staining was utilized to determine the Aβ plaques as mentioned in previous study [32]. In brief, paraffin-embedded brain sections (4-μm thickness) were stained with 1% Thioflavin S in the dark for 10 min, followed by washing with 70% ethanol and three washes with PBS. Thereafter, Thioflavin S-stained Aβ plaques were detected using a fluorescence microscope. The areas of Aβ plaques in the hippocampus of the APP/PS1 mice were counted using Image Pro Plus 6.0 software.
Nissl staining
Nissl staining was used to evaluate the survival neurons in hippocampus as described previously [33]. In brief, after the behavioral tests, mice or rats were anaesthetized and then transcardially perfused with saline followed by 4% paraformaldehyde (PFA) in PBS (0.1 mol/L, pH 7.4). Brain tissues were collected and post-fixed in 4% PFA at 4 °C overnight, followed by a graded series of sucrose solution and stored at −20 °C. Subsequently, the frozen brains were sectioned (4-μm thickness) using a Leica vibrating blade microtome for Nissl staining. Image Pro Plus 6.0 software was used to analyze the survival neurons.
Immunofluorescence histochemistry (IHC)
IHC was applied to detect the activation of microglia and astrocyte by ionized calcium binding adapter molecule-1 (Iba-1) staining and glial fibrillary acidic protein (GFAP) staining as reported in our previous study [25]. Briefly, mice or rats were anaesthetized and transcardially perfused as mentioned above. Frozen brain sections were cut into 3-μm thickness in a coronal plane by a cryostat microtome, and IHC staining was conducted. The slices were incubated with the primary antibodies against Iba-1 and GFAP overnight at 4 °C, and then incubated with secondary antibodies for 1 h at room temperature. Pixel counts were derived from the average of five adjacent sections per mice or rats, then the Iba-1 and GFAP-positive cells were counted using Image Pro Plus 6.0 software.
Determination of redox status
The levels of ROS, malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), 8-isoprostane, the activities of superoxide dismutase (SOD) and its subtypes SOD1–3, glutathione peroxidase (GSH-Px), catalase (CAT) were measured to determine the redox status. Briefly, the hippocampal tissues were collected and homogenized with PBS (0.1 mol/L, pH 7.4). Then the samples were centrifuged at 3000 × g for 20 min at 4 °C, then the levels of ROS, MDA, 4-HNE, 8-isoprostane and the activities of SOD, SOD1, SOD2, SOD3, GSH-Px, CAT were measured using appropriate detection kits. Dihydroethidium (DHE) staining was used to detect endogenous superoxide in the hippocampus in vivo as described previously [34]. In brief, mice or rats were administered DHE (20 mg/kg) by single i.p. injections. After 18 h, mice or rats were anesthetized and then perfused with 4% PFA, then the DHE fluorescence of hippocampal sections was detected using fluorescence microscopy and the intensity of red fluorescence was quantified using Image Pro Plus 6.0 software.
Liquid chromatography-mass spectrometry (LC-MS) analysis of TLB-penetrated into the blood-brain barrier (BBB)
In brief, 0.5 mg of TLB was dissolved with 1 mL methanol, then 500 μg/mL TLB standard solution was obtained. Next, a 180 μL aliquot of brain tissue homogenates was deproteinized by 600 μL methanol, and centrifuged at 12,000 × g for 15 min at 4 °C. A 60 μL aliquot of the supernatant was analyzed using LC-MS analysis. The chromatographic condition was listed as following: Agilent ZORBAX SB-C18 column (4.6 mm × 150 mm, 5 μm); mobile phase A: 0.1% formic acid acetonitrile, mobile phase B: 0.1% formic acid aqueous solution; flow rate: 0.8 mL/min; gradient elution: 0–5 min, 75% A; temperature of column: 25 °C; injection of volume: 1 μL. Mass spectrometry condition was listed as following: negative ionization multiple reaction monitoring Q1: 435.0, Q3: 273.1; declustering voltage: −104 V, collision energy: −24 V; curtain gas 40 psi; ionization voltage −4500 V; temperature of ion source: 550 °C; spray gas: 60 psi; auxiliary heating gas: 60 psi.
Cell culture and drug treatments
Experiments in vitro were carried out using the murine microglial immortalized cell line BV2, because BV2 cells were used to mimic Aβ-induced AD pathology as described previously [35, 36] and manipulation of gene expression could be achieved in BV2 cells in contrast to primary microglial at a very low transfection efficiency [37]. The BV2 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and incubated in a humidified incubator at 37 °C with 5% CO2. Thereafter, BV2 cells were simultaneously treated with Aβ25-35 and different concentrations of TLB (12.5, 25, 50 μM) or its analog (50 μM) for 48 h.
Determination of cell viability and lactate dehydrogenase (LDH)-based cytotoxicity assay
Cell viability was evaluated using Cell counting kit (CCK)-8. In brief, BV2 cells were treated as mentioned above. Then the cells were incubated with 10 µL of CCK-8 solution for 4 h at 37 °C with 5% CO2, and then the absorbance was detected at 490 nm by a microplate reader. In parallel, the cytotoxicity was measured by quantitating the release of LDH. Following incubation, the media was collected and centrifuged at 400 × g for 5 min. The amount of LDH released from BV2 cells was lysed in 1% Triton X-100 according to the manufacturer’s protocols.
Determination of pro-inflammatory factors
The levels of pro-inflammatory factors in the hippocampus of mice and rats or the cell culture medium were determined using enzyme-linked immune-sorbent assay (ELISA) as described previously [38]. In brief, the hippocampal tissues or cells were collected and homogenized with PBS (0.1 mol/L, pH 7.4). Then the samples were centrifuged at 3000 × g for 20 min at 4 °C, the levels of interleukin (IL)−1β, IL-6, tumor necrosis factor (TNF)-α and HMGB1 level were evaluated using ELISA kits following the manufacturer’s protocols.
Detection of mitochondrial ROS (mtROS)
mtROS generation was detected using flow cytometer (Navios, Beckman Coulter) after staining with Mito-SOX, a specific fluorescent probe for evaluating mtROS production. In brief, the cells were treated as mentioned above. Thereafter, the cells were resuspended in Hanks’ balanced salt solution and stained with Mito-SOX (2.5 μM at 37 °C for 20 min). The percentages of Mito-SOX-positive cells were analyzed using flowing software.
Measurement of the oxygen consumption rate (OCR)
OCR was measured using the Seahorse XFe24 Extracellular Flux Analyzer (Seahorse Biosciences, USA) as described in previous study [39]. OCR was assessed in the presence of oligomycin (1 μM), carbonylcyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP; 1 μM), a mixture of rotenone and antimycin A (1 μM), and the XFe Wave software (Seahorse Biosciences) was applied to quantify the basal respiration, ATP production, respiratory capacity.
Western blot
Western blot analysis was performed as described previously [25]. In brief, the hippocampal tissues of mice or rats, and BV2 cells were homogenized using RIPA buffer. Then equal amounts of protein and 10 μg protein from tissue lysates or 20 μg protein of cell lysates were separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a PVDF membrane. Thereafter, the membrane was blocked with 5% nonfat milk in TBST at room temperature for 1 h. Subsequently, the membranes were incubated with corresponding primary antibodies including HMGB1 (1:1000), TLR4 (1:1000), myeloid differential protein (MyD)88 (1:1000), TNF receptor-associated factor (TRAF) 6 (1:1000), p-NF-κBp65 (1:1000), NF-κBp65 (1:1000), Sirt3, acetyl-SOD2 (ac-SOD2) and SOD2 overnight at 4 °C. Then, species-specific HRP-conjugated secondary antibody was used to mark membrane-bound antibodies, and β-actin or Histone H3 was applied as a loading control. ECL Western blot detection reagents were used to visualize representative bands and the band optical intensity was quantified by ImageJ software.
Co-immunoprecipitation (Co-IP)
The SIRT3 antibody, HMGB1 antibody or HMGB1 antibody (Acetyl-Lys12) was incubated with a hippocampal tissue extract to form an antibody/antigen complex in solution at 4 °C overnight. Then the complex was pulled out of the sample by protein A/G agarose beads (10 μL). Thereafter, agarose beads were washed with lysis buffer for three times, and then the supernatants were analyzed by Western blot analysis. The acetylation of HMGB1 was determined using anti-lysine-acetylation antibody, then the acetylated level and site of HMGB1 were detected using anti-acetyl-HMGB1 (Lys12) antibody.
Overexpression (OE) of HMGB1
HMGB1-OE BV2 cells were generated using lentivirus. The lentivirus-based HMGB1-OE plasmid, pHBLV-CMV-MCS-3FLAG-EF1-ZsGreen-T2A-PURO, with the HMGB1 sequence (NM_001313893.1). In brief, BV2 cells were transfected with lentiviral-packed HMGB1-OE for 48 h, and then the OE of HMGB1 by lentivirus was confirmed by Western blot. Thereafter, the transfected cells were cultured and treated with or without TLB after Aβ25–35 insults for further analysis.
Generation of Sirt3-knockout (KO) BV2 cells
Sirt3-KO BV2 cells were generated using a CRISPR/Cas9 system as previously described [25]. In brief, BV2 cells were transfected with lentiviral-packed CRISPR/Cas9 Sirt3 at an MOI of 100 for 48 h. Thereafter, the knockout of endogenous Sirt3 by CRISPR/Cas9 Sirt3 lentivirus was verified using Western blot. Thereafter, the transfected BV2 cells were cultured and treated with or without TLB after Aβ25-35 insults for following analysis.
Sirt3 activity and NF-κBp65 DNA-binding activity assay
The BV2 cells were treated with or without TLB as described above. Then the protein was extracted by SIRT3 assay buffer and the activity of SIRT3 was determined with the CycLex Sirt3 Deacetylase Fluorometric Assay Kit according to the manufacturer’s protocols.
Nuclear extracts of hippocampal tissue were prepared by the NE‐PER Nuclear and Cytoplasmic Extraction Reagents. Afterward, the NF-κB binding activity was determined using the NF-κBp65 Transcription Factor Assay Kit according to the manufacturer’s instructions.
Molecular docking
Molecular docking was performed by Autodock 4.2 software to predict the affinity of TLB and HMGB1, TLR4, MyD88, TFRA6 or NF-κB. The human X-ray crystal structures of HMGB1 (PDB code: 2LY4), TLR4 (PDB code: 3FXI), MyD88 (PDB code: 4EO7), TFRA6 (PDB code: 3HCS) and NF-κB (PDB code: 1TNR) were retrieved from the Protein Data Bank to complete the docking simulation. Thereafter, the resulting conformations and their energies were wrote to a molecular database file, and the ligand/protein complex were generated by the PyMOL molecular graphics system. The interplay between HMGB1 and SIRT3 (PDB code: 3GLS) was detected using ZDOCK and RDOCK, which was a widespread admissive method to implement detailed prediction of protein/protein docking as described in previous study [31].
Surface plasmon resonance (SPR)
Biacore X100 instrument (GE Healthcare, Uppsala, Sweden) with Biacore X100 and sensor chip CM5 (BR-1003-99; GE Healthcare) was performed to further confirm the interaction between HMGB1 and TLB. Briefly, HMGB1 (ab167781; Abcam) was dissolved in 10 mM sodium acetate (pH 5.5) at a concentration of 20 μg/mL, and absorbed as immobilization. TLB was diluted at 0, 3.9, 7.81, 15.625, 31.25, 62.5, 125, 250, and 500 μM with PBS-P buffer. The protein interaction time was set to 120 and 300 s for dissociation. Glycine-HCl (pH 2.0, BR-1003-55; GE Healthcare) was applied for regeneration. The Biacore evaluation 3.1 analysis software (GE Healthcare) was used to analyze the data for evaluating binding affinity between TLB and HMGB1. Equilibrium dissociation constant (KD) was evaluated using global fitting of the affinity data from different concentrations of TLB.
Data and statistical analysis
Statistical analysis was undertaken only for studies where each group size was at least n = 5. All data were expressed as the mean ± standard error of the mean and analyzed using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). Two or multiple groups were compared using Student’s unpaired t test or one-way ANOVA. For the MWM test, two-way repeated-measures ANOVA. Bonferroni’s post hoc tests were run only when F achieved P < 0.05 and there was no significant variance in homogeneity. P < 0.05 was set as indicating statistical significance.
Materials
TLB and its analog (purity ≥ 98%) were from Chengdu Push Bio-technology Medical Technology Corporation (Chengdu, China). The nuclear magnetic resonance spectra and mass spectrum data of the TLB and its analog were shown in Supplementary Fig. S1. Aβ25-35 (Cat# A4559) and Thioflavin S (Cat# T1892) were purchased from Sigma-Aldrich (St. Louis, MO, USA). DHE (Cat# D23107) and NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Cat# 78835) were purchased from Thermo Fisher scientific (Barrington, IL, USA). LDH (Cat# RJ16172), IL-1β (Cat# RJ16944), IL-6 (Cat# RJ16958), TNF-α (Cat# RJ14858), ROS (Cat# RJ15780), MDA (Cat# RJ15503), 4-HNE (Cat# RJ20901), 8-hydroxy-2′-deoxyguanosine (Cat# RJ 11170), SOD (Cat# RJ17004), SOD1 (Cat# RJ28487), SOD2 (Cat# RJ28488), SOD3 (Cat# RJ28489), GSH-Px (Cat# RJ25745), and CAT (Cat# RJ15737) assay kits were obtained from Shanghai Renjie Bioengineering Institute (Shanghai, China). HMGB1 ELISA kit (Cat# ST51011) was purchased from Shino Test Corporation (Kanagawa, Japan). CycLex SIRT3 Deacetylase Fluorometric Assay Kit (Cat# CY-1153V2) was obtained from MBL life science (Tokyo, Japan). CCK-8 (Cat# ab228554), and the primary antibodies were used in present study, including Iba-1 (Cat# ab178846), GFAP (Cat# ab68428), HMGB1 (Cat# ab79823), TLR4 (Cat# ab13867), MyD88 (Cat# ab219413), TRAF6 (Cat# ab33915), NF-κBp65 (Cat# ab16502), p-NF-κBp65 (Cat# ab76302), SIRT3 (Cat# ab189860), ac-SOD2 (Cat# ab137037), SOD2 (Cat# ab68155), NF-κBp65 transcription factor assay kit (Cat# ab133112) and GAPDH activity assay kit (Cat# ab204732) were purchased from Abcam (Cambridge, UK). Mito-SOX (Cat# M36008) and protein A/G magnetic beads (Cat# 88803) were purchased from Thermo Fisher scientific (Barrington, IL, USA). Anti-acetylated-Lysine antibody (Cat# 9441) was from Cell Signaling Technology (Danvers, MA, USA) and anti-acetyl-HMGB1 (Lys12) (Cat# OASG03545) antibody was from Aviva Systems Biology (San Diego, CA, USA).
Results
TLB rescues cognitive impairment in APP/PS1 mice and Aβ25-35-induced AD rats
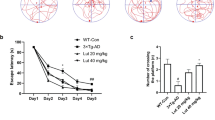
To understand the effects of TLB on AD, two AD-like models, APP/PS1 mice and Aβ25-35-induced AD rats, were utilized. First, we examined the spatial learning and memory of mice through MWM. Compared with WT group, APP/PS1 mice showed significant spatial learning and memory deficits as evidenced by longer escape latency time during the training trails, a lower crossing number on the platform, and less time spent in the target quadrant in the probe trail. However, TLB treatment significantly reversed these defects in APP/PS1 mice (Fig. 1a–e). Furthermore, the percentage of correct spontaneous alternations and the discrimination index of novel from familiar objects were also decreased in APP/PS1 mice, while, TLB significantly reversed these changes (Fig. 1f, g), which indicated that TLB effectively attenuated cognitive impairment in APP/PS1 mice. Moreover, the swimming speed among all groups did not display any alteration during training time, which suggested there was no motor disturbance in the treated mice (Fig. 1h). Especially, due to Aβ promoted formation of senile plaque that is a crucial hallmark of AD, we subsequently determined whether TLB treatment decreased amyloid plaque formation in APP/PS1 mice using thioflavin S staining. The results showed that TLB remarkably reduced Aβ deposition in hippocampus than that of APP/PS1 group (Fig. 1i, j). Intriguingly, to corroborate the results above, we further investigated these researches in another animal model of AD, Aβ25-35-induced AD rats. The results distinctly showed that TLB (2.5, 5, 10 mg/kg) effectively ameliorated neuronal pathological alteration in hippocampus, and similar results were obtained (Supplementary Fig. S2).

TLB at doses of 4 or 8 mg/kg was administered to APP/PS1 mice or age-matched WT mice (starting from 6 months old) for 3 months. a Experimental procedures for TLB administration. The MWM, Y-maze and NOR were performed for determining the learning and memory abilities of mice in different groups. b Escape latency (n = 10 or 11). c Representative track images of mice in the probe test. d The number of crossing platform (n = 10 or 11). e Time spent in the target quadrant. f Alternation rates (%) in the Y-maze task (n = 10 or 11). g Discrimination index in the NOR test (n = 10 or 11). h Swim speed (n = 10 or 11). i Representative images of thioflavin S staining in hippocampus. j Quantification of the areas of Aβ plaques (n = 5). Analysis of transcriptomics. k The fold change of DEGs of WT vs. APP/PS1 and APP/PS1 + TLB vs. APP/PS1 were summed up using volcano Plot. l The number of different and overlapped DEGs from WT vs. APP/PS1 and APP/PS1 + TLB vs. APP/PS1 were summed up using venn diagram. m Analysis of KEGG pathway. n PPI network was generated by Cytoscape 3.6.0. The size of respective node indicated the degree of connectivity in PPI network. Larger node (HMGB1, TLR4, NF-κB) shares more connection to other nodes. Scale bar = 500 μm. The data were presented as the mean ± SEM. **P < 0.01 vs. WT group; #P < 0.05, ##P < 0.01 vs. APP/PS1 group.
Furthermore, according to the results of genome-wide RNA-seq analysis, up-regulated DEGs and down-regulated DEGs were identified in comparison of WT vs. APP/PS1 and APP/PS1 + TLB vs. APP/PS1, and the FC of these DEGs was visualized by volcano plot (Fig. 1k). Thereafter, a venn diagram to visual depiction of the similarities and differences between the DEGs was created. The results showed that there were 350 overlapped DEGs between WT vs. APP/PS1, 163 overlapped DEGs between APP/PS1 + TLB vs. APP/PS1, and only 58 overlapped DEGs among the three groups (Fig. 1l). Furthermore, KEGG pathway analysis ascertained a considerable enrichment of genes involved in TLR signaling pathway and NF-κB signaling pathway, et al. (Fig. 1m). Interestingly, protein-protein interactions in the DEGs identified after TLB treatment were depicted. There were 485 interaction pairs among these DEGs-encoded proteins, the size of circles represented the degree of protein connection to others. Notably, RNA-seq analysis disclosed that hippocampus of mice expressed high levels of HMGB1, TLR4, NF-κB, which might be the dominant relevant genes (Fig. 1n). These results suggest that TLB, in deed, triggers an anti-inflammatory response contributing to retardation of neuroinflammation in AD. Most interestingly, TLB signal was significantly detected in the brain homogenates of rats (Supplementary Fig. S3), suggesting that TLB could penetrate the BBB. Together, these results reveal that TLB effectively rescues cognitive deficits of AD animal models, and HMGB1 might be a pivotal factor involved in the neuroprotective effects of TLB on AD.
TLB inhibits activation of microglia and astrocytes in hippocampus of AD models via suppression of HMGB1/TLR4/NF-κB signaling pathway
Next, to explore the effects of TLB on activation of microglia and astrocytes in hippocampus of AD models, we performed immunolabeling for microglia and astrocytes. The results showed that a significant increase in Iba-1 and GFAP expressions in the CA1, CA3 and DG hippocampal regions of APP/PS1 mice or rats in Aβ25-35 group than those of age-matched WT mice or sham group. However, a significant decrease in Iba-1 and GFAP expressions in the CA1, CA3 and DG hippocampal regions of TLB group was observed than those of APP/PS1 group (Fig. 2a–h) or Aβ25-35 group (Supplementary Fig. S4a–d), which suggested that TLB efficiently inhibited activation of microglia and astrocytes in hippocampus of APP/PS1 mice or Aβ25-35-induced AD rats. These findings indicate that TLB is able of halting neuroinflammation at early stage of AD. Moreover, the levels of IL-1β, IL-6, TNF-α and HMGB1 were measured by ELISA assay. The results showed that the levels of IL-1β, IL-6, TNF-α and HMGB1markedly increased in APP/PS1 mice or rats of Aβ25-35 group than those of age-matched WT group or sham group. However, TLB significantly decreased the levels of inflammatory cytokines of IL-1β, IL-6, TNF-α and HMGB1, which suggested that TLB hampered neuroinflammation in APP/PS1 mice or Aβ25-35-induced AD rats (Fig. 2i–l and Supplementary Fig. S4e–h). Furthermore, the expressions of the crucial proteins in HMGB1/TLR4/NF-κB signaling pathway and NF-κBp65 activity were determined using Western blot and NF-κBp65 transcription factor assay, respectively. The results showed that HMGB1 of cytoplasm, TLR4, MyD88, and TRAF6 protein expressions, phosphorylation of NF-κBp65 protein level, as well as NF-κBp65 DNA-binding activity were observably increased, while, HMGB1 of nucleus was decreased in hippocampus of APP/PS1 mice or AD rats than those of age-matched WT group or sham group; however, TLB significantly reversed these changes (Fig. 2m–y and Supplementary Fig. S4i–u). These findings indicate that the protective effect of TLB on the rodent models of AD, at least partly, is due to mediation of HMGB1/TLR4/NF-κB signaling pathway.

The expressions of the microglia marker (Iba-1) or astrocyte marker (GFAP) in CA1, CA3 and DG hippocampal regions were observed using IHC. a Representative images of Iba-1 expression in hippocampus. b Iba-1-positive cell numbers in the CA1 region (n = 5). c Iba-1-positive cell numbers in the CA3 region (n = 5). d Iba-1-positive cell numbers in the DG region (n = 5). e Representative images of GFAP expression in hippocampus. f GFAP-positive cell numbers in the CA1 region (n = 5). g GFAP-positive cell numbers in the CA3 region (n = 5). h GFAP-positive cell numbers in the DG region (n = 5). i IL-1β level (n = 6). j IL-6 level (n = 6). k TNF-α level (n = 6). l HMGB1 level (n = 6). m Representative Western blot of cytoplasm-HMGB1 protein expression. n Quantitation of cytoplasm-HMGB1 protein expression (n = 5). o Representative Western blot of nucleus-HMGB1 protein. p Quantitation of nucleus-HMGB1 protein expression (n = 5). q Representative Western blot of TLR4 protein expression. r Quantitation of TLR4 protein expression (n = 5). s Representative Western blot of MyD88 protein expression. t Quantitation of MyD88 protein expression (n = 5). u Representative Western blot of TRAF6 protein expression. v Quantitation of TRAF6 protein expression (n = 5). w Representative Western blot of phosphorylation of NF-κBp65 protein level. x Quantitation of phosphorylation of NF-κBp65 protein level (n = 5). y NF-κBp65 binding activity determined at OD 450 nm (n = 6). The data were presented as the mean ± SEM. **P < 0.01 vs. WT group; #P < 0.05, ##P < 0.01 vs. APP/PS1 group. (magnification ×40 and ×400, scale bar = 500 μm and 50 μm).
TLB mitigates oxidative stress in the hippocampus of AD models through activation of SIRT3/SOD2 pathway
Considering on the overproduction of oxidative stress triggering increased Aβ plaque deposition and neuroinflammation, we determined the oxidative status in the APP/PS1 mice and the Aβ25-35-induced AD rats with treatment of TLB. The results showed that the levels of oxidative stress markers including ROS, MDA, 4-HNE, 8-isoprostane were significantly higher in APP/PS1 mice or Aβ25-35-induced AD rats than those of age-matched WT group or sham group; while, TLB prominently reversed these changes (Fig. 3a–d and Supplementary Fig. S5a–d). Moreover, activities of the antioxidant markers including SOD and its subtypes SOD1–3, GSH-Px, CAT were lower in APP/PS1 mice or Aβ25-35-induced AD rats than those of age-matched WT group or sham group; while, TLB prominently reversed these changes (Fig. 3e–j and Supplementary Fig. S5e–j). Moreover, the intensity of superoxide dye was significantly increased in the hippocampus of APP/PS1 mice or Aβ25-35-induced AD rat brains than those of age-matched WT group or sham group; whereas, TLB effectively reduced the increase in the intensity of superoxide dye in the hippocampus than those of APP/PS1 mice or Aβ25-35-induced AD rats (Fig. 3k–n and Supplementary Fig. S5k, l). Furthermore, TLB significantly increased the SIRT3 protein expression and decreased the acetylation level of SOD2 than those of APP/PS1 mice or Aβ25-35-induced AD rats (Fig. 3o–q and Supplementary Fig. S5m–p). These findings suggest that TLB mitigates oxidative stress in the hippocampus of AD models through activation of SIRT3/SOD2 pathway.

a ROS level. b MDA level (n = 5). c 4-HNE level (n = 5). d 8-isoprostane level (n = 5). e SOD activity (n = 5). f SOD1 activity (n = 5). g SOD2 activity (n = 5). h SOD3 activity (n = 5). i GSH-Px activity (n = 5). j CAT activity (n = 5). k DHE signal (red) indicated the levels of superoxide in the hippocampus of APP/PS1 mice or age-matched WT mice. l DHE intensity in CA1 region of hippocampus (n = 5). m DHE intensity in CA3 region of hippocampus (n = 5). n DHE intensity in DG region of hippocampus (n = 5). o Representative Western blots of SIRT3, ac-SOD2 and SOD2. p Quantitation of SIRT3 protein expression. q Quantitation of ac-SOD2 level (n = 5). The data were presented as the mean ± SEM. **P < 0.01 vs. WT group; #P < 0.05, ##P < 0.01 vs. APP/PS1 group.
TLB is predicted to bind with HMGB1
Based on the results of RNA-seq analysis and since TLB inhibited the activation of HMGB1, we next asked whether TLB could serve as a ligand of HMGB1. The results showed that the binding energy of TLB with HMGB1, TLR4, MyD88, TRAF6 and NF-κB was −5.06, −3.44, −3.42, −1.37 and −3.14 kcal/mol, which indicated that among HMGB1/TLR4/NF-κB pathway, TLB selectively bound to HMGB1, due to the standard threshold for a ligand to bind with target protein is considered to be greater than or equal to −5 kcal/mol. We thereafter explored the assumptive binding modes and interactions within the amino acid pocket, including Ser14, Tyr15, Met12, Asp49, Trp48, and Ile50 (Fig. 4a). Similarly, we also implemented molecular docking analyses to detect the affinity of structural analog of TLB with HMGB1. Intriguingly, our results showed that the binding energy of structural analog of TLB with HMGB1 was −4.15 kcal/mol, which suggested that analog of TLB displayed weak interaction with HMGB1. Furthermore, SPR was utilized to verify the ability of TLB to bind HMGB1 protein in vitro binding. Full kinetics performed at different concentrations of TLB corroborated strong affinity for HMGB1 in the molar range when fitting the curves with a 1:1 binding kinetic model displayed, KD = 8.541 × 10−4 M (Fig. 4b). These findings indicate that TLB displays a stronger affinity and directly toward to HMGB1.

a Hypothetical complex of HMGB1 protein (blue) with TLB (green). a1 The whole view of the TLB dimer and HMGB1 displayed the molecular binding surface. a2 The substrate binding sites of TLB with HMGB1. a3 A close-up enlargement of the substrate binding pocket. a4 Two-dimensional interaction map for TLB onto HMGB1 protein binding site. b Sensor gram showed full kinetics between TLB and HMGB1. HMGB1 was directly immobilized in the sensor chip and TLB was flowed over the protein-coated chip at different analyte concentrations (from highest to lowest, 500, 250, 125, 62.5, 31.25, 15.625, 7.81, 3.9, and 0 μM) with single-cycle kinetics. The experiment was performed using a Biacore X100 system, and the Biacore evaluation analysis software was used to determine the accuracy of the analysis. A binding behavior was displayed with KD = 8.541 × 10−4 M. RU represents response unit.
TLB inhibits Aβ25-35-induced cytotoxicity in BV2 cells via targeting HMGB1 through SIRT3/SOD2/mtROS signaling pathway
The effect of Aβ25-35 on BV2 cells was initially determined, and the results showed that Aβ25-35 (6.25–100 μM) significantly reduced cell viability and enhanced LDH release within 72 h (Supplementary Fig. S6a, b). Since exposed to 50 μM Aβ25-35 for 48 h, the cytotoxicity was about up to 50% in BV2 cells, thus this concentration and time point were accepted to mimic in vitro AD pathology in the following experiments. We next determined the cell cytotoxicity of TLB and its analog on BV2 cells via MTT assay. The results showed that TLB or its analog did not exert any effect on BV2 cells up to 50 μM, however, 100 μM TLB evidently promoted cell proliferation of BV2 cells (Supplementary Fig. S6c), while, 100 μM TLB analog showed cytotoxicity on BV2 cells (Supplementary Fig. S6d). Thus, the concentrations below 50 μM of TLB or its analog were used to determine the effects of TLB on Aβ25-35-induced cytotoxicity in BV2 cells. Furthermore, the results showed that TLB (12.5, 25, 50 μM) concentration-dependently increased Aβ25-35-induced the cell viability of BV2 cells, however, 50 μM TLB analog did not increase the cell viability of BV2 cells after Aβ25-35 insults (Fig. 5a). Additionally, TLB also effectively decreased the level of LDH in a concentration-dependent manner. However, 50 μM TLB analog did not decrease the LDH level of BV2 cells after Aβ25-35 insults (Fig. 5b). Also, TLB significantly decreased the level of IL-1β, IL-6 and TNF-α (Fig. 5c–e). Furthermore, the expressions of the crucial proteins in HMGB1/TLR4/NF-κB signaling pathway were determined using Western blot. The results showed that HMGB1 of cytoplasm, TLR4, MyD88 and TRAF6 protein expressions as well as phosphorylation of NF-κBp65 protein were observably increased in Aβ25-35-treated group than those of control group, and HMGB1 protein expression of nucleus was decreased in Aβ25-35-treated group than that of control group; in contrast, TLB significantly reversed these changes (Fig. 5f–k). Our findings indicates that TLB, but not its structural analog, suppresses Aβ25-35-induced cytotoxicity through HMGB1/TLR4/NF-κB signaling pathway, in agreement with the beneficial effect of TLB in vivo. Meanwhile, TLB also reduced mtROS production than that of Aβ25-35-treated group (Fig. 5l, m), as well as increased the protein expression of SIRT3, decreased the acetylation level of SOD2 (Fig. 5n, o). Moreover, to confirm the effects of TLB on mitochondrial metabolism during Aβ25-35 insults, the mitochondrial respiration was measured by the Seahorse XFe24 Extracellular Flux Analyzer. Oligomycin, a complex V inhibitor, was used to determine the proportion of ATP-linked OCR in the basal respiration. FCCP, a mitochondrial uncoupler, was used to determine the maximal respiration and calculate the spare respiratory capacity. Rotenone and antimycin, inhibitors of complex I and III, were used to block complex I and III of the electron transport chain. The results showed that the Aβ25-35 elicited a remarkable decline in basal respiration, ATP production-linked oxygen consumption rate, the maximal respiration, and the spare respiratory capacity than those of the control group in of BV2 cells, whereas, TLB significantly reversed these changes (Fig. 5p, q). Furthermore, HMGB1-OE substantially abrogated the beneficial effects of TLB on Aβ25-35-induced cytotoxicity and the inhibitory effects of TLB on HMGB1 level and NF-κBp65 activity after Aβ25-35 insults (Fig. 5r–v), which further verified that HMGB1 was the potential therapeutic target of TLB.

BV2 cells were treated with or without TLB (12.5, 25, 50 μM) or its analog (50 μM) for 48 h after Aβ25-35 insults. a Cell viability (n = 6). b LDH level (n = 6). c IL-1β level (n = 6). d IL-6 level (n = 6). e TNF-α level (n = 6). f Representative Western blot for HMGB1 in cytoplasm. g Quantification of HMGB1 in cytoplasm (n = 5). h Representative Western blot for HMGB1 in nucleus. i Quantification of HMGB1 in nucleus (n = 5). j Representative Western blots for TLR4, MyD88, TRAF6 and p-NF-κBp65 in BV2 cells. k Quantification of TLR4, MyD88, TRAF6 and p-NF-κBp65 in BV2 cells (n = 5). l mtROS generation was detected using flow cytometer after staining with Mito-SOX. m The percentages of Mito-SOX-positive cells analyzed using a flow analysis software (n = 5). n Representative Western blots for SIRT3, ac-SOD2 and SOD2. o Quantification of SIRT3 and ac-SOD2 (n = 5). p OCR was measured using Seahorse to determine the mitochondrial function. q Quantification of OCR. r HMGB1-OE BV2 cells were generated using lentivirus (n = 6). Treatment with or without 50 μM TLB for 48 h after Aβ25-35 insults in WT and HMGB1-OE BV2 cells. s Cell viability (n = 6). t LDH level (n = 6). u HMGB1 level. v NF-κBp65 binding activity (n = 6). The data were presented as the mean ± SEM. **P < 0.01 vs. control group (WT); #P < 0.05, ##P < 0.01 vs. Aβ25-35 group (WT); ††P < 0.01 vs. control group (HMGB1-OE); §P < 0.01 vs. Aβ25-35 group (HMGB1-OE).
Interaction between HMGB1 and SIRT3 is involved in the neuroprotective effect of TLB on cognitive impairment of AD
Molecular docking and Co-IP were used to explore the relationship between HMGB1 and SIRT3 in the neuroprotective effect of TLB on cognitive impairment in APP/PS1 mice. The results showed that there was a strong interaction between HMGB1 and SIRT3 as evidenced by ZDOCK and RDOCK method (Fig. 6a). Furthermore, the direct interaction between HMGB1 and SIRT3 was verified using SPR. The results indicated that HMGB1 directly bound to SIRT3 in a concentration-dependent manner with a KD value of 2.635 × 10−2 M (Fig. 6b). These findings presented that there existed strong affinity between HMGB1 and SIRT3. Based on these predicted results, the Co-IP assays further showed that TLB inhibited the acetylation of HMGB1 than that of APP/PS1 group (Fig. 6c, d); also, we revealed that HMGB1 acetylation formed a direct complex with SIRT3 in APP/PS1 group, while, TLB induced direct interplay between HMGB1 and SIRT3 (Fig. 6e, f). Furthermore, SIRT3-KO largely offset the inhibitory effects of TLB on the HMGB1 level and NF-κBp65 binding activity (Fig. 6g–i). These findings suggests that interaction between HMGB1 and SIRT3 is involved in the beneficial effect of TLB on cognitive impairment in APP/PS1 mice.

a Reciprocity of HMGB1 between SIRT3. a1 The substrate binding sites. a2 The substrate binding surface. a3 Interactions of HMGB1 with active amino acid sites and Pi of SIRT3. The HMGB1 protein was shown in red color solid ribbon, while SIRT3 protein was shown in blue. b binding affinity of HMGB1 with SIRT3 was detected by SPR assay. c, d Co-IP analysis indicated that a reduction in HMGB1 acetylation following treatment of TLB after Aβ25-35 insults (n = 5). e, f TLB induced direct interactions between HMGB1 and SIRT3 (n = 5). g SIRT3-KO BV2 cells were generated using lentivirus-based CRISPR/Cas9 system (n = 6). Treatment with or without 50 μM TLB for 48 h after Aβ25-35 insults in WT and SIRT3-KO BV2 cells. h HMGB1 level (n = 6). i NF-κBp65 binding activity determined at OD 450 nm (n = 6). The data were presented as the mean ± SEM. **P < 0.01 vs. control group (WT); ##P < 0.01 vs. Aβ25-35 group (WT); ††P < 0.01 vs. control group (SIRT3-KO); §P < 0.01 vs. Aβ25-35 group (SIRT3-KO).
Discussion
Here we reveal, for the first time, in vivo and in vitro evidence that (1) TLB, a naturally occurring food additive, effectively rescues cognitive impairment in two animal models of AD with an excellent safety profile and BBB permeability. (2) TLB directly binds to HMGB1, retarding neuroinflammation and oxidative stress dependent on SIRT3/SOD2/mtROS signaling pathway (Fig. 7). Collectively, our findings discover the potential therapeutic target of TLB against AD, and put forward “proof-of-concept” for TLB treatment in AD.

TLB, a naturally occurring food additive, rescues cognitive impairment of AD via targeting HMGB1 through activation of SIRT3/SOD2/mtROS signaling pathway, leading to reduced levels of inflammatory factors and oxidative stress.
Accumulating evidence suggests that neuroinflammation initiates at the very early stages, probably prior to the deposit of amyloid plaque burden in the AD pathology, indicating that inhibition of neuroinflammation may be a promising therapeutic strategy for AD [40, 41]. Intriguingly, recent study provides preliminary evidence that TLB could alleviates cognitive deficits in 3 × FAD AD animal model [28], however, its detailed mechanism is lacking. Thus, the above scenario and pharmacological profile drove us to excavate the therapeutic potential target of TLB against AD in two complementary AD animal models: APP/PS1 mice and Aβ25-35-induced AD rats. Among them, APP/PS1 mice were used to evaluate the inhibitory effects of TLB on Aβ production and neuroinflammation, while, Aβ25-35-injected rats were used to explore the capacity of TLB to challenge the acute neurotoxicity of amyloid. At last, we intended to correlate the observed in vivo effects of TLB with a promising neuroprotective effect detected in cultures of BV2 cells, exposed to Aβ25-35. Our findings demonstrated that both chronic administration and subchronic administration of TLB exerted neuroprotective effects in two AD models, in keeping with the results in vitro. Additionally, at the highest concentration of TLB, there was no toxicity both in animal AD models or glia cells, which corroborated the safety profile of TLB.
Furthermore, transcriptome analysis was utilized to explore which factor is involved in the beneficial effect of TLB on AD. Our results suggested that HMGB1, TLR4, and NF-κB were identified as DEGs, and the TLR4/NF-κB signaling pathway might be inhibited in APP/PS1 mice after treatment with TLB, which indicated that TLB might interfere HMGB1-mediated AD pathological conditions. Based on the data of RNA-seq, we next determined whether TLB treatment could reduce neuroinflammation in animal AD models. Of note, neuroinflammation is characterized by the presence of activated microglia and astrocytes surrounding amyloid plaques [42]. Aβ promotes microglia activation, then leads to activation of astrocytes, thereby secrets pro-inflammatory cytokines including IL-1β, IL-6, and TNF-α, paralleled through up-regulating NF-κB activity. As expected, TLB significantly restrained the activation of microglia and astrocytes, as well as reduced the pro-inflammatory cytokines and NF-κB activity [43, 44]. Notably, targeting to HMGB1 could halt AD progression through mitigating neuroinflammation, reducing Aβ accumulation, and hence improve cognitive deficits [45]. Recently evidence demonstrates that HMGB1 exerts its biological activity mainly through binding to the TLR4, which is a transmembrane protein and enriches in microglia and astrocyte [46]. Activation of TLR4 is implicated in the induction of neuroinflammation and Aβ aggregation. The binding of HMGB1 to TLR4 activates its downstream genes such as MyD88 and TFAR6, and the NF-κB signaling axis, leading to the secretion of inflammatory factors [47, 48]. Our findings demonstrated that, TLB evidently prevented the activation of HMGB1/TLR4/NF-κB axis in APP/PS1 mice or Aβ25–35-induced AD rats, which suggested the inhibitory effect of TLB on neuroinflammation in AD, at least partly, through suppressing HMGB1/TLR4/NF-κB signaling pathway.
It is worth noting that oxidative stress is a crucial pathogenic feature of AD that may provoke Aβ plaques accumulation and neuroinflammation [49]. Oxidative stress happens when there is a disbalance between the generation of ROS and anti-oxidative enzyme ability of living system, ultimately leads to neuronal death [50]. Besides, ROS not only triggers HMGB1 secretion, but also evokes HMGB1 release during cell injury or death. Thus, we next focused on the role of oxidative stress in the beneficial effects of TLB on AD. Our findings demonstrated that TLB effectively decreased the disequilibrium of redox status in hippocampus of the APP/PS1 mice or Aβ25–35-induced AD rats through upregulation of the SIRT3 protein expression and inhibition of the acetylated process of mitochondria-specific SOD2 protein. These findings indicates that, TLB efficiently restores redox homeostasis, at least partially, through SIRT3/SOD2 signaling pathway.
Intriguingly, TLB failed to bind with TLR4, MyD88, TRAF6, as well as NF-κB, which drove us to excavate what was the therapeutic target of TLB on AD. As expected, molecular docking analysis disclosed that TLB strongly bound to HMGB1, and the SPR affinity data further confirmed that TLB effectively bound to HMGB1 protein directly. Therefore, we hypothesized that TLB behaved as a specific and stable binder with HMGB1. Furthermore, to decipher whether HMGB1 can be the potential target of TLB on AD, and considering on that TLR4 is expressed on microglia, Aβ25–35-induced oxidative injury and inflammation in BV2 cells in vitro was used. Consistent with the findings in vivo, TLB exerted the beneficial effects on Aβ25–35-induced cytotoxicity. Intriguingly, a structural analog of TLB that did not bind to HMGB1, and showed no protective effect on Aβ25–35-induced cytotoxicity in vitro, which corroborated that TLB might target HMGB1. Also, in keeping with the findings in vivo, the neuroprotective effects of TLB on neuroinflammation and oxidative stress in vitro through HMGB1/TLR4/NF-κB axis and SIRT3/SOD2/mtROS signaling pathway. Most importantly, lentiviral overexpression studies further confirmed that TLB rescued AD via directly targeting HMGB1. Intriguingly, due to HMGB1 translocates from nucleus into the cytoplasm by acetylation, and SIRT3 is a typical type III HDAC of sirtuins family that have potent deacetylase activity [51]. Thus, we hypothesized that TLB induced deacetylation of HMGB1 at the site of K12 through activating SIRT3. As expected, acetylation of HMGB1 elicited it to dissociate from SIRT3, which indicated that HMGB1 might be a novel deacetylation target of SIRT3; while, TLB affected directly on the interaction between HMGB1 and SIRT3. Interestingly, SIRT3-KO tremendously abolished the inhibitory effects of TLB on the HMGB1 level and NF-κBp65 binding activity, which further confirmed that there is a crosstalk between HMGB1 and SIRT3. These findings suggests that SIRT3 hinders HMGB1 transcription and extracellular release by maintaining HMGB1 in an inactive (deacetylated) state which subsequently inhibits the inflammatory response, and the anti-neuroinflammatory effect of TLB by targeting HMGB1 was dependent on SIRT3 activity.
Of note, we discover that TLB not only directly targets HMGB1, but also indirectly mediates nuclear translocation of HMGB1 through activation of SIRT3. However, what is the detailed mechanism of crosstalk between HMGB1 and SIRT3, and which is the dominant way (direct or indirect) of TLB on HMGB1 remain still unclear. Thus, these mysteries will be unveiled in our next story. Moreover, the safety and effectiveness of TLB supplementation also will be determined in other types of AD or other species, and eventually by elaborately designed, large cohort clinical trials in humans in the future.
In summary, we reveal new tricks from food additive and achieve striking results that TLB rescues cognitive impairment of AD via targeting HMGB1, at least partly, through mediating SIRT3/SOD2/mtROS signaling pathway. Thus, we validate TLB might be a potent weapon to combat AD, and open up a new avenue for prospective clinical trials to evaluate the beneficial effects of TLB on AD.
References
Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med. 2020;26:1256–63.
Wang P, Wang F, Ni L, Wu P, Chen J. Targeting redox-altered plasticity to reactivate synaptic function: A novel therapeutic strategy for cognitive disorder. Acta Pharm Sin B. 2021;11:599–608.
Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature. 2018;554:249–54.
Jiang X, Lu H, Li J, Liu W, Wu Q, Xu Z, et al. A natural BACE1 and GSK3beta dual inhibitor Notopterol effectively ameliorates the cognitive deficits in APP/PS1 Alzheimer’s mice by attenuating amyloid-beta and tau pathology. Clin Transl Med. 2020;10:e50.
Zhang F, Gannon M, Chen Y, Yan S, Zhang S, Feng W, et al. Beta-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3beta/tau cascade. Sci Transl Med. 2020;16:e044769.
Rajendran L, Paolicelli RC. Microglia-mediated synapse loss in Alzheimer’s disease. J Neurosci: Off J Soc Neurosci. 2018;38:2911–9.
Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217:459–72.
Tao CC, Cheng KM, Ma YL, Hsu WL, Chen YC, Fuh JL, et al. Galectin-3 promotes Abeta oligomerization and Abeta toxicity in a mouse model of Alzheimer’s disease. Cell Death Differ. 2020;27:192–209.
Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22:719–28.
Ding B, Lin C, Liu Q, He Y, Ruganzu JB, Jin H, et al. Tanshinone IIA attenuates neuroinflammation via inhibiting RAGE/NF-kappaB signaling pathway in vivo and in vitro. J Neuroinflammation. 2020;17:302.
Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2019;130:1802.
Tanaka H, Homma H, Fujita K, Kondo K, Yamada S, Jin X, et al. YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat Commun. 2020;11:507.
Huang CM, Cai JJ, Jin SW, Lin QC, Fang QJ, Nan K, et al. Class IIa HDAC downregulation contributes to surgery-induced cognitive impairment through HMGB1-mediated inflammatory response in the hippocampi of aged mice. J Inflamm Res. 2021;14:2301–15.
Peng Y, Gao P, Shi L, Chen L, Liu J, Long J. Central and peripheral metabolic defects contribute to the pathogenesis of Alzheimer’s disease: targeting mitochondria for diagnosis and prevention. Antioxid Redox Signal. 2020;32:1188–236.
Ruggeri FS, Habchi J, Chia S, Horne RI, Vendruscolo M, Knowles TPJ. Infrared nanospectroscopy reveals the molecular interaction fingerprint of an aggregation inhibitor with single Abeta42 oligomers. Nat Commun. 2021;12:688.
Park MH, Lee M, Nam G, Kim M, Kang J, Choi BJ, et al. N,N’-Diacetyl-p-phenylenediamine restores microglial phagocytosis and improves cognitive defects in Alzheimer’s disease transgenic mice. Proc Natl Acad Sci USA. 2019;116:23426–36.
Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, et al. Author correction: Metformin reverses established lung fibrosis in a bleomycin model. Nat Med. 2018;24:1627.
Patel D, Roy A, Kundu M, Jana M, Luan CH, Gonzalez FJ, et al. Aspirin binds to PPARalpha to stimulate hippocampal plasticity and protect memory. Proc Natl Acad Sci USA. 2018;115:E7408–17.
Zhu Y, Wan L, Li W, Ni D, Zhang W, Yan X, et al. Recent advances on 2’-fucosyllactose: physiological properties, applications, and production approaches. Crit Rev Food Sci Nutr. 2020;12:1–10.
Shang A, Liu HY, Luo M, Xia Y, Yang X, Li HY, et al. Sweet tea (Lithocarpus polystachyus rehd.) as a new natural source of bioactive dihydrochalcones with multiple health benefits. Crit Rev Food Sci Nutr. 2020;29:917–34.
Smith RL, Waddell WJ, Cohen SM, Fukushima S, Gooderham NJ, Hecht SS, et al. GRAS favoring substances 25. Food Technol. 2011;65:44–75.
Yin S, Zhang X, Lai F, Liang T, Wen J, Lin W, et al. Trilobatin as an HIV-1 entry inhibitor targeting the HIV-1 Gp41 envelope. FEBS Lett. 2018;592:2361–77.
Wang L, Liu M, Yin F, Wang Y, Li X, Wu Y, et al. Trilobatin, a novel SGLT1/2 inhibitor, selectively induces the proliferation of human hepatoblastoma cells. Molecules. 2019;24:3390.
Fan X, Zhang Y, Dong H, Wang B, Ji H, Liu X. Trilobatin attenuates the LPS-mediated inflammatory response by suppressing the NF-kappaB signaling pathway. Food Chem. 2015;166:609–15.
Gao J, Chen N, Li N, Xu F, Wang W, Lei Y, et al. Neuroprotective effects of trilobatin, a novel naturally occurring Sirt3 agonist from lithocarpus polystachyus rehd., mitigate cerebral ischemia/reperfusion injury: involvement of TLR4/NF-kappaB and Nrf2/Keap-1 signaling. Antioxid Redox Signal. 2020;33:117–43.
Li N, Li X, Shi YL, Gao JM, He YQ, Li F, et al. Trilobatin, a component from Lithocarpus polystachyrus Rehd., increases longevity in C. elegans through activating SKN1/SIRT3/DAF16 signaling pathway. Front Pharmacol. 2021;12:655045.
Chen N, Wang J, He Y, Xu Y, Zhang Y, Gong Q, et al. Trilobatin protects against Abeta25-35-induced hippocampal HT22 cells apoptosis through mediating ROS/p38/Caspase 3-dependent pathway. Front Pharmacol. 2020;11:584.
Ding J, Huang J, Yin D, Liu T, Ren Z, Hu S, et al. Trilobatin alleviates cognitive deficits and pathologies in an Alzheimer’s disease mouse model. Oxid Med Cell Longev. 2021;2021:3298400.
Lv J, Wang W, Zhu X, Xu X, Yan Q, Lu J, et al. DW14006 as a direct AMPKalpha1 activator improves pathology of AD model mice by regulating microglial phagocytosis and neuroinflammation. Brain Behav Immun. 2020;90:55–69.
Gong QH, Wang Q, Pan LL, Liu XH, Xin H, Zhu YZ. S-propargyl-cysteine, a novel hydrogen sulfide-modulated agent, attenuates lipopolysaccharide-induced spatial learning and memory impairment: involvement of TNF signaling and NF-kappaB pathway in rats. Brain Behav Immun. 2011;25:110–9.
Gao J, Long L, Xu F, Feng L, Liu Y, Shi J, et al. Icariside II, a phosphodiesterase 5 inhibitor, attenuates cerebral ischaemia/reperfusion injury by inhibiting glycogen synthase kinase-3beta-mediated activation of autophagy. Br J Pharmacol. 2020;177:1434–52.
Yagensky O, Kohansal-Nodehi M, Gunaseelan S, Rabe T, Zafar S, Zerr I, et al. Increased expression of heme-binding protein 1 early in Alzheimer’s disease is linked to neurotoxicity. Elife. 2019;8:e47498.
Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A, et al. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci. 2018;21:72–80.
Wang CY, Zhang Q, Xun Z, Yuan L, Li R, Li X, et al. Increases of iASPP-Keap1 interaction mediated by syringin enhance synaptic plasticity and rescue cognitive impairments via stabilizing Nrf2 in Alzheimer’s models. Redox Biol. 2020;36:101672.
Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, et al. LC3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell. 2019;178:536–51.e14.
Lim SL, Tran DN, Zumkehr J, Chen C, Ghiaar S, Kieu Z, et al. Inhibition of hematopoietic cell kinase dysregulates microglial function and accelerates early stage Alzheimer’s disease-like neuropathology. Glia. 2018;66:2700–18.
Milton RH, Abeti R, Averaimo S, DeBiasi S, Vitellaro L, Jiang L, et al. CLIC1 function is required for beta-amyloid-induced generation of reactive oxygen species by microglia. J Neurosci: Off J Soc Neurosci. 2008;28:11488–99.
Liu MB, Wang W, Gao JM, Li F, Shi JS, Gong QH. Icariside II attenuates cerebral ischemia/reperfusion-induced blood-brain barrier dysfunction in rats via regulating the balance of MMP9/TIMP1. Acta Pharmacol Sin. 2020;41:1547–56.
Ge X, Tang P, Rong Y, Jiang D, Lu X, Ji C, et al. Exosomal miR-155 from M1-polarized macrophages promotes EndoMT and impairs mitochondrial function via activating NF-kappaB signaling pathway in vascular endothelial cells after traumatic spinal cord injury. Redox Biol. 2021;41:101932.
Lee JY, Han SH, Park MH, Song IS, Choi MK, Yu E, et al. N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat Commun. 2020;11:2358.
Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29:787–803.
Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, Haass C. Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron. 2020;108:801–21.
Hou K, Zhao J, Wang H, Li B, Li K, Shi X, et al. Chiral gold nanoparticles enantioselectively rescue memory deficits in a mouse model of Alzheimer’s disease. Nat Commun. 2020;11:4790.
Gunesch S, Hoffmann M, Kiermeier C, Fischer W, Pinto AFM, Maurice T, et al. 7-O-Esters of taxifolin with pronounced and overadditive effects in neuroprotection, anti-neuroinflammation, and amelioration of short-term memory impairment in vivo. Redox Biol. 2020;29:101378.
Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, Michels M, et al. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-beta accumulation, Tau phosphorylation, and cognitive impairment. J Biol Chem. 2018;293:226–44.
Xu B, Zang SC, Li SZ, Guo JR, Wang JF, Wang D, et al. HMGB1-mediated differential response on hippocampal neurotransmitter disorder and neuroinflammation in adolescent male and female mice following cold exposure. Brain Behav Immun. 2019;76:223–35.
Xu X, Piao HN, Aosai F, Zeng XY, Cheng JH, Cui YX, et al. Arctigenin protects against depression by inhibiting microglial activation and neuroinflammation via HMGB1/TLR4/NF-kappaB and TNF-alpha/TNFR1/NF-kappaB pathways. Br J Pharmacol. 2020;177:5224–45.
Moghadam F, LeGraw R, Velazquez JJ, Yeo NC, Xu C, Park J, et al. Synthetic immunomodulation with a CRISPR super-repressor in vivo. Nat Cell Biol. 2020;22:1143–54.
Pan RY, Ma J, Kong XX, Wang XF, Li SS, Qi XL, et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-beta clearance. Sci Adv. 2019;5:eaau6328.
Zrzavy T, Schwaiger C, Wimmer I, Berger T, Bauer J, Butovsky O, et al. Acute and non-resolving inflammation associate with oxidative injury after human spinal cord injury. Brain. 2021;144:144–61.
Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang XJ, Wang H, et al. NAD(+) improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1alpha pathway. J Neuroinflammation. 2021;18:207.
Acknowledgements
This work was supported by Science and Technology Support Plan of Guizhou Province (No. [2020]1Y010), Innovative Research Team of comprehensive utilization with Lithocarpus polystachyus Rehd. sweet tea in Zunyi City (No. [2021]4), Zunyi Science and Technology Project (No. [2021]187), Program for Changjiang Scholars and Innovative Research Team in University, China (No. IRT_17R113).
Author information
Authors and Affiliations
Contributions
JMG, GTS, XZ, NNC, YGL, and YW performed the experiments. FX and YQH helped with bioinformatics analysis and molecular docking analysis. JYZ and FL helped with LC-MS analysis. All authors were involved in the analysis of data. JMG wrote the manuscript. JSS and QHG designed the experiments and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Gao, Jm., Zhang, X., Shu, Gt. et al. Trilobatin rescues cognitive impairment of Alzheimer’s disease by targeting HMGB1 through mediating SIRT3/SOD2 signaling pathway. Acta Pharmacol Sin 43, 2482–2494 (2022). https://doi.org/10.1038/s41401-022-00888-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00888-5
Keywords
This article is cited by
-
GR/Ahi1 regulates WDR68-DYRK1A binding and mediates cognitive impairment in prenatally stressed offspring
Cellular and Molecular Life Sciences (2024)
-
HMGB1 mediates synaptic loss and cognitive impairment in an animal model of sepsis-associated encephalopathy
Journal of Neuroinflammation (2023)
