
Abstract
Cyclic guanosine monophosphate-adenosine monophosphate adenosine synthetase (cGAS) is a DNA sensor that detects and binds to cytosolic DNA to generate cyclic GMP-AMP (cGAMP). As a second messenger, cGAMP mainly activates the adapter protein STING, which induces the production of type I interferons (IFNs) and inflammatory cytokines. Mounting evidence shows that cGAS is extensively involved in the innate immune response, senescence, and tumor immunity, thereby exhibiting a tumor-suppressive function, most of which is mediated by the STING pathway. In contrast, cGAS can also act as an oncogenic factor, mostly by increasing genomic instability through inhibitory effects on DNA repair, suggesting its utility as an antitumor target. This article reviews the roles and the underlying mechanisms of cGAS in cancer, particularly focusing on its dual roles in carcinogenesis and tumor progression, which are probably attributable to its classical and nonclassical functions, as well as approaches targeting cGAS for cancer therapy.
Similar content being viewed by others
Introduction
Cyclic GMP-AMP synthase belongs to the nucleotidyltransferase superfamily responsible for transferring the phosphate group on nucleoside triphosphates to other nucleic acids or proteins to generate nucleoside monophosphates [1]. The presence of foreign DNA in mammalian cells induces the production of cyclic GMP-AMP, which triggers downstream pathways to cause immune responses. Chen et al. discovered cGAS in fractionated cytosolic extracts [2]. As a cytoplasmic DNA receptor, cGAS plays a key role in the immune response by linking DNA and cGAMP. A large number of studies have suggested the important mechanisms of cGAS in innate immunity and virus defense. Furthermore, given the autoimmune and inflammatory diseases triggered by cGAS, the development of cGAS inhibitors for treatment has attracted much attention [3].
Cancer is a disease with high mortality and morbidity worldwide. The complicated and largely unknown mechanisms of cancer make therapy challenging [4]. Cumulative evidence has revealed that cGAS is involved in carcinogenesis and the development of cancer; for example, cGAS performs functions including sensing free DNA and the micronuclei of tumor cells to induce inflammation, antitumor immunity, regulation of DNA repair, and tumor metastasis [5,6,7]. The dual role played by cGAS in cancer deserves more attention and will affect strategies and approaches for cancer therapy. In this review, we will focus on the dual role of cGAS in tumor suppression and promotion and summarize the recent advances in cGAS-targeting agents as cancer therapy.
Overview of the human cGAS protein: from structure to function
Functional domains and structures of the human cGAS protein
Human cGAS (hcGAS), also named Mab-21 domain-containing protein 1 (MB21D1), is a 60 kDa protein comprising an unstructured and non-conserved amino terminus with 160 amino acids and a highly conserved Mab21 domain at the carboxyl terminus with 522 amino acids [8]. In the Mab21 domain, the three hydrophobic amino acids (E225, D227, and D319) coordinate with divalent ions (mainly Mg2+) to form an active site of cGAS, which can influence the catalytic generation of cGAMP from ATP and GTP [9, 10]. The opposite molecular surface platform of the active site has a zinc-finger protrusion formed by highly conserved histidine and cysteine residues (H390, C396, C397, and C404) with Zn2+, contributing to the specific recognition of B-type dsDNA [11]. In addition, Lys187 and Leu195, as key amino acids in the protein spine, make hcGAS more sensitive to long DNA fragments than murine cGAS. This allows hcGAS to distinguish some of the DNA fragments produced by the normal metabolism of the cell and avoid overactivation of the autoimmune response. Key substitutions on the binding surface will determine the preference of cGAS for different lengths of DNA [12]. As such, the two ionic binding sites of Zn2+ and Mg2+ are critical for cGAS to recognize and bind DNA and to exert catalytic activity in a DNA sequence-independent manner (Fig. 1).

The human cGAS primary sequence contains 522 amino acids, with a non-conserved N terminus and a conserved Mab21 domain in the C terminus. The protein spine (K187 and L195 in yellow), catalytic sites (E225, D227, and D319 in green) and zinc ribbon (H390, C396, C397, and C404 in red) contribute to the function of cGAS. The structure of human cGAS is from the Protein Data Bank, https://www.rcsb.org/3d-view/6CTA
Insights into the classical and nonclassical functions of human cGAS
Substantial studies have enriched the understanding of the functions of cGAS, which can be divided into two parts: one is its classic biological function [2], and the other is its newly discovered nonclassical biological function in the nucleus [5].
The classical biological function of cGAS serves as an indispensable bridge between the recognition of extrinsic pathogen dsDNA and immune defense. The cGAS dimer associates with two molecules of dsDNA, triggering liquid–liquid phase separation to form a ladder shape structure [13, 14]. This allows cGAS to be allosterically activated, opening the catalytic pocket to catalyze the cyclization of ATP and GTP to form cGAMP [2]. cGAMP was the first circular dinucleotide found in multicellular animals and acts as an endogenous second messenger to trigger downstream cascade signals [15]. In addition to exogenous pathogen DNA, autologous DNA damage and nuclease inactivation mutations also lead to the enrichment of cytoplasmic DNA [16]. The improper and excessive activation of cGAS caused by such self-DNA molecules is reported to result in the development of autoimmune diseases and autoinflammatory reactions [17]. In summary, cGAS acts as an external or self-dsDNA recognizer to stimulate immune cascades through the production of cGAMP.
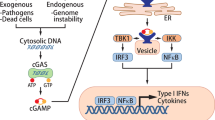
The most dominant and typical downstream target of cGAS is the STING signaling pathway [2, 15]. STING is an endoplasmic reticulum localization adapter that contains an N-terminal transmembrane helix and a large C-terminal cytoplasmic domain [18]. cGAMP binds to the small pocket of dimeric STING, changes the conformation of STING, and promotes the transport of STING from the endoplasmic reticulum to the Golgi apparatus [19]. During this process, the Cys89 and Cys91 residues in the exposed C-terminus of STING are palmitoylated, recruiting TANK-binding kinase 1 (TBK1) to activate its autophosphorylation. Activated TBK1 can phosphorylate STING in turn and enhance the interaction between STING and interferon regulatory factor 3 (IRF3), thereby further promoting the phosphorylation of IRF3 by TBK1. Phosphorylated IRF3 dimerizes and enters the nucleus, where it induces the transcription of type I interferons (IFNs) and other cytokines [20]. IκB kinase (IKK) can be activated simultaneously via STING and phosphorylates Ser32 and Ser36 of the NF-κB inhibitory protein IκBα, resulting in its polyubiquitination and degradation, after which NF-κB enters the nucleus [21, 22]. The cGAS-STING-IKK signaling pathway induces classic NF-κB functions in the nucleus together with IRF3, initiates the transcription of immune-stimulatory genes, and participates in other vital activities through nonclassical NF-κB pathways [23] (Fig. 2).

As a DNA sensor, cGAS recognizes foreign, and self-dsDNA to form a 2:2 complex and converts ATP and GTP into cGAMP. cGAMP stimulates STING and promotes the transport of STING from the ER to the Golgi. Then, STING initiates a series of downstream kinase cascade reactions to activate IRF3 and NF-κB. In the nucleus, these reactions stimulate the transcriptional expression of SASP-related molecules, type I interferon, cytokines, etc. The function of cGAS is regulated by a variety of posttranslational modifications: AKT and BLK phosphorylate cGAS; TTLL4/TTLL6 and CCP5/CCP6 regulate glutamylation of cGAS; and RNF185 and TRIM 38 mediate ubiquitination and SUMOylation
In addition to the role of cGAS in catalyzing cGAMP synthesis, researchers found that in the case of DNA damage caused by hydrogen peroxide treatment, phosphorylation at Tyr215 of cGAS is downregulated, which promotes its nuclear translocation. In the nucleus, the interaction between cGAS and polyadenylate diphosphate ribosyltransferase-1 (PARP1) further prevents the formation of PARP1-Timeless complexes and inhibits homologous recombination repair in a cGAMP-independent manner [5]. In addition, when nuclear cGAS binds to chromatin, it encounters and interacts with a group of replication fork components, including replication factor C1 and proliferation cell nuclear antigen, and ultimately slows replication forks as a “roadblock”. Chen et al. showed that cGAS deficiency in U2OS and BJ cell lines causes replication stress and increases genomic instability, making the cell lines more sensitive to IR and chemotherapy [24, 25].
Posttranslational modification: critical for the regulation of human cGAS
Due to the critical and diverse functions of cGAS, pathogens and host cells have developed a variety of ways to regulate the enzymatic activity, subcellular localization, and protein stability of cGAS. Posttranslational modification is one of the strategies used to achieve tight and dynamic control. For example, Akt, one of the most vital and versatile protein kinases in eukaryotes, can phosphorylate hcGAS at Ser305 to inhibit its activity [26, 27]. The tyrosine kinase BLK phosphorylates Tyr215 to maintain cytosolic localization of cGAS. Conversely, dephosphorylation of this site facilitates cGAS shuttling into the nucleus, where it can function [5]. TTLL4-mediated monoglutamylation of cGAS will prevent its synthase activity, while TTLL6-mediated polyglutamylation of cGAS will hinder its DNA binding ability. In contrast, CCP5 hydrolyzes the monoglutamylation of cGAS, and CCP6 removes the polyglutamylation of cGAS, which together lead to the activation of cGAS. Therefore, glutamylation and deglutamylation of cGAS tightly regulate the immune response to DNA virus infection [28, 29]. To inactivate cGAS early in viral infection while maintaining its proper protein levels in the cytoplasm to initiate innate immune signal, some cGAS proteins are SUMOylated at Lys217 by TRIM 38, which weakens cGAS activation. TRIM 38 also catalyzes the SUMOylation of STING to inhibit it [30]. RNF185, the first discovered E3 ubiquitin ligase of cGAS, specifically catalyzes K27-linked polyubiquitination to enhance cGAS enzymatic activity and increase the yield of cGAMP [31]. In addition to the several posttranslational modifications mentioned above, the acetylation of lysines 384, 398, and 414 can also keep cGAS in an inactive state. As such, aspirin, which has high acetylation activity, is used to inhibit cGAS and achieve the purpose of reducing inflammatory factors and treating autoimmune diseases [32] (Fig. 2). Different types of posttranslational modifications of cGAS have been continuously discovered and studied in-depth, and the upstream pathways related to their respective modifications have also been exposed, providing various strategies for disease treatment that interfere with posttranslational modifications of cGAS.
The multifaceted roles of human cGAS in cancer
Chromosome instability and DNA damage easily lead to the occurrence and malignant progression of tumors [33]. These processes easily cause the generation of micronuclei and enrichment in the cytoplasm. When the micronucleus ruptures, it activates cGAS and causes a series of downstream reactions, such as the immune response [34]. With the increased use of tumor immunotherapy, the relationship between cGAS and cancer has attracted more attention. The function of cGAS is being continuously explored, and its role in tumor inhibition and promotion has become controversial.
Tumor-suppressive roles of cGAS
cGAS facilitates anticancer immune signaling
The main function of cGAS in immune responses is to promote the upregulation of type I interferons through the downstream cascade in a STING-dependent manner. The three main functions of type I IFN are to restrict the spread of infectious agents, regulate the innate immune response, and activate the adaptive immune system [35].
Some cancer cells die naturally or are phagocytized by antigen-presenting cells (dendritic cells and macrophages). Tumor-derived DNA escapes from phagocytic vesicles, accumulates in the cytoplasm of dendritic cells, and activates the cGAS-STING signaling pathway, producing large amounts of type I IFN and inflammatory cytokines (tumor necrosis factor-α and interleukin-6) [36]. Type I IFN can stimulate the proliferation and activation of NK cells, negatively regulate cancer cell proliferation and induce apoptosis through the upregulation of p53 [7, 37]. Type I IFN stimulates the maturation of DCs and the expression of class I major histocompatibility complex (MHC I) and costimulatory molecules. Then, mature DCs migrate to lymph nodes to activate tumor-targeting CD8+ T cells through MHC I [38]. CD8+ T cells are activated and expanded and are then trafficked to kill tumor cells. In turn, dying cancer cells release more antigens that are further captured by DCs to form a positive feedback loop [39]. In this way, DCs and type I IFN connect the innate immune response with the adaptive immune response. Moreover, cGAMP produced by cancer cells is taken up by relevant cells through paracrine signaling, enhancing the activation of STING and its downstream pathways (Fig. 3).

① Tumor-derived cGAMP and DNA can activate APCs, which triggers the cGAS-STING pathway and promotes activation of immune cells that function against tumors. ② Upregulation of SASP molecule expression enhances senescence and changes the tissue immune microenvironment to eliminate tumors. ③ Increased levels of LC3 and p62 puncta improve autophagy in tumors through activation of the cGAS-STING pathway. ④ DNA damage and chromosome instability in tumor cells activate the cGAS–STING pathway, and IRF3 induces apoptosis in a manner dependent on the proapoptotic proteins Bax and MOMP.
Seng-Ryong Woo et al. implanted B16 melanoma cells subcutaneously. One day later and 7 days later, tumor-infiltrating CD45+ cells and CD11c+ cells, respectively, showed more pIRF3 expression and nuclear localization. RT–PCR showed significant induction of IFN-β transcripts in CD45+ cells from WT mice but not in CD45+ cells from STING-deficient mice [40]. Immune checkpoint blockade through inhibition of negative regulators of T cells, such as PD-1 and PD-L1, has emerged as a successful therapy for cancers, the effectiveness of which depends on intrinsic antitumor immunity. A large number of studies have shown that the efficacy of immunotherapy is highly dependent on the abundance of antigen expression, tumor immunogenicity, and tumor immune infiltration [41]. Defects in the cGAS-STING signaling pathway will reduce tumor immunogenicity, resulting in a decreased response to and decreased efficacy of immune checkpoint blockers. A recent study using the highly aggressive B16F10 melanoma model to investigate the role of cGAS in antitumor immunity found that there was no significant difference in B16 tumor growth among mice of different genotypes without treatment. However, in response to anti-PD-L1 antibody treatment, WT tumor volumes in mice were reduced sixfold, but cGAS−/− or STINGgt/gt [42] tumors were not. Furthermore, reduced cGAS and STING expression is associated with poor survival of patients with invasive breast ductal carcinoma [43].
cGAS promotes senescence of cancer cells
Cellular senescence is an irreversible state induced by a variety of internal or external factors (such as telomere shortening, DNA damage, and oncogenic signals) and is a natural barrier to tumorigenesis [44]. Senescent cells show characteristics of flattening, enlargement, nuclear protrusion, and cell cycle arrest [45]. In response to the abovementioned stress-inducing cytosolic DNA aggregation, growth-arrested cells alert cGAS, which in turn activates STING-mediated induction of senescence-associated pathways in an NF-κB-dependent manner [46]. Deletion of cGAS in different mouse or human cells abrogates the expression of senescence-associated inflammatory genes in response to DNA-damaging agents, including etoposide and ionizing irradiation. Subsequent studies have further revealed that mouse embryonic fibroblasts (MEFs) from cGAS knockout mice have significantly reduced signs of senescence compared with MEFs from WT mice, and they undergo faster spontaneous immortalization [47]. Furthermore, a cGAS-/- mouse B16F10 melanoma model constructed with CRISPR also showed the same results [48]. The above results prove that cGAS is involved in normal and cancer cell senescence. Senescence is characterized by stable cell cycle arrest and complex proinflammatory secretions, including cytokines, chemokines, and proteases, also known as the senescence-associated secretory phenotype (SASP). cGAS triggers the SASP by activating the downstream STING pathway based on cytosolic chromatin fragments (CCFs) in the cancer cell cytoplasm [49]. As the key components of the SASP, IL-6 and IL-8 can strengthen the growth stagnation of senescent cells, recruit immune cells to change the tissue microenvironment, and cause the clearance of senescent cells. Normal senescent liver cells and tumor cells attract NK cells and neutrophils to kill tumor cells via the SASP [50] (Fig. 3). Another study found that conditioned medium (CM) from WT MEFs exposed to oxidative stress promoted a senescence response in both WT MEFs and cGAS KO MEFs. In contrast, CM from cGAS KO MEFs cultured under the same conditions failed to arrest MEFs [47]. Senescent cells regulate their own behavior via autocrine SASP-related mechanisms and mediate the senescence of surrounding cells via paracrine SASP-related mechanisms [48]. In addition, type I IFN can induce DNA damage and elevate the p53 level, which is a form of p53-p21WAF1 classical signaling that synergistically promotes cGAS-dependent senescence. Restoring IFN signaling in IFN-deficient melanoma cells can induce senescence and inhibit the progression of melanoma [51, 52]. Hence, cGAS can prevent carcinogenesis and tumor progression through downstream signals to promote cellular senescence and timely cell clearance.
cGAS induces autophagy to exert antitumor effects
Autophagy is a highly conserved intracellular degradation process for eliminating damaged organelles and invading pathogens. Autophagy will present tumor antigens to promote the T-cell response, and the removal of such cytotoxic elements reduces the likelihood of tumor formation. STING, which is a downstream protein of cGAS, harbors LC-3 interacting regions (LIRs) that enable the STING-containing reticulum–Golgi intermediate compartment (ERGIC) to serve as a membrane source for LC3 lipidation, which is a key step in autophagosome biogenesis [53]. The expression of STING markedly increased the levels of LC3 and p62 puncta in both HeLa and wild-type MEF cells [54]. For instance, autophagy has an antitumor function in HCC, and mice deficient in autophagy genes develop spontaneous liver tumors. In the model of HCC induced by diethylnitrosamine, STING-deficient mice bearing HCC show decreased phospho-STAT1, autophagy, and cleaved caspase3 in the liver [55]. Beclin-1, an important autophagy regulator, interacts with cGAS in a manner dependent on the DNA binding site of cGAS. Beclin-1 inhibits the NTase activity of cGAS, reduces the synthesis of cGAMP, and produces a negative regulatory effect. Interestingly, the cGAS-Beclin-1 combination competitively dissociates the negative autophagy factor Rubicon from the Beclin-1 complex, thereby enhancing PI3KC3 kinase activity and increasing autophagy [56] (Fig. 3). However, due to the paradoxical role of autophagy in tumor progression, cGAS may also promote tumor progression by causing tumor cells to adapt to stressful environments.
cGAS activation contributes to apoptotic cell death in cancer
Studies have found that cGAS also acts as a tumor suppressor in other ways. It has recently been reported that cGAS can induce apoptosis in long-term mitotic arrest cells and play a tumor-inhibiting role [57, 58]. The nuclear membrane is destroyed during mitosis, and chromosomal DNA is exposed to the cytoplasm to form a spindle structure. The presence of a large number of nucleosomes can competitively inhibit the ability of DNA to activate cGAS and prevent the inflammatory response [34]. However, if mitosis is blocked for a long time, phosphorylated IRF3 can slowly accumulate through weak cGAS signals. Because transcription is turned off during mitosis, IRF3 induces apoptosis independently of transcription by directly activating the proapoptotic protein Bax and inhibiting the antiapoptotic protein XIAP [59]. Furthermore, MOMP is a crucial step in apoptosis mediated by the Bax protein family, and Bcl-xL and Mcl1 are the major inhibitors of Bax during mitosis. Mitotic stress before apoptosis contributes to cGAS activation. cGAS operates upstream of Bcl-xL but independently of Mcl1 and promotes apoptosis by indirectly regulating MOMP. Based on these findings, HeLa cells were injected subcutaneously into NSG mice, and Taxol delayed the growth of wild-type tumors and showed larger areas of apoptosis, but these patterns were not seen in cGAS-depleted tumors. In addition, researchers found that taxane-treated non-small-cell lung cancer patients who had high levels of cGAS displayed prolonged survival compared to patients with low cGAS levels [57] (Fig. 3).
Disruption of the cGAS-STING pathway may promote cancer development. At present, inhibition of the cGAS-STING pathway has been observed in lung adenocarcinoma, colorectal carcinoma, melanoma, liver cancer, and gastric cancer and in cancer cells lacking telomerase [48, 60,61,62,63,64].
Tumor-promoting roles of human cGAS
cGAS promotes tumor progression in a STING-dependent manner
As mentioned above, cGAS-STING mediates tumor immunity for immune surveillance and clearance, but the presence of chronic inflammation also participates in the occurrence and metastasis of malignant tumors [65]. 7,12‐Dimethylbenz(a)anthracene (DMBA), as a carcinogen, induces the production of inflammatory cytokines and skin inflammation and further promotes the occurrence of skin tumors in mice [66]. STING knockout mice are resistant to DMBA-induced skin cancer, though wild-type mice are not. In addition, the induced type I interferon also promotes STAT3 activity, which mediates the mobilization of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) and reduces the chemotaxis of NK cells, T cells, and macrophages, which is harmful to the formation of the tumor immune microenvironment [67]. In human squamous cell carcinomas or MC38 colon tumors, cGAS-STING signaling eliminates tumor immunogenicity by recruiting regulatory T cells or mobilizing myeloid suppressor cells [68]. Similar to the above immunosuppressive mechanism, cGAS-STING activates indoleamine 2,3-dioxygenase (IDO) for immune regulation in Lewis lung cancer (LLC). It promotes the immune tolerance of cancer cells and ultimately promotes the proliferation of LLC cells [69]. In addition, studies have found that protocadherin seven can promote the formation of gap junctions between carcinoma and astrocytes in the brain. Cancer cells transfer cGAMP to astrocytes via these channels to activate the STING pathway to support tumor growth and brain metastasis [6]. Abnormal nonclassical NF-κB activity in the cGAS-STING-IKK pathway is related to a variety of malignancies and can promote epithelial-mesenchymal transition (EMT) in prostate cancer and upregulate TERT in glioblastoma to promote proliferation, etc [70]. In contrast to the acute cGAS-induced SASP, the cGAS-STING signaling pathway is continuously activated at a low intensity, resulting in a large accumulation of IL-6 and other SASP molecules, which will induce the invasiveness of epithelial cells, transformation, and metastasis and promote tumor angiogenesis together with vascular endothelial growth factor [71, 72] (Fig. 4). Self-DNA sensing has emerged as a key contributing response in the pathogenesis of cancer and autoimmune diseases. Activation of cGAS by self-DNA triggers autoimmune diseases, which in turn form a continuous chronic inflammatory environment and induce the occurrence of cancer, and this notion can be inferred based on the classic functions of cGAS-STING. However, the positive or negative correlation between autoimmune diseases and cancer is still unconfirmed. For example, Crohn’s disease comes with a higher risk of several gastrointestinal cancers and extraintestinal malignancies, while multiple sclerosis appears to reduce the risk of gastrointestinal cancers [73, 74]. Therefore, further experimental research is needed to determine whether cGAS plays a certain role.

① Continuous and chronic stimulation, in turn, increases the expression of IDO and proinflammatory cytokines to form an immunosuppressive tumor microenvironment. ② The cGAMP produced in tumor cells can be exported to adjacent astrocytes, ultimately leading to metastasis and tumor growth. ③ In classic functions, chronic activation of cGAS-STING upregulates nonclassical NF-κB signaling and the SASP, and chronic stimulation promotes EMT. ④ Nuclear cGAS inhibits homologous recombination repair by competing with DNA repair elements to promote tumor progression
The tumor-promoting effect of cGAS is basically related to its classic STING-dependent functions. Tumor immunity, inflammation, tumor senescence, autophagy, and apoptosis are all ways in which cGAS participates in tumor suppression. On the other hand, these processes have also been implicated in cGAS promotion of tumor progression. The main reason is the dual role of downstream response factors in the cGAS-STING signaling pathway. The duality is significantly showcased in three contexts: the spatial activation of cGAS in host cells and the indirect activation of surrounding cells; the acute and chronic cGAS response; and different tumor microenvironments and tumor types. The dichotomous effect makes it more difficult to recognize the role of cGAS in the classical pathways involved in tumors.
cGAS drives tumorigenesis through STING-independent functions
Improper repair after double-stranded DNA breaks (DSBs) can easily lead to chromosomal translocation or deletions and eventually lead to pathological effects related to genomic instability, including tumorigenesis and accelerated senescence [75]. It has been shown that cGAS, as a DNA repair inhibitor, can be recruited to DNA damage sites. It interacts with PARP1 to prevent PARP1-timeless complex formation and inhibits homologous recombination (HR) DNA repair, which seriously affects genome stability. Further animal experiments have demonstrated that overexpression of cGAS makes PC-9 cells more sensitive to DNA damage and increases their proliferation rate [5]. In addition, studies have been performed to explain the cGAS HR inhibitory function from another perspective [76]. The RAD51 recombinase forms protein filaments at one of the “3” single-strand broken DNA tails, termed RAD51-ssDNA filaments. The filaments will target a homologous duplex and exchange ssDNA with the latter to form a displacement loop (D-loop). Finally, DNA intermediate repair is accomplished by DNA synthesis and resolution [77]. The researchers found that nuclear cGAS compresses the template dsDNA to a higher-order state to prevent RAD51-mediated ssDNA exchange and ultimately achieves inhibition of HR repair, accelerating genomic instability and micronucleus generation [78]. This process is related to the DNA binding and oligomerization of cGAS, rather than its enzymatic function, so that it can be regarded as a nonclassical function of cGAS [79]. This notion suggests that cGAS promotes the development of tumors independent of STING functions. On the other hand, whether inhibiting homologous recombination repair forces cells to die is still unclear (Fig. 4).
Targeting cGAS for antitumor therapy
cGAS-STING agonists in the treatment of cancer
Given that cGAS can inhibit tumors by enhancing tumor immune surveillance, accelerating cellular senescence, and promoting apoptosis and that approximately 50% of tumors have aberrant expression of molecules in the cGAS-STING pathway, cGAS-STING agonists have been used to treat tumors [80]. The second messenger cGAMP, which is an enzymatic product of cGAS and stimulating factor of STING, is an ideal signaling pathway agonist [39]. cGAMP was injected intratumorally into wild-type and STING-deficient mice inoculated with B16F10 melanoma. The CD8+ T cell response was completely eliminated in STINGgt/gt mice, while the tumor growth of wild-type mice was effectively delayed. The use of cGAMP far away from the tumor can also cause a systemic antitumor response and control the growth of distant colon cancer [81, 82]. Although cGAMP can be synthesized directly in vitro without direct toxicity and side effects, its application is limited due to transmembrane-related difficulties. Therefore, some natural cyclic dinucleotides (CDNs), such as 3′5′-c-di-GMP [83], 3′3′-cGAMP [84], or CDN analogs, and small molecule inhibitors designed for efficient delivery, such as MIW815 and DMXAA, have been discovered and developed [85]. Due to the differences between human and mouse STING structures, DMXAA exerted strong antitumor activity in mice but failed in human clinical experiments. Another agonist—a pH-sensitive polymer bearing a seven-membered ring with a tertiary amine (PC7A)—binds to a noncompetitive STING surface site (including E269 and D297 in the α5 helix) that is distinct from the cGAMP binding pocket and acts through polymer-induced phase condensation to activate STING [86].
It is believed that the tumor-suppressive effect of cGAS can synergize with other cancer treatments while activating the cGAS-STING pathway [87]. One therapy method is synergistic chemotherapy. Ovarian cancers are susceptible to recurrence during later chemotherapy owing to resistance to platinum drugs, resulting in shortened overall survival [88]. Compared with mice treated with carboplatin alone, tumor-bearing mice treated with carboplatin and the STING agonist 2′3′-c-di-AM (PS) 2 (Rp, Rp) had significantly longer overall survival. It is possible that immunogenic cell death due to carboplatin further enhanced the response to STING agonists by amplifying the IFN response in antigen-presenting cells [89]. The second treatment method is coordinated radiotherapy. It is difficult to produce a systemic antigen-specific response with irradiation alone, but irradiation combined with the STING agonist RR-CDG drives two stages of non-T cell-dependent necrosis via TNFα- and CD8+ T-cell-dependent immune responses. The latter transforms radiation-mediated cell death into an endogenous vaccine to enhance local and systemic tumor control [90]. The application of cGAS-STING agonists can regulate the tumor microenvironment by promoting antigen presentation, activating DCs and antitumor T cells, and transforming immune “cold” tumors into “hot” tumors. Ultimately, these effects will improve tumor immunogenicity, promote cancer cell killing and enhance the tumor response to immunotherapy. Some studies have shown that PD-L1 inhibitors lose their antitumor effect in mice lacking cGAS, and the combination of cGAMP and PD-L1 inhibitors can rescue this effect [42]. A completed phase 1 clinical trial of the combination of cGAS agonists and anti-PD-1 antibody therapy showed promising efficacy, exhibiting type I IFN induction and revived responses to immunotherapy in 90.9% of patients with advanced metastatic solid tumors [91]. CTLA-4, PD-1, and CD47 inhibitors combined with cGAS-STING pathway agonists have been continuously revealed to have antitumor effects [89, 92], strongly suggesting that the pathway is necessary for the treatment of immune checkpoint blockade therapy and that the combination of the two therapies is feasible.
Inhibitors of cGAS-STING as anticancer therapeutics
Intracellular cGAS can be targeted to inhibit tumor promotion as a method to cure tumors. The small molecular inhibitors Ru.332, Ru.365, and Ru.521, which are similar to cGAMP, can be better inserted into the catalytic pocket to competitively inhibit the catalytic function of cGAS because of their benzimidazole structure. These inhibitors not only occupy the catalytic pocket after activation of the cGAS conformational transitions but also interact with two highly conserved amino acid residues, Arg364 and Tyr421, which are conducive to binding to cGAS and exert its inhibitory activity [93]. Hydroxychloroquine (HCQ) and quinacrine (QC) are different from the above three kinds of cGAS catalytic active site inhibitors. They bind within the dsDNA-cGAS interface, which hinders the recognition function of cGAS [94]. The traditional Chinese medicinal compound Astin C binds to the C-terminal binding domain of STING, and H232 and R238 are indispensable for binding. The structural domain is involved in cGAMP binding activation, and blocking this site with Astin C can antagonize the cGAS-STING pathway. In addition, Astin C also disrupts the interaction between STING and SCAP to hinder the IRF33 cascade [3]. These inhibitors were mainly developed for autoimmune diseases, and whether they can be directly used in the treatment of cancers remains to be verified. Nonclassical cGAS-mediated inhibition of DNA homologous recombination repair in the nucleoplasm and carcinogenesis represents a new direction in the development of cGAS inhibitors; for example, drugs that can control cGAS nuclear shuttling can be used. Of course, the use of cGAS-STING inhibitors comes at the cost of local immunosuppression, which may damage the tumor clearance rate, so the advantages and disadvantages should be fully considered in the process of treatment.
Other anticancer therapies
If the cGAS-STING signaling pathway is defective in the expression of key proteins, the entire pathway will be disrupted. So far, the use of some agonists has not been effective due to the lack of target proteins, which means that the cells lack protection against the invasion of viruses, bacteria, and other microorganisms. Natural or modified oncolytic viruses that dissolve cancer cells without affecting normal cells provide another option for cancer treatment that will spare normal cells [95]. cGAS-STING-deficient cancer cells are susceptible to infection and death by viruses. At the same time, dying tumor cells can be used as tumor-specific immune response targets to generate systemic antitumor immunity [96]. The absence of the STING signal makes ovarian cancer cells highly susceptible to oncolytic virus γ34.5-deleted-HSV1 (a herpes simplex virus) infection in vivo and in vitro. Replication and cytolysis of oncolytic HSV-1 in melanoma cells enhance tumor sensitivity and reduce tumor volume [60]. The further engineered strain of HSV-1 talimogene laherparepvec (T-VEC) had higher lasting remission and objective response rates in patients with advanced melanoma in phase III clinical trials. It has become the first oncolytic virus approved by regulatory authorities in the United States, Europe, and Australia.
Tumor vaccines can provide tumor antigens and immunostimulatory signals to antigen-presenting cells, leading to tumor-specific T-cell immune responses, which are used to train the immune system to selectively destroy cancer cells and provide better prevention. With the continuous discovery of new tumor antigens, tumor vaccines have become an attractive strategy. However, tumor vaccines are limited by the slow speed of vaccine-induced responses, the immunosuppression of the tumor microenvironment, and the lack of suitable adjuvants [97]. Because cGAS-STING agonists can induce effective and long-lasting antitumor immunity, they have been increasingly used and developed as anticancer vaccine adjuvants. STINGVAX is a STING-based tumor vaccine containing granulocyte-macrophage colony-stimulating factor (GM-CSF) and cyclic dinucleotides (CDNs). STINGVAX induces a higher frequency of T cells in tumor tissues and inhibits tumor size in a dose-dependent manner compared to a tumor vaccine without CDN adjuvants [98]. Another tumor vaccine, the polymer PC7A, can be used as a vaccine adjuvant as well as a delivery system to present OVA antigens and exert a more effective antitumor effect [86]. Appropriate cGAS agonists, as adjuvants of tumor vaccines, play an important role in overcoming tolerance and enhancing tumor-specific immunity (Table 1).
Conclusions and perspectives
cGAS was first discovered as a cytoplasmic DNA receptor involved in innate immunity, and now, the knowledge of its functions has been greatly enriched and extended, suggesting that further efforts aiming to explore cGAS and its associated pathways as intervention targets for the treatment of a variety of diseases, including cancer, are warranted. cGAS has an obvious DNA binding domain and catalytic pocket, and structure-targeted drugs have also been discovered and developed. In addition, posttranslational modifications such as phosphorylation, acetylation, and glutamylation also regulate cGAS activity, which means that intervention methods targeting cGAS can be diverse and selective. However, due to its indirect regulation and dual roles affecting diverse downstream regulatory factors, cGAS has multiple functions in cancers. Acute and chronic inflammation, immunosuppression, immune escape, dual effects on senescence, opposite effects on autophagy, and DNA repair inhibition are diverse cellular responses influenced by cGAS, and these heterogenous effects make it challenging to precisely and comprehensively describe the role of cGAS. Therefore, when we choose an intervention, the activation status of cGAS and its functions in a particular tumor should be completely understood, and the corresponding measures should be taken to determine which of its dual roles is more prominent. In addition, as mice and other experimental animals mostly serve as the preclinical models for drug evaluation, close attention should be given to the difference between human and mouse cGAS during the development of cGAS-targeting strategies for cancer therapy.
References
Aravind L, Koonin EV. DNA polymerase beta-like nucleotidyltransferase superfamily: identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999;27:1609–18.
Sun LJ, Wu JX, Du FH, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic dna sensor that activates the type I interferon pathway. Science. 2013;339:786–91.
Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, et al. The cyclopeptide astin C specifically inhibits the innate immune CDN sensor STING. Cell Rep. 2018;25:3405–21.
Nunes SC. Tumor microenvironment - selective pressures boosting cancer progression. Adv Exp Med Biol. 2020;1219:35–49.
Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. 2018;563:131–6.
Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–8.
Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019;29:1236–48.
Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–7.
Kuchta K, Knizewski L, Wyrwicz LS, Rychlewski L, Ginalski K. Comprehensive classification of nucleotidyltransferase fold proteins: identification of novel families and their representatives in human. Nucleic Acids Res. 2009;37:7701–14.
Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C, et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015;349:1232–6.
Kranzusch PJ, Lee ASY, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013;3:1362–8.
Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, et al. Structure of the human cGASDNA complex reveals enhanced control of immune surveillance. Cell. 2018;174:300–11.
Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014;6:421–30.
Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704–9.
Wu JX, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–30.
Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, et al. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011;26:125–32.
Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun. 2017;8:2176.
Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–8.
Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N, et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe. 2015;18:157–68.
Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5:ra20.
Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–6.
Sharma S, TenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–51.
Hong C, Tijhuis AE, Foijer F. The cGAS paradox: contrasting roles for cGAS-STING pathway in chromosomal instability. Cells. 2019;8:1228.
Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, et al. cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv. 2020;6:eabb8941.
Basit A, Cho MG, Kim EY, Kwon D, Kang SJ, Lee JH. The cGAS/STING/TBK1/IRF3 innate immunity pathway maintains chromosomal stability through regulation of p21 levels. Exp Mol Med. 2020;52:643–57.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74.
Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 2015;13:440–9.
Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol. 2016;17:369–78.
Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe. 2018;24:234–248 e5.
Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity. 2016;45:555–69.
Wang Q, Huang L, Hong Z, Lv Z, Mao Z, Tang Y, et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017;13:e1006264.
Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell. 2019;176:1447–60 e14.
Bakhoum SF, Landau DA. Chromosomal instability as a driver of tumor heterogeneity and evolution. Cold Spring Harb Perspect Med. 2017;7:a029611.
Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–65.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49.
Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein alpha signaling. Immunity. 2017;47:363–73.
Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34:67–73.
Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci Transl Med. 2020;12:eaay9013.
Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ, et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–4.
Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42.
Paglialunga L, Salih Z, Ricciuti B, Califano R. Immune checkpoint blockade in small cell lung cancer: is there a light at the end of the tunnel? ESMO Open. 2016;1:e000022.
Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA. 2017;114:1637–42.
Wu MZ, Cheng WC, Chen SF, Nieh S, O’Connor C, Liu CL, et al. miR-25/93 mediates hypoxia-induced immunosuppression by repressing cGAS. Nat Cell Biol. 2017;19:1286–96.
Campisi J, di Fagagna FD. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40.
Wolter K, Zender L. Therapy-induced senescence - an induced synthetic lethality in liver cancer? Nat Rev Gastroenterol Hepatol. 2020;17:135–36.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602.
Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat cell Biol. 2017;19:1061–70.
Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114:E4612–E4620.
Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34:428–45.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60.
Katlinskaya YV, Katlinski KV, Yu Q, Ortiz A, Beiting DP, Brice A, et al. Suppression of Type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep. 2016;15:171–80.
Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11:785–97.
Fischer TD, Wang C, Padman BS, Lazarou M, Youle RJ. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J Cell Biol. 2020;219:e202009128.
Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019;26:1735–49.
Thomsen MK, Skouboe MK, Boularan C, Vernejoul F, Lioux T, Leknes SL, et al. The cGAS-STING pathway is a therapeutic target in a preclinical model of hepatocellular carcinoma. Oncogene. 2020;39:1652–64.
Liang QM, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–38.
Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H, et al. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. 2019;178:302–315 e23.
Li C, Liu W, Wang F, Hayashi T, Mizuno K, Hattori S, et al. DNA damage-triggered activation of cGAS-STING pathway induces apoptosis in human keratinocyte HaCaT cells. Mol Immunol. 2021;131:180–90.
Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci USA. 2013;110:16544–9.
Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76:6747–59.
Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14:282–97.
Bu Y, Liu F, Jia QA, Yu SN. Decreased expression of TMEM173 predicts poor prognosis in patients with hepatocellular carcinoma. PLoS One. 2016;11:e0165681.
Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep. 2017;7:39858.
Raaby Gammelgaard K, Sandfeld-Paulsen B, Godsk SH, Demuth C, Meldgaard P, Sorensen BS, et al. cGAS-STING pathway expression as a prognostic tool in NSCLC. Transl Lung Cancer Res. 2021;10:340–54.
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA, et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–71.
Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014;5:1–9.
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8:1736.
Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76:2076–81.
Tegowski M, Baldwin A. Noncanonical NF-kappaB in cancer. Biomedicines. 2018;6:66.
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Coppe JP, Patil CK, Rodier F, Sun YU, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
Nadeem MS, Kumar V, Al-Abbasi FA, Kamal MA, Anwar F. Risk of colorectal cancer in inflammatory bowel diseases. Semin Cancer Biol. 2020;64:51–60.
Freeman HJ. Colorectal cancer risk in Crohn’s disease. World J Gastroenterol. 2008;14:1810–1.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8.
Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, et al. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. Embo J. 2019;38:e102718.
Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64.
Murga M, Jaco I, Fan Y, Soria R, Martinez-Pastor B, Cuadrado M, et al. Global chromatin compaction limits the strength of the DNA damage response. J Cell Biol. 2007;178:1101–8.
Hengel SR, Spies MA, Spies M. Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chem Biol. 2017;24:1101–19.
Pepin G, Gantier MP. cGAS-STING activation in the tumor microenvironment and its role in cancer immunity. Adv Exp Med Biol. 2017;1024:175–194.
Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA. 2015;112:15408–13.
Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, et al. Antitumor activity of cGAMP via stimulation of cGAS-cGAMPSTING- IRF3 mediated innate immune response. Sci Rep. 2016;6:19049.
Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901–10.
Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 2016;76:2137–52.
Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–30.
Li S, Luo M, Wang Z, Feng Q, Wilhelm J, Wang X, et al. Prolonged activation of innate immune pathways by a polyvalent STING agonist. Nat Biomed Eng. 2021;5:455–66.
Yum S, Li MH, Frankel AE, Chen ZJJ. Roles of the cGAS-STING pathway in cancer immunosurveillance and immunotherapy. Annu Rev Cancer Biol. 2019;3:323–44.
Oronsky B, Ray CM, Spira AI, Trepel JB, Carter CA, Cottrill HM. A brief review of the management of platinum-resistantplatinum-refractory ovarian cancer. Med Oncol. 2017;34:103.
Ghaffari A, Peterson N, Khalaj K, Vitkin N, Robinson A, Francis JA, et al. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br J Cancer. 2018;119:440–49.
Baird JR, Friedman D, Cottam B, Dubensky TW, Kanne DB, Bambina S, et al. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res. 2016;76:50–61.
Lv MZ, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020;30:966–79.
Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA, et al. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. 2017;5:676–84.
Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750.
An J, Minie M, Sasaki T, Woodward JJ, Elkon KB. Antimalarial drugs as immune modulators: new mechanisms for old drugs. Annu Rev Med. 2017;68:317–30.
Lee J, Ghonime MG, Wang R, Cassady KA. The antiviral apparatus: STING and oncolytic virus restriction. Mol Ther Oncolytics. 2019;13:7–13.
Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother Cancer. 2016;4:1–8.
Lopes A, Vandermeulen G, Preat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38:146.
Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced Type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–52.
Zawit M, Swami U, Awada H, Arnouk J, Milhem M, Zakharia Y, et al. Current status of intralesional agents in treatment of malignant melanoma. Ann Transl Med. 2021;9:1038.
Zhu X, Han W, Liu Y, Wang H, Lin D, Fu Z, et al. Rational design of a prodrug to inhibit self-inflammation for cancer treatment. Nanoscale. 2021;13:5817–25.
Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017;12:e0184843.
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–8.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (81830107) to Qiao-jun He., a grant from the National Natural Science Foundation of China (81773753) to Bo Yang, and a grant from the Natural Science Foundation of Zhejiang Province (LR19H310002) to Hong Zhu. No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Contributions
HZ and QL conceived and designed the concept of this review article. QJH and BY amended the manuscript. JMD wrote the manuscript. MJQ, TY, and RHC collected the related research articles and reviews. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Du, Jm., Qian, Mj., Yuan, T. et al. cGAS and cancer therapy: a double-edged sword. Acta Pharmacol Sin 43, 2202–2211 (2022). https://doi.org/10.1038/s41401-021-00839-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00839-6
