
Abstract
The nicotinamide adenine dinucleotide (NAD+/NADH) and nicotinamide adenine dinucleotide phosphate (NADP+/NADPH) redox couples function as cofactors or/and substrates for numerous enzymes to retain cellular redox balance and energy metabolism. Thus, maintaining cellular NADH and NADPH balance is critical for sustaining cellular homeostasis. The sources of NADPH generation might determine its biological effects. Newly-recognized biosynthetic enzymes and genetically encoded biosensors help us better understand how cells maintain biosynthesis and distribution of compartmentalized NAD(H) and NADP(H) pools. It is essential but challenging to distinguish how cells sustain redox couple pools to perform their integral functions and escape redox stress. However, it is still obscure whether NADPH is detrimental or beneficial as either deficiency or excess in cellular NADPH levels disturbs cellular redox state and metabolic homeostasis leading to redox stress, energy stress, and eventually, to the disease state. Additional study of the pathways and regulatory mechanisms of NADPH generation in different compartments, and the means by which NADPH plays a role in various diseases, will provide innovative insights into its roles in human health and may find a value of NADPH for the treatment of certain diseases including aging, Alzheimer’s disease, Parkinson’s disease, cardiovascular diseases, ischemic stroke, diabetes, obesity, cancer, etc.
Similar content being viewed by others

Introduction
One of the prominent features of eukaryotic cell metabolism is the compartmentalization of reactions in different organelles. Cell compartmentalization is essential as an additional control mechanism that utilizes pyridine nucleotide-based cofactors (NAD+, NADP+, NADH, and NADPH) to switch electrons between metabolites to undergo biosynthesis, redox homeostasis, signal transmission, and ATP production [1, 2]. NADPH is a coenzyme [3], essential for regular cellular function [4], as it provides reducing equivalents [3]. The oxidative phase of the pentose phosphate pathway has long been considered the main pathway for the generation of cytosolic NADPH. However, cytosolic NADP+-dependent dehydrogenases utilizing isocitrate and malate as substrates have been known to contribute [5, 6]. These substrates originate from the mitochondria, where they also take part in the TCA cycle. But in the case NADPH is highly consumed, proliferating cells have been known to utilize both the pentose phosphate and mitochondria-linked pathways to generate the substantial levels of NADPH necessary for anabolic reactions [6, 7].
Due to the vital importance of NAD+/NADH and NADP+/ NADPH redox pairs, various approaches such as enzymatic cycling assay [8], capillary electrophoresis [9], isotope-labeling techniques [10, 11], high-performance liquid chromatography, and mass spectrometry [12, 13] were developed. However, all these methods require cell lysis and cannot be used for live-cell studies. Pyridine dinucleotides exist in either oxidized or reduced forms, however, only reduced coenzymes possess native fluorescence. NADH and NADPH are weakly fluorescent; in contrast, their oxidized counterparts NAD+ and NADP+ are not [14]. Based on its optical property, the weak endogenous fluorescence of NADPH has been studied by single-photon or multiphoton excitation to monitor metabolic states in living cells or in vivo. However, these methods have drawbacks of low sensitivity and cell injury resulting from ultraviolet irradiation [14,15,16].
In contrast to these traditional methods, genetically encoded fluorescent sensors have been continuously invented and improved to detect these two pairs of redox molecules [17,18,19,20,21,22,23]. For pyridine dinucleotides, currently there are several genetically encoded fluorescent sensors available such as the NADH sensor Frex [21]; NADPH sensor iNAP sensor [24]; the NAD+/NADH ratio sensors Peredox [20], RexYFP [17], and SoNar [23]; the NAD+ sensor cpVenus [18] and FiNad [25]; and the NADP+ sensors Apollo-NADP+ [19] and NADP+ sensor NADPsor [22]. These sensors provide real-time tracking and accurate measures of cellular NAD(H) and NADP(H) levels and their compartmental pools [26,27,28]. Very recently, semisynthetic biosensors namely NAD(P)-Snifits [25, 29] and NAD-Luc family [30] were introduced as new, powerful tools to study the role of NAD(P)(H) in metabolism and signaling in healthy and diseased cells. This review will mainly discuss the NADPH synthesis, transportation of redox couple, and the role of NADPH (beneficial or detrimental) in various health conditions.
Synthesis of NADPH
Synthesis of NAD+
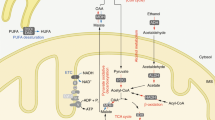
Traditionally, niacin (also known as nicotinic acid) was assumed to be the single external source for de novo synthesis of NAD+. But later, it was found that niacin alone is inadequate to sustain intracellular NAD+ [31]. Three different pathways, namely the De novo pathway, the Preiss-Handler pathway, and the Salvage pathway, utilize four different precursors, i.e., tryptophan, nicotinic acid, nicotinamide, and nicotinamide riboside, to synthesize NAD+ in mammalian cells (Fig. 1). Compared to de novo biosynthesis, the salvage pathway predominates in most cell types [32, 33]. Mitochondrial NAD+ transporters have been identified in yeast and plants [34, 35], but their existence in mammals remains controversial. It had been generally thought that NAD+ cannot be transported across the plasma membranes of any cell types. However, studies have suggested that NAD+ can be transported across the plasma membranes of at least certain types of cells. Connexin 43 hemichannels could mediate transmembrane NAD+ fluxes in a Ca2+‐dependent manner in 3T3 fibroblasts [31] and across the plasma membranes of astrocytes [36, 37]. These channels have been long known to mediate the exchange between coupled cells of many small molecules, including second messengers and signal metabolites. However, few data are available on the role of connexin hemichannels as potential pores in individual, non-coupled cells [38, 39]. Recently, SLC25A51 has been identified as a mammalian mitochondrial NAD+ transporter. It was found that the loss of SLC25A51 decreases mitochondrial but not whole-cell NAD+ content, impairs mitochondrial respiration, and blocks the uptake of NAD+ into isolated mitochondria. Conversely, overexpression of SLC25A51 or SLC25A52 increases mitochondrial NAD+ levels and restores NAD+ uptake into yeast mitochondria lacking endogenous NAD+ transporters [40].

NAD+ synthesis via salvage pathway using NAM or NR, de novo pathway using tryptophan and preiss-handler pathway using NA as the precursors. NAD+, nicotinamide adenine dinucleotide, NAM, nicotinamide; NMN, nicotinamide mononucleotide; NR, nicotinamide riboside; NRK, NR kinase; NA, nicotinic acid; TDO, tryptophan 2,3-dioxygenase; IDO, indoleamine 2,3-dioxygenase; KFase, kynurenine formamidase; K3H, kynurenine‐3‐hydroxylase; AMS, 2‐amino‐3‐muconate‐semialdehyde; ACMS, 2‐amino‐3‐carboxy‐muconate‐semialdehyde; ACMSD, ACMS decarboxylase; NAMN, nicotinic acid mononucleotide; QPRT, quinolinate phosphoribosyltransferase; PRPP, phosphoribosyl pyrophosphate; NAAD, nicotinic acid adenine dinucleotide; NAPRT, nicotinic acid phosphoribosyltransferase; NMNAT, nicotinamide mononucleotide adenylyltransferase; NADSYN, NAD+ synthetases.
In the de novo pathway, NAD+ biosynthesis begins with the essential amino acid L-tryptophan, which is acquired from the diet [41]. The first and rate-limiting step is the conversion of tryptophan to N-formylkynurenine. In mammals, tryptophan 2,3-dioxygenase is superior to indoleamine 2,3-dioxygenase (IDO) in the liver, whereas in extrahepatic tissues like the lung, spleen, and small intestine, the cytosolic IDO plays a key role [41]. Moreover, N‐ formylkynurenine is converted into kynurenine by kynurenine formamidase, which is further hydroxylated into 3‐hydroxy‐kynurenine by kynurenine‐3‐hydroxylase. Once formed, hydroxylated kynurenine also undertakes two enzymatic reactions forming an unstable intermediate, 2‐amino‐3‐carboxy‐muconate‐semialdehyde (ACMS). ACMS can be eliminated from the NAD+ synthetic pathway by its decarboxylation into 2‐amino‐3‐muconate‐ semialdehyde catalyzed by ACMS decarboxylase. This process eventually leads to the development of picolinic acid or CO2 and H2O. ACMS can also endure spontaneous cyclization to form quinolinic acid (QA), which is then converted to nicotinic acid mononucleotide (NAMN) by quinolinate phosphoribosyltransferase (QPRT) using phosphoribosyl pyrophosphate (PRPP) as a co‐substrate [33]. Of note, the QPRT reaction is not significant and arises only when the level of QA exceeds the enzymatic capacity of ACMS decarboxylase, which renders this reaction as a second rate-limiting step in de novo NAD+ synthesis and helps to explain why Trp‐dependent synthesis is less efficient than the other two NAD+ biosynthetic pathways. NAMN is then adenylated to form nicotinic acid adenine dinucleotide (NAAD) with the catalysis of ATP by NMN adenyltransferases (NMNATs). The ultimate step in this pathway is to convert NAAD into NAD+ by ATP‐dependent NAD+ synthetases, which catalyze an amidation reaction using glutamine or ammonia as an amide donor [32].
In the Preiss‐Handler pathway, at first, nicotinic acid from dietary sources is metabolized to NAMN by nicotinic acid phosphoribosyltransferase (NAPRT) at the expense of PRPP. Consequently, NMNAT adenylates NAMN to form NAAD. NAD synthetase catalyzes the ATP‐dependent amidation of NAAD to yield NAD+ [33]. ATP is a dual allosteric regulator that can stimulate or inhibit NAPRT activity at low (<100 μM) or high (100–640 μM) concentrations, respectively. As described in the previous section, NAMN serves as the converging point for the Preiss‐Handler pathway and the de novo pathway, with NAMN undergoing the same reactions for NAD+ synthesis [32].
The salvage pathway utilizes NAM, a product of niacin, and SIRT enzymes to synthesize NAD+. In mammals, NAM phosphoribosyltransferase (NAMPT), a rate‐limiting enzyme, catalyzes the conversion of NAM and PRPP into NMN, which is then converted into NAD+ by NMNATs [33, 42]. NAMPT mRNA is ubiquitously expressed in all tissues, with higher levels present in bone marrow, liver, and muscle than other tissues [43]. Currently, two isoforms of Nampt are identified.
The intracellular Nampt (iNampt) contributes as a rate limiting enzyme for NAD+ biosynthesis. The extracellular Nampt (eNAMPT) was initially recognized as an immunomodulatory cytokine that synergizes with interleukin 7 and stem cell factor to enhance pre-B cell colony formation [44]. It is upregulated upon activation in innate and adaptive immune cells, including neutrophils, monocytes, macrophages, and epithelial and endothelial cells [45, 46]. Furthermore, eNAMPT was found in human circulation, and leukocytes were also identified as a source for eNAMPT [43]. Revollo and colleagues reported that mouse adipocytes also secreted eNAMPT, exhibiting even higher NAD+ biosynthetic activity than the intracellular iNAMPT [47]. Similarly, Yoon et al. [48] demonstrated that deacetylation of iNAMPT by the SIRT1 deacetylase enhances eNAMPT secretion and activity in murine adipocytes. And, adipocyte‐specific knock‐out or knock‐in of NAMPT systemically affected plasma eNAMPT levels, hypothalamic NAD+ biosynthesis, SIRT1 function, and exercise capacity [48]. It was revealed that increasing eNAMPT promotes NAD+ synthesis, counteracts aging, and extends healthspan in mice [49]. Interestingly, the overexpression of iNAMPT, as well as increased circulating levels of eNAMPT, were reported in conditions including obesity, type 2 diabetes, atherogenic inflammatory diseases, therefore supporting eNAMPT as a potential biomarker of cardio-cerebro-vascular disorders [50,51,52].
NR is a newly discovered NAD+ precursor that also feeds into the salvage pathway. NR is first phosphorylated into NMN by NR kinases, after which NMNATs catalyze the production of NAD+. It is reported that overexpression of NRK1 in NIH3T3 cells and hepatocytes elevates cellular NAD+ levels in response to NR addition. Similarly, in mice, the administration of NR augmented NAD+ levels in muscle, liver, brain, and brown adipose tissue, and such effect was abolished in NRK1 knockout mice [32, 53].
Synthesis of NADP+
NAD kinases (NADKs) are homo-oligomers of 2–8 subunits. This enzyme exists in the prokaryotes such as E. coli, Mycobacterium tuberculosis, Bacillus subtilis, Salmonella enterica, and the yeast S. cerevisiae [2]. In S. cerevisiae, NADK-3 is located in the mitochondrial matrix and favors NADH as a substrate, whereas NADK-1 and NADK-2 are cytosolic proteins and show substrate specificity with preference particularly towards NAD+ [2]. Both cytoplasmic and mitochondrial NADKs transfer a phosphate group most frequently from ATP to the 2′-hydroxyl group of the adenosine ribose moiety of NAD+ and thus, phosphorylate NAD+ to NADP+ [54, 55].
In mammalian cells, for many years, only a single human NADK has been recognized, and it is localized to the cytosol. The sources of mitochondrial NADP+ remained obscure. Ohashi et al. discovered that the uncharacterized human gene C5ORF33, named MNADK, encodes a novel mitochondrion localized NAD kinase that catalyzes the formation of NADP+ from NAD+ and ATP. The MNADK is predominantly expressed in the liver, brown fat, heart, muscle, and kidney in mice [54, 56, 57]. MNADK facilitates fatty acid oxidation, prevents oxidative stress, regulates mitochondrial sirtuin activity, and protects metabolic stress-induced non-alcoholic fatty liver disease in mice [58]. The cytoplasmic NADK silencing with shRNA in HEK293 cells led to a threefold decrease of NADPH while overexpression increased NADPH 4–5‐fold [59]. Likewise, with overexpression of cytoplasmic NADK, a remarkable increase in cytoplasmic and mitochondrial NADPH was observed [60, 61]. An MNADK-deficient patient exhibited symptoms characteristic of mitochondrial disease [56]; fibroblasts from MNADK mutant patients had decreased dienoyl‐CoA reductase (DECR) activity and reduced mitochondrial NADPH levels with no change in cytosolic NADP(H) levels. Whereas, overexpression of the wild‐type MNADK restored DECR activity in patient fibroblasts, suggesting that MNADK might be an appealing therapeutic target for this disorder [62]. Identification of MNADK proposes a model in which NADK and MNADK are important for the NADP+ synthesis in cytosol and mitochondria, respectively. It provides a key clue to the mechanism involved in mitochondrial NADP+ and NADPH production and the maintenance of redox balance in mammalian cells [56, 57].
Synthesis of NADPH
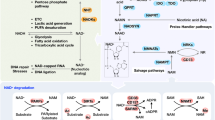
Cells consume two major energy packages: phosphate bonds and electrons. The phosphate bonds in ATP form are produced in a substantial amount by glycolysis and oxidative phosphorylation. In contrast, high-energy electrons in the form of NADPH are produced by a wide variety of pathways [63] by different enzymes, including glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase in the oxPPP; cytosolic and mitochondrial NADP-dependent isocitrate dehydrogenases (IDHc and IDHm) [64]; cytosolic and mitochondrial NADP-dependent malic enzymes (MEc and MEm); proton-translocating NAD(P)+ transhydrogenase [65, 66]; glutamate dehydrogenase [67]; methylenetetrahydrofolate dehydrogenase (MTHFD) and aldehyde dehydrogenases (ALDHs) in folate metabolism (Fig. 2) [63].

Biosynthesis of NADPH from NAD+. NADK, NAD kinase; MNADK, mitochondrial NADK; G6PD, glucose-6-phosphate dehydrogenase; IDHc, cytosolic NADP-dependent isocitrate dehydrogenase; MEc, cytosolic NADP-dependent malic enzymes; IDHm, mitochondrial NADP-dependent isocitrate dehydrogenase; MEm, mitochondrial NADP-dependent malic enzymes; GDH, glutamate dehydrogenase; NNT, Nicotinamide nucleotide transhydrogenase; ETC, electron transport chain; ALDH1L1/ALDH1L2, aldehyde dehydrogenase (cytosol/mitochondrial); MTHFD1/2, methylenetetrahydrofolate dehydrogenase (cytosol/mitochondrial).
Glucose 6-phosphate dehydrogenase (G6PD)
Previously, the pentose phosphate pathway, a metabolic pathway parallel to glycolysis, was considered the main source of NADPH production. G6PD catalyzes the translation of glucose-6-phosphate to 6-phosphogluconolactone and proceeds the NADPH generation from NADP+ [4, 68,69,70]. Both G6PD and 6PGD reduce NADP+ to NADPH, but the reaction catalyzed by G6PD is a rate-limiting one. It resembles unique significance to numerous cellular processes that consume NADPH [4], such as safeguarding the cell against oxidative stress for cell growth and proliferation [71, 72]. Diminishing G6PD activity depresses NADPH levels [68], and by altering the NADPH concentration, G6PD activity can also influence NADPH-dependent superoxide production [68,69,70, 73, 74]. Traditionally, it was thought that the NADPH/NADP+ ratio modulates G6PD. As the ratio decreases, G6PD activity increases to supply additional NADPH. It is assumed that G6PD deficiency decreases myocardial antioxidant capability that exacerbates the adverse cardiac effects such as ischemia-reperfusion injury [75]. It has been determined that eNOS stimulation and basal production of ·NO by eNOS were significantly lower when G6PD activity was inhibited, illustrating that eNOS is dependent on G6PD derived NADPH [76]. An additional system that is dependent on G6PD mediated NADPH is NADPH oxidases. These enzymes have diverse physiological (cell growth, proliferation, and migration, immune functions) and many pathophysiological (diabetes, renal failure, and cardiovascular disease) roles [77].
Isocitrate dehydrogenase (IDH)
Using isocitrate as an electron donor, isocitrate dehydrogenases reduce NADP+ to NADPH [64, 78]. In eukaryotic organisms, three different isomers of IDH catalyze the reversible oxidative decarboxylation of isocitrate to 2-oxoglutarate. NAD+-dependent IDH localizes in mitochondria, whereas NADP+-dependent IDH exists in the mitochondria (IDH1/mIDH) and the cytosol or peroxisomes (IDH2/cIDH) [79, 80].
The expression of mIDH and cIDH differs strongly between different tissues. mIDH and cIDH are predominately expressed in the heart and the liver, respectively [64, 79]. In rat liver, the IDH contribution for NADPH production was shown to be 16–18-fold higher compared to G6PD [81]. One of the studies considered mIDH as the most important NADPH producer in the mitochondria that contributes to cellular defense against oxidative stress-mediated mitochondrial loss [82]. However, isoforms of these enzymes also can switch reducing equivalents between the cytosol and the mitochondria. For instance, IDH2 consumes mitochondrial NADPH to mediate the reductive carboxylation of alpha-ketoglutarate to isocitrate. It is followed by the subsequent transport of citrate/isocitrate to the cytosol, where it is oxidized by IDH1 to yield cytosolic NADPH [83].
Malic enzymes
Two NADP+-dependent ME isoforms (cytosolic and mitochondrial) have been defined in mammalian tissues. Cytosolic malic enzymes (ME1/MEc) catalyze the reversible oxidative decarboxylation of L-malate to produce pyruvate and CO2 with the reduction of NAD+ or NADP+ to NADPH [84]:
NADPH produced from the MEc is utilized for the long-chain fatty acids synthesis. It might also supply reducing equivalents for detoxification processes in chronic inflammation of the liver [85]. Similarly, in mitochondria of the bovine adrenal cortex, ME is the main source of NADPH to meet the requirements of steroidogenesis [86]. In S. cerevisiae, ME commonly localizes in the mitochondria. It converts malate, an intermediate of the Kreb’s cycle, into pyruvate. The only anaplerotic reaction in S. cerevisiae during its growth, i.e., carboxylation reaction catalyzed by pyruvate carboxylase resulting in oxaloacetate formation, also needs pyruvate. As mentioned above, in S. cerevisiae, the ME is in mitochondria. If it can accomplish the cytosolic requirement for pyruvate as proposed by several studies, then transference of pyruvate from the mitochondrial matrix to the cytosol must occur [87].
Proton-translocating NAD(P)+ transhydrogenase
Proton-translocating NAD(P)+ transhydrogenase (also known as nicotinamide nucleotide transhydrogenase (NNT)) is localized in the plasma membranes of bacteria and the inner mitochondrial membranes of eukaryotes. It catalyzes the hydride transfer between NADH and NADP+ and couples the transfer of protons across the inner membrane from the cytosol to the mitochondrial matrix [88]. The electrochemical proton gradient (∆p) maintained across the IMM drives the forward NNT reaction to establish an NADPH/NADP+ ratio at least 500-fold higher than the NADH/NAD+ ratio in the matrix [65, 89, 90]. This reaction is reversible under physiological conditions and permits the maintenance of appropriate cellular NADH and NADPH redox levels [91].
NNT demonstrates a dominant contribution during non-phosphorylating respiration or electron transport inhibition-induced respiratory failure. It emerges as a key anti-oxidative enzyme based on its ability to restore NADPH from NADH [92]. Thus, formed NADPH is utilized for biosynthesis, detoxification, and retaining a reduced glutathione pool [89]. Transhydrogenase mutation leads to severe cortisol deficiency in humans, suggesting that mitochondrial transhydrogenase sustains mitochondrial NADPH levels not only for antioxidant defense but also to support steroid hormone biosynthesis [93]. The NADPH/NADP+ ratio in differentiated PC12 cell lysate was significantly decreased in NNT silencing, suggesting that NNT is a powerful source of NADPH in mitochondria, and its availability can influence the total cellular NADPH and NADP+ pools [94]. Likewise, NNT makes a major contribution to peroxide metabolism, and lack of NNT activity leads to compromised peroxide metabolism in intact mitochondria [95].
Glutamate dehydrogenase
Glutamate dehydrogenase (GDH) is a homo-hexameric enzyme found in all living organisms that catalyzes the reversible oxidative deamination of glutamate to α-ketoglutarate while reducing NAD(P)+ to NAD(P)H [10, 96,97,98]. GDH catalyzes the reaction:
Lower life forms, such as bacteria or yeasts, exhibit distinct GDH isoenzymes that show strict specificity for NAD+ or NADP+ [99]. The NAD+ dependent GDH is involved in the metabolic role, whereas the NADP+-specific enzyme serves in biosynthetic functions [100]. In contrast, mammalian GDH1 (hGDH1 in the human) shows dual coenzyme specificity. It utilizes both NAD+ and NADP+ for catabolic and synthetic functions, respectively [99]. hGDH2 is co-localized with hGDH1 in the human brain, kidney, testis, and steroidogenic organs, but not in the liver [97, 101]. In steroid-producing cells, the hGDH1 and hGDH2 mediated glutamate flux is thought to generate NADPH needed for the biosynthesis of steroidal hormones [99, 102]. As compared to control cells, the NADPH levels and the GSH/GSSG ratio were significantly decreased, whereas mitochondrial ROS and intracellular H2O2 levels were increased in GDH1 knockdown cells [67]. For cancer metabolism, it is a chief enzymatic pathway because it yields nitrogen for nucleotide and amino acid synthesis; it provides an alternative carbon source to supply TCA cycle intermediates, and as a byproduct, NADPH is formed redox homeostasis [103]. GDH-derived NADPH is consumed to support the reductive carboxylation of α-KG by IDH2. The compensatory increase in the expression of GDH1 or GDH2 promotes the growth of IDH-mutant glioma cells [104]. In KRAS-driven pancreatic adenocarcinoma cells and colorectal cancer cells, a pathway involving glutamine-dependent NADPH production is essential for redox balance and growth. In these cells, glutamine is used to produce aspartate in the mitochondria. This aspartate is then transported to the cytosol, where it is deaminated to produce oxaloacetate and then malate. And then malate is converted into pyruvate, apparently increasing the NADPH, which can potentially maintain the cellular redox state [105, 106].
Folate metabolism
Folate metabolism is a common metabolic practice that aids in initiating and transferring 1C units for biosynthetic processes, including purine and thymidine synthesis and homocysteine methylation [10, 11]. First, the 1C units predominantly move in the system as 5,10-methylene-THF, which can be prepared from serine and glycine. As 1C-loaded folates could not enter intracellular membranes, 5,10-methylene-THF must be produced in the mitochondria and the cytosol [107]. The cytosolic formyl-THF can be produced from methylene-THF by the cytosolic methylene-THF dehydrogenase MTHFD1 with simultaneous cytosolic NADPH production. NADP+ is used as a cofactor in this reaction and is reduced to NADPH. In contrast, in mitochondria, there are two enzymes with bifunctional dehydrogenase/cyclohydrolase activity (MTHFD2 and MTHFD2L) and a separate formate-THF ligase enzyme (MTHFD1L) [108]. The mitochondrial MTHFD2/MTHFD2L also utilizes NADP+ as a cofactor to transform serine-derived methylene-THF to formyl-THF, which results in the collateral production of mitochondrial NADPH [11, 63, 109,110,111]. ALDH1L2 also contributes to NADPH production by the oxidation of formyl-THF to CO2 and THF [112]. The mitochondrial 10-formyl-THF might also help to generate cytosolic 1C units. In the case when carbon-containing THF species do not enter the mitochondrial membrane, mitochondrial 10-formyl-THF can be cleaved by methylenetetrahydrofolate dehydrogenase 1-like (MTHFD1L) into free formate, which can readily cross the mitochondrial membrane and be adjusted in the cytosol by methylenetetrahydrofolate dehydrogenase (MTHFD1) [112]. Recent work suggests that ALDH1L2 activity supports mitochondrial redox homeostasis and, in that way, contributes to melanoma metastasis (Fig. 3) [113, 114]. Likewise, the mitochondrial respiratory chain generates ROS whose deactivation involves THF-mediated mitochondrial NADPH production [115].

Folate-mediated 1C metabolism in NADPH production. THF, tetrahydrofolate; MTHFD1, cytosolic methylene tetrahydrofolate dehydrogenase; MTHFD2, mitochondrial methylenetetrahydrofolate dehydrogenase; ALDH1L1, cytosolic aldehyde dehydrogenase; ALDH1L2, mitochondrial aldehyde dehydrogenase; SHMT1, cytosolic serine hydroxymethyltransferase; SHMT2, mitochondrial serine hydroxymethyltransferase.
It has recently been shown that serine-derived 1-carbon metabolism generates NADPH to the same extent as the PPP. MTHFD2 is overexpressed in cancers [116]; MTHFD2 and ALDH1L2 produced NADPH contribute to multiple downstream signaling that substantially impact cancer cell survival [10]. Genetic silencing of MTHFD2 or MTHFD1 reduces NADPH/NADP+ and GSH/GSSG ratios [11, 112]. Similarly, anti-folate medications such as methotrexate and 5-FU target one-carbon metabolic pathways. However, these drugs have many adverse effects as these pathways are significant in healthy proliferating cells. Future therapeutics with specificity and selectivity in targeting one-carbon metabolism are required to obstruct individual one-carbon pathway enzymes [117]. A recent study revealed that the NADPH produced through the folate cycle supported melanoma cells to endure oxidative stress during metastasis. Knockdown of MTHFD1L successfully lowered NADPH concentration and triggered ROS and ROS-induced cell cycle delay, as well as hepatocellular carcinoma growth limitation in vitro and in vivo [113].
Transportation of redox couple
All the NADP-dehydrogenases commonly have different isozymes, which are confined in the different subcellular compartments to confirm the formation and utilization of the cofactors NADH and NADPH at the required site. Thus, the subcellular compartmentation of redox couples critically influences the functional state of the cell and, ultimately, cellular survival. The interconversion between NADP+ and their reduced forms, i.e., NADPH, can occur in multiple metabolic and biosynthetic pathways such as glycolysis, the pentose phosphate pathway, the tricarboxylic acid cycle, and mitochondrial oxidative phosphorylation [32]. It is clear that NADPH plays a much broader role; however, detection of NADPH metabolism in living cells remains technically challenging. Indeed, the past few years have seen the development of genetic tools for measuring and manipulating NADH and NADPH metabolism, which had better assisted these investigations. The iNap sensors were designed by engineering SoNar’s binding site to switch ligand selectivity from NADH to NADPH [23]. These sensors enabled the quantification of cytosolic and mitochondrial NADPH pools and exhibited cellular NADPH dynamics under glucose-induced oxidative stress [24]. At present, iNap is one of the most effective genetically encoded sensors for tracking subtle differences in cellular NADPH or NADP+ redox state in living cells and in vivo models due to its characteristics such as intense fluorescence, rapid responsiveness, pH insensitivity, large dynamic range, targeting to subcellular organelles, and ratiometric imaging [24]. Yet, more investigations and measurements are required to acknowledge NADPH redox ratios and pools sizes in different compartments, even within the mitochondria and the cytosol [118]. It is reported that neither NADH nor NADPH can be transported across intracellular membranes [59, 119]. Thus indirect multistep shuttles encompassing compartmentalized redox processes are utilized to transfer electrons between the mitochondria and the cytosol [32, 120, 121].
Malate-pyruvate shuttle
In the bovine adrenal cortex, two isoforms of ME (cytosolic and mitochondrial) were studied. The mitochondrial malic enzyme is a primary source of NADPH for mitochondrial mixed-function oxidations in this tissue. The reaction catalyzed by the cytosolic ME showed kinetic reversibility, whereas, in mitochondria, the reaction was irreversible. On this basis, a concept of “malate shuttle” was proposed, affecting the net transfer of NADPH reducing equivalents into the mitochondria by the operation of the cytosolic malic enzyme to produce malate from pyruvate and carbon dioxide (Fig. 4) [86]. This metabolic shunt comprises pyruvate carboxylase and malate dehydrogenase (catalyzing a reversible reduction of oxaloacetate to malate). In this shuttle, NADPH is formed at the expense of one ATP used by pyruvate carboxylase and one NADH utilized by malate dehydrogenase. Malate dehydrogenase subsists as three isozymes: one mitochondrial, a cytosolic form, and a peroxisomal form. Based on isozymes participating in the metabolic shunt, either oxaloacetate or malate has to be transported across the mitochondrial membrane [87].

Pyruvate malate shuttle and pyruvate decarboxylation.
The pyruvate malate shuttle is considered much more efficient than the PPP in generating NADPH in the cytosol. The PPP can produce NADPH only as rapidly as glucose enters and ends its transits through the pathway. In contrast, the shuttle is not controlled and can replace its reactants to function at a very rapid pace. It is reported that in the pancreatic islet, less than 3% of glucose oxidation emerges via the PPP pathway [122]. It was demonstrated that when pyruvate was applied to the rat mitochondria, malate and citrate were formed, and 98% of these metabolites were recovered outside the mitochondria. It is also evident that pyruvate malate shuttle, i.e., malate derived from pyruvate in the mitochondria, can produce NADPH in the cytosol by malic enzyme [122, 123].
Malate aspartate shuttle (MAS)
Malate-aspartate shuttle includes combined cytosolic and mitochondrial transamination of aspartate and glutamate by aspartate aminotransferase and inter-conversion of malate and oxaloacetate by malate dehydrogenase (MDH) [66]. MAS shares enzyme intermediates with the TCA cycle and the ETC, confirming a close relation between shuttle activity and mitochondrial respiration. Passage of MAS metabolites across the inner mitochondrial membrane occurs through two antiporters, the reversible α-ketoglutarate/malate carrier and the energy-dependent aspartate/glutamate carrier (AGC) which facilitates the efflux of aspartate in exchange for mitochondrial approval of glutamate, driven by the membrane potential. AGC directs flux through MAS and aids in an uneven NADH supply between the cytosolic and mitochondrial compartments [124]. The electron carrier malate is introduced from the cytosol into the mitochondrial matrix through α‐KG/malate antiporter (encoded by SLC25A11 gene) in conjunction with the export of α‐KG into the cytosol. Once in the matrix, malate is oxidized into oxaloacetate (OAA) by MDH, transferring electrons to NAD+, forming NADH. OAA is then transaminated into aspartate by mitochondrial glutamic-oxaloacetic transaminase (GOT2). It transports an amino group from glutamate to oxaloacetate, yielding aspartate and α‐ketoglutarate. Subsequently, the aspartate‐glutamate antiporter (encoded by SLC25A13 gene) exports aspartate into the cytosol where cytosolic glutamic-oxaloacetic transaminase (GOT1) transfers an amino group from aspartate to α‐ketoglutarate to produce glutamate and oxaloacetate to complete the cycle, which is, in turn, reduced to malate in conjunction with oxidizing NADH to NAD+ by cytosolic MDH (Fig. 5). Thus, the malate‐aspartate shuttle is reversible and requires multiple enzymes [42, 120]. In this shunt, NADH is oxidized to NAD+ in the cytosol, and NAD+ is reduced to NADH in mitochondria. NAD+ is used as an electron acceptor during glycolysis, whereas NADH is used by mitochondrial complex I to drive the mitochondrial electron transport chain [32].

Outline of the malate aspartate shuttle. ASPAR, aspartate; AST, aspartate aminotransferase; α-KGLUT, α-ketoglutarate; GLUT, glutamate; MDH, malate dehydrogenase; OAA, oxaloacetate. The net shuttle result is a transfer of the cytosolic NADH to the mitochondrial matrix.
Similarly, the malate-aspartate shuttle (MAS) is deliberated to be the central redox shuttle system shifting, reducing equivalents from the cytosolic NAD+/NADH to the mitochondria that functions in the brain, mainly essential in neuronal cells for the oxidative metabolism of glucose [121]. Evolving evidence suggests MAS as an imperative modulator of the cytosolic and mitochondrial calcium homeostasis in the heart. In the isolated rat heart, MAS suppression by the aminotransferase inhibitor aminooxyacetate persuades infarct limitation, recovers hemodynamic responses, and modifies glucose metabolism, similar to effects observed in classical ischemic preconditioning [124]. Similarly, the malate-aspartate shuttle activity was critical for the lactate metabolism in the two-cell embryo. Besides, the mouse zygote’s failure to utilize lactate as an energy source was found to be due to deficient malate-aspartate shuttle activity [125].
Isocitrate dehydrogenase shuttle
As mentioned earlier, the mitochondrial inner membrane is impermeable to NADPH. Hence, communication between the cytosolic and the mitochondrial NADPH pools is conducted by the isocitrate‐α‐KG shuttle [32]. The alpha-ketoglutarate could directly pass into the mitochondria or go through transamination with alanine or aspartate to generate glutamate, which might easily enter the mitochondria and be metabolized or take part in a malate aspartate shuttle [126]. In this shuttle, NADP+‐dependent IDH2 converts α‐KG into isocitrate by oxidizing NADPH to NADP+ in the mitochondrial matrix. Isocitrate is then forced into the cytosol in exchange for malate by the citrate carrier protein (encoded by the SLC25A1 gene). In the cytosol, IDH1 catalyzes the reverse reaction by converting isocitrate to α‐KG and NADP+ to NADPH. Afterward, α‐KG is transferred into the mitochondrial matrix by the α‐KG/malate antiporter as a carrier in the malate‐aspartate shuttle (Fig. 6) [32]. As the reducing equivalents are transported into the matrix in this shuttle, the reduced NADP+/NADPH in the matrix triggers glutathione reductase to reduce the GSSG/GSH and stimulates thioredoxin reductase to reduce thioredoxin. As soon as the mitochondrial GSSG/GSH and thioredoxin pools become significantly reduced, O2− and H2O2 are produced by the mitochondrial glutathione reductase and thioredoxin reductase enzymes as flavins in these two enzymes yield ROS in the absence of their GSSG and oxidized thioredoxin substrates [60]. Thus, the isocitrate‐α‐KG shuttle plays a fundamental role in retaining cellular NADPH levels [32].

Isocitrate dehydrogenase shuttle. GLUD, glutamate dehydrogenase; NNT, Nicotinamide nucleotide transhydrogenase; α-KG, alpha-ketoglutarate.
Reduced nicotinamide adenine dinucleotide phosphate: a friend or a foe?
The question, “Is NADPH beneficial or a detriment to health”? has been discussed for more than decades without resolution. Unfortunately, certain gaps in knowledge preclude our ability to accurately label NADPH as good or bad. The following segment will elucidate this perplexing question by deliberating the (1) essential functions of NADPH and (2) association between NADPH and diseases
Functions of NADPH
Reductive biosynthesis
Reductive biosynthesis refers to the anabolic pathway that requires hydride ions to reduce carbon atom in metabolic intermediates. It is the synthesis of large molecules from smaller ones used in the formation of cellular components. NADPH acts as a significant reductant for the anabolic reactions such as biosynthesis of fatty acid and amino acids like proline and glutamate [91, 127]. The fatty acid synthase enzyme complex catalyzes the saturated fatty acid production through the combination of acetyl-CoA and malonyl‐CoA to form palmitate in a series of reactions requiring the consumption of ATP and input of reducing potential from NADPH [127]. Numerous systems, for instance, enzymes such as 3-hydroxy-3-methylglutaryl-CoA (HMG‐CoA) reductase and the NADPH-cytochrome P450 oxidoreductase that are essential for lipid production and steroid biosynthesis, respectively, are dependent on NADPH. The HMG-CoA reductase utilizes two NADPH molecules and ultimately aids in converting HMG‐CoA to mevalonate [128]. NADPH is a vital cofactor for reducing ribonucleotide to deoxyribonucleotide by ribonucleotide reductase and indirectly supports DNA synthesis [61]. The biosynthesis of phospholipids and steroids, ubiquinol (coenzyme Q10), dolichol, heme, and vitamin D also requires NADPH as a reducing entity at several steps [129].
Detoxification
Detoxification is the physiological or medicinal removal of toxic substances from a living organism, including the human body. The deactivation and excretion of xenobiotics such as drugs, toxins, carcinogens, and other probable destructive components are performed by cytochrome P-450-dependent enzymes in the liver. Phase I detoxification, i.e., P450 catalyzed monooxygenation of substrates resulting in the addition of one or more hydroxyl groups, converts insoluble organic compounds into hydrophilic ones, and facilitates their breakdown as well as the elimination of a wide variety of endogenous and exogenous compounds. The cytochromes are renewed with the help of NADPH-dependent cytochrome P450 reductases (CPRs). The NADPH donates an electron to its FAD-FMN electron transport chain by CPRs before providing them to the heme moiety of cytochrome P450 [61, 130].
Antioxidant
Oxidative stress arises due to the difference between the ROS generation and the cellular defenses [131]. Cells contain various antioxidant mechanisms, including glutathione, thioredoxin, catalase, and superoxide dismutase, that contribute to the inhibition and the elimination of oxidative damage under physiological situations [132]. Once oxidized, glutathione is restored to the reduced state via a series of redox‐coupled, enzyme‐catalyzed reactions in which NADPH donates electrons to the active site of glutathione reductase. Glutathione reductase then reduces oxidized glutathione disulfide dimers back to the monomeric form with a free sulfhydryl group that can then act as a direct antioxidant as well as serve as the substrate for glutathione‐S‐transferase, glutathione peroxidase, and other enzymes involved in antioxidant defense to protect against free radical-mediated cytotoxicity [132,133,134,135]. Besides, the NADPH-dependent thioredoxin system similarly utilizes NADPH [136, 137]. Likewise, catalase has an allosteric binding site for NADPH to sustain itself in an active state [4]. Also, if catalase or glutathione do not scavenge the free radicals effectively, the increased H2O2 levels will hinder the superoxide dismutase enzyme activity. Thus, although catalase and SOD do not utilize NADPH directly to convert H2O2 to water, it requires NADPH for their action. In summary, NADPH is considered the sole source of the reducing power of the antioxidant systems, and the preservation and reinforcement of all these antioxidants eventually require NADPH [4, 138].
ROS generation-immune response
Interestingly, besides being a vital antioxidant, NADPH also generates reactive oxygen species (ROS) through NADPH oxidases (Nox) [139]. Under physiologic conditions, ROS are involved in signaling pathways that regulate vascular tone as well as cellular processes like proliferation, migration, and differentiation. However, high doses of ROS, which are produced after induction or activation of NADPH oxidases, contribute to the development of endothelial dysfunction and vascular disease [140]. The mammalian Nox family comprises seven members: Nox1, Nox2 (formerly gp91phox), Nox3, Nox4, Nox5, Duox1, and Duox2 [132]. NADPH oxidase was first described in phagocytes of the immune system. The NADPH oxidase in phagocytes plays critical role in innate immunity by generating microbicidal ROS [141]. An immune response is a reaction that ensues within an organism to shield against foreign pathogens, including viruses, bacteria, parasites, and fungi, that could cause severe complications if not eliminated from the host body. The neutrophils and monocyte/macrophages are the first responders of the innate immune system. They act together along with cells of the adaptive immune system, T and B lymphocytes, and reach the inflammation site within minutes to support a quick and precise response to subsequent infection [142]. These immune cells must have sufficient energy to reach the infected area, then utilize NADPH as a cofactor to produce ROS, and eventually destroy pathogens and impaired tissue [143]. Basically, Nox1–5, using NADPH as a substrate, produces superoxide anions that can be quickly converted to other ROS, whereas DUOX1 and DUOX2 specifically produce hydrogen peroxide [144]. A lot of inflammatory receptors in macrophages, including TLR4, are attached to Nox2. TLR4 recognizes bacterial lipopolysaccharide and mediates proinflammatory cytokines production necessary for the development of immune response. After TLR4 receptor stimulation, the oxidative burst rises, driven by NADPH [143]. As the Nox is turned on, the oxygen requirement by neutrophils increases by 100 folds. In the case of hypoxia, limited oxygen is available and therefore limits the full activation of oxidase. With rapid oxygen utilization, NADPH consumption also ramps up. When the cell lacks sufficient NADPH supplies to fulfill the need for the oxidative burst, then new NADPH produced by the hexose-monophosphate shunt is a must. Depleted NADPH pools can fail Nox2 stimulation. Similar is the case of G6PD insufficiency, where cells cannot support the requirements of the Nox abundantly. Under 1% of normal G6PD functions, neutrophils’ antibacterial abilities are compromised, resulting in chronic granulomatous disease-like symptoms [145].
In mammals, nNOS, inducible NOS (iNOS), and eNOS are the three isoforms of nitric oxide synthases (NOS) that contribute to the conversion of L‐arginine to L‐citrulline and ·NO. Nitric oxide is secreted as free radicals by phagocytes (monocytes, macrophages, and neutrophils) as part of the human immune response. It is toxic to bacteria and intracellular parasites, including Leishmania [146] and malaria [147]. In macrophages, NADPH obtained from the PPP is crucial to sustaining redox homeostasis to synthesize ·NO [6]. Moreover, the histological study showed that some neurons express high NNT levels [148]. Interestingly, neurons producing ·NO (nNOS labeled) as neurotransmitters have been found localized in the areas with high NNT expressions [148, 149]. The double-labeling of NNT and nNOS demonstrated that most of the nNOS-positive neurons from the lateral tegmental nucleus, pedunculopontine tegmental nuclei, and lateral lemniscus colocalize with NNT. These data together suggest that NNT contributes to ·NO biosynthesis in these areas [148]. Likewise, the relationship between NAPDH oxidase and iNOS contribute to inflammation-induced cytotoxicity: Activated iNOS can produce a huge toxic amount of ·NO in a sustained manner [150]. Thus produced ·NO can rapidly interact with the NADPH oxidase-generated superoxide that results in peroxynitrite formation, ultimately directing to DNA damage, mitochondrial respiration inhibition, and PARPs activation [151].
Numerous studies have suggested that NADPH oxidase and iNOS can generate synergistic effects in inducing cell death [152, 153]. Tightly coupled NOSs consume about 1.5 moles of NADPH for each mole of ·NO [154]. NADPH, which serves as the vital electron donor for ·NO generation; however, when NOS isoforms are uncoupled, the electrons from NADPH allow one-electron reduction of oxygen to generate reactive oxygen species [154]. Moreover, augmented NADPH levels urge to produce ROS through Nox activity and proceeds towards the phagocytosis mechanism.
Circadian clock regulation
Circadian rhythms are inherent mechanisms that perfuse all levels of biology to regulate behavioral and functional events, for instance, the sleep-wake cycle, mobility, body temperature, and hormone secretion [155]. In mammals, the central clock is also recognized as the “master pacemaker” that resides in the hypothalamic suprachiasmatic nucleus (SCN). The SCN coordinates peripheral clocks distributed in most tissues and organs to modulate proper tissue-specific functions [156,157,158,159]. Primarily, dual positive stimulators, circadian locomotor output cycles kaput (CLOCK) and brain and muscle Arnt-like protein 1 (BMAL1), proceed transcription/translation of the Period1/2 (Per1/2), Cryptochrome1/2 (Cry1/2), retinoic acid receptor-related orphan receptor, and Rev-erb/genes. Once the expression levels of Per and Cry get to a certain threshold, they shift into the nucleus and hinder further transcription of CLOCK-BMAL1 [160,161,162].
Neuronal PAS domain protein 2 (NPAS2), a homolog of CLOCK, establishes a heterodimer with BMAL1 that couples to E-box or E-box-like elements in the regulatory regions of several clock genes to fuel the gene expression [156]. The bHLH domain of NPAS2 functions as a fundamental DNA binding and a dimerization site with other bHLH proteins. The NPAS2/ BMAL1 heterodimer’s DNA binding ability is found to be improved by NADH and NADPH, signifying regulation by the redox cofactors [155, 163]. O’Neill and Reddy reported that variations in NADPH and NADH redox couples modify the circadian rhythms of peroxiredoxin redox forms in erythrocytes. Similar NADPH-controlled peroxiredoxin oscillatory systems have been identified in several organisms, including archaeal bacteria [164]. Likewise, a study by Rey et al. revealed the PPP activity, one of the chief producers of NADPH, as a fundamental element of circadian mechanism [165], as its downregulation affected circadian rhythms in fruit flies, mouse tissues, and human cells. Along with PPP inhibition by 6-aminonicotinamide, BMAL1 and CLOCK association was highly elevated, DNA-binding activity was improved, and expression of several BMAL1/CLOCK targets was enhanced [165].
While all the above evidence indicates that NADPH is somehow beneficial, in some situations, its role could also be harmful. Below, we will discuss the role of NADPH on health and diseases that will further put some light on its controversial characteristics.
NADPH and diseases
Aging
Reactive oxygen species are continuously formed by cells, and ROS-driven impairment results in aging. Aging results in oxidative stress-mediated cell dysfunction and exhaustion of endogenous antioxidants. The Redox Stress Theory of Aging [166] and the Redox Theory of Aging [167] interrelated life expectancy with redox changes, including variations in the NADP+/NADPH, GSSG/GSH, and oxidized thioredoxin/reduced thioredoxin ratios. It is reported that in healthy cells, both of the cytoplasmic and the mitochondrial NAD+/NADH couples are ~99.9% and 87% oxidized, respectively, and they turn into a further reduced state with aging. However, the cytoplasmic and the mitochondrial NADP+/NADPH couple, which are roughly 99% reduced in healthy cells, become more oxidized with aging [168, 169]. NADP+/NADPH is considered as the most significant redox couple against aging-induced cellular oxidation. The redox couple was diminished by 12% in aged cells [170].
Aging-related deprivation of NAD+ [171] might be one of the reasons for the NADPH level decline with age [172]. The increase in NAD+ degradation and reduced NADP/NADPH ratio occurred as a result of oxidative stress due to altered mitochondrial electron transport chain [173, 174] and decreased expression of nicotinamide phosphoribosyltransferase [175]. Among the cellular elements that can inhibit free radical damage, glutathione reductase, NADPH, and NADPH generating hexose monophosphate shunt (HMS) is the pivotal system [170]. It has been demonstrated that the transgenic mouse model with modest G6PD overexpression has higher NADPH concentrations, lower ROS-derived loss, and better protection from aging-associated functional decline [176]. It has been shown that between NADPH and GSH, NADPH was more efficient and essential for neuron survival in aging. Higher neuron loss due to NADPH decline was directly associated with the increase in aging [177]. It is also reported that vitamin C or E depends upon NADPH to recycle into their active state and defends cells from aging [60]. Moreover, NADPH and GSH that actively scavenged H2O2 were found to be reduced in aged rats [178].
On the contrary, the increased NADPH production could also act as a burden. It could boost NADPH-oxidase-induced ROS generation that unfortunately enhances aging. The patients with G6PD deficiency were found to have a longer lifespan [179]. The reason behind this finding might be accounted for the decreased NAPDH oxidase-dependent ROS generation resulting from decreased NADPH generation.
Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive syndrome that causes neurons to degenerate and die. It causes dementia, memory impairment, neuropsychiatric disturbances, and a continuous decline of behavioral and social skills that disturbs a person’s capability to work independently. Literature review from experimental and human studies has proven that NADPH is more efficient than GSH for neuron survival; in comparison to GSH diminution, advanced neurodegeneration was detected when NADPH was extrapolated to zero in the 3×Tg-AD mice neurons [177]. Theoretically, any metabolic change that alters NADPH concentration, such as mitochondrial dysfunction and oxidative stress, will lower the antioxidant activity and greatly affect neurodegenerative processes [180,181,182]. It has been evident that diminished NADPH causes failure to replenish endogenous antioxidants such as GSH, which weakens the ROS scavenging, favoring oxidative stress, and AD development [183].
Parkinson’s disease
Parkinson’s disease (PD) is a progressive nervous system syndrome that disturbs movement. Disturbed dopaminergic neurotransmission initiates it in the basal ganglia, commonly due to an increase in dopaminergic neurons damage, arising first in the substantia nigra, and subsequently over the progression of the disease in other areas as well. Lately, NADH and NADPH and their pharmaceutically suitable derivatives have been proposed to prepare a medicament appropriate for Parkinson’s disease treatment [184, 185]. Interestingly, clinical tests showed that intravenous NADH administration showed a substantial effect on improving motricity even in patients with advanced Parkinsonism [185]. Targeting the PPP, a major source of NADPH, in neuronal cell models has shown to summarize many of the cellular phenotypes concomitant with PD, such as increased oxidative stress, mitochondrial dysfunction, diminished GSH levels, and neuronal apoptosis [186]. Numerous studies have demonstrated that NADPH shows potent neuroprotection against oxidative stress. A study by Zhou et al. investigated that exogenous supplementation of NADPH revealed neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by inhibiting oxidative stress, diminishing motor dysfunction, and glia-mediated neuroinflammation both in vitro and in vivo [187].
In comparison to most other cell types, neurons specially metabolize glucose by the PPP to sustain their antioxidant activity. Thus, PPP inhibition in neuronal cell models and increasing levels of oxidative stress contribute to the pathogenesis of PD. A study by Dunn et al. using postmortem human brain tissue showed that AD brains exhibited an increased NADPH production in areas affected by the disease. In PD, however, the increase in NADPH production was only observed in the affected areas of end-stage cases. This result suggests that the suppression of PPP enzymes and a failure to increase antioxidant reserve is an initial event in the pathogenesis of sporadic PD [188].
Cardiovascular disorders (CVDs)
Cardiovascular diseases (CVDs) represent the main cause of mortality in the world. Numerous studies have evidenced that oxidative stress modifies various functional responses of vascular endothelial cells and is considered to play a critical role in developing CVDs such as hypertension, atherosclerosis, myocardial infarction, heart failure, and so on. It is reported that high blood pressure patients, when administered with NADH or NADPH, exhibited a substantial reduction of blood pressure with time, lacking any side effects [189]. The contraction and relaxation of the cardiac muscles demand a large amount of energy and, NADPH supplies that energy in the appropriate and demanded way, thus resulting in a reduced diastolic blood pressure that opposes to endothelial cells;[189] aids vascular smooth muscle cell survival, which is beneficial for vascular repair after injury and decreases the risk of atherosclerotic plaque rupture [190]. A study by Jain et al. stated that the protection of various reductant pools (GSH, Trx, and NADPH) might reduce reperfusion injury caused by increased oxidative stress. It was observed that deficiency of G6PD worsens cardiac dysfunction followed by ischemia-reperfusion, and these hearts show decreased restoration of glutathione compared to control hearts [191]. Recent studies indicate that G6PDX mice are moderately more susceptible to age-associated cardiac hypertrophy and ventricular dilation in response to myocardial infarction or pressure overload-induced heart failure [68]. One of the principal objectives of IDH2 is to switch the mitochondrial redox balance and thus facilitate the cellular resistance against oxidative damage via NADPH production [192]. The hearts of IDH2-deficient mice showed augmented heart failure, increased apoptosis, senescence, and hypertrophy, and exhibited mitochondrial dysfunction, which was associated with a loss of redox homeostasis [78, 192].
Similarly, it is demonstrated that IDH2 inhibition via the mitochondrial recruitment of Ca2+/calmodulin-dependent protein kinase II decreases NADPH synthesis, thereby reducing the antioxidant capabilities of the cardiomyocytes [193]. It signifies that IDH2 has a significant role in maintaining both mitochondrial function and cardiac contractile function in pressure-overload hypertrophy by upgrading antioxidant capacity and preventing oxidative stress. Recently, Boslett et al. [194] and Reyes et al. [195] elucidated that in the post-ischemic heart, when the eNOS cofactor tetrahydrobiopterin (BH4) is exhausted, its repletion only partially repaired eNOS-mediated coronary vasodilation, specifying additional critical elements that trigger endothelial dysfunction. It has been revealed that eNOS substrate NADPH was depleted, prompting compromised eNOS activity and limiting BH4 release through NADPH-dependent salvage pathways. With mutual NADPH and BH4 preservation, a full repair of NOS-dependent coronary flow was observed. It has been shown that ischemia and reperfusion of the heart cause CD38 activation with significant depletion of the cardiac NADPH pool. The suppression of CD38 disallowed NADPH diminution and well-preserved endothelium-dependent relaxation with the increased recovery of contractile function and decreased infarction [194, 195]. Similarly, the plasma NADPH level and the NADPH/NADP+ ratio were significantly lower, whereas the NADPH/NADP+ redox potentials were significantly higher in the acute myocardial infarction group. The plasma GSH level and the GSH/GSSG ratio were markedly lower, whereas the plasma GSSG level and the GSH/GSSG redox potentials were significantly higher in the AMI group [196]. Reyes et al. found that depletion of cardiac NADPH pool was observed in isolated rat hearts subjected to myocardial I/R [195]. Similarly, Zhu et al. exhibited that exogenous NADPH, through the AMPK/mTOR pathway stimulation, inhibited mitochondrial dysfunction and cardiomyocyte apoptosis. Thus, NADPH is a promising candidate for cardio-protection against myocardial ischemic diseases in both in vitro and in vivo models [197].
However, NADPH also acts as a source of electrons in the Nox reactions that generate O2− and H2O2, subsequently leading to hypertension and aortic smooth muscle hypertrophy [198], and suppression of NADPH exerts protection against reperfusion injury associated with revascularization [199, 200]. Under certain exercise conditions, there occurs an increase in energy demand, i.e., higher cardiac workload. It increases both ADP and Ca2+ uptake by the mitochondria to increase ATP production to match the demand. Such energized mitochondria exhibit higher ATP and NADPH levels and lesser electron flow, thereby increasing the possibility of ROS generation in the electron transport chain [201, 202]. Wang et al. studied a case of mutation (G6PD activity limiting to ~20% that of normal mice) and found that G6PD deficiency mice had declined cardiac hypertrophy, signifying a role for excess reducing equivalents in the pathobiology of aggregation-induced cardiomyopathies [203]. Moreover, boosting G6PD activity results in overexpression of NADPH, which stimulates Nox and eventually superoxide anion production in heart failure [204] and myocardial ischemia [205]. To date, although findings advised that G6PD deficiency may lessen free radicals production in failing myocardium and may decrease the risk of coronary heart disease progression, an increase in oxidative stress along with heart failure is also observed in G6PD deficient failing myocardium [206]. Thus, the role of G6PD in CVDs is contradictory and needs further investigation.
Ischemic stroke
Ischemic stroke, the most frequent type of stroke, is the leading cause of death and disability in the aging society. NADPH might be considered a potential candidate for the treatment of ischemic stroke as the administration of exogenous NADPH not only reduced infarct volume but also noticeably improved recovery of neurological activity against ischemia/reperfusion-induced injury in stroke models [207]. It is also believed that during the early ischemic reperfusion, NADPH oxidase acts as the main source of free radicals, and the upregulation of NADPH oxidase leads to neuronal damage. Thus, suppression of G6PD activity followed by diminished Nox activity and superoxide anion generation during the early reperfusion period could be preventive mechanisms [208]. Analogous to this concept, in another study, a co-treatment of apocynin and NADPH was used on ischemia reperfusion-induced brain inflammation and neuronal injury. NADPH distinctly attenuated I/R induced phosphorylation and degradation of IκBα, expression of cyclooxygenase (Cox2), iNOS, and ROS production. It is reported that the combination of NADPH and apocynin [139] and NADPH and NAD+ [209] remarkably lowered infarct volume, reduced long-term mortality, enhanced post-stroke survival, inhibited signaling pathways involved in apoptosis and necroptosis, and recovery of neurological functions in stroke models. These studies propose that, besides the antioxidant effect, NADPH might also have anti-inflammatory effects, and synergism of NADPH and Nox inhibitors or NADPH and NAD+ could produce greater neuroprotection in both in vitro and in vivo models of ischemic stroke [139, 209].
Similarly, the arrangement of NADPH with NAD+ significantly improved the ATP level and reduced the levels of ROS and oxidative damage of macromolecules [209]. It is supposed that cerebral ischemia might shift glucose metabolism into the PPP by inhibition of aerobic glycolysis and hence, raise G6PD activity, which results in NADPH accumulation during ischemia. As a result, the raised intracellular NADPH functions as a crucial source for NADPH oxidase activation following ischemic-reperfusion.
Diabetes
Preclinical and clinical investigation shows that hyperglycemia increases oxidative stress and plays an important role in the pathogenesis of diabetes and its complications. In moderate and severe hyperglycemia, the NADPH/NADP+ ratio and GSH concentration were decreased in the liver and pancreas [210]. Similarly, mitochondrial NADPH produced by isocitrate dehydrogenase is determined to regulate insulin secretion [211]. A common inhibitor of PPP, 6-aminonicotinamide, lessened insulin secretion in rat islets [212]. A study by Zhang et al [213]. showed that NADPH maintained proper ROS levels in the cytoplasm and provided reducing power to mitochondrial shuttle reactions to maintain insulin production. Even a modest decrease in G6PD activity that alters NADPH activity and increases ROS production leads toward cell damage and death [213]. Hyperglycemia results in raised polyol pathway flux leading to sorbitol accumulation, a decrease of the NADPH/ NADP, and increases the NADH/NAD+ ratio. Lately, it has been stated that G6PDH activity and expression, along with NADPH and GSH levels, were decreased in the rat kidney cortex during streptozotocin-induced diabetes, suggesting a close association between these parameters [214]. Neutrophils exposed to high glucose showed decreased G6PD activity leading to diminished NADPH level and depressed NADPH oxidase activity in neutrophils. Hence, enhancing G6PD activity might be the vital therapeutic method to prevent and treat diabetic complexities [215].
A study by Zhang et al. observed the role of G6PD in pancreatic beta-cell survival and its physiological function. They reported that pancreatic islets comprise lesser NADPH-dependent antioxidants. They found that in cultured beta cells and isolated mouse and human islets, increased glucose in the medium decreased G6PD activity and reduced beta-cell survival. The silencing of G6PD with antisense oligonucleotides also produced beta cell death, whereas overexpression of G6PD rescued the cells from the increase in ROS caused by increased glucose in the medium [213]. In contrast, it is evident that the enrichment of G6PD over-produces NADPH, fuels NADPH oxidase, and superoxide anion production, augmenting liver dysfunction in type 2 diabetic (T2D) rats [216]. Quantitative metabolomics proved that hepatic NADP+ and NADPH levels were considerably collapsed in prediabetes and T2D but were mostly protective when mice were supplemented with NR [217].
Obesity
Numerous reasons, including genetic and environmental, contribute for the progression of obesity. One study discovered the possibility that increased intracellular NADPH availability could exacerbate the enzymatic activity [218]. The NADP+/NADPH pool also moderates several oxidoreductase enzymes, including 11β-HSD1. All the factors related to the transformation of cortisone into cortisol are obscure, and the intracellular concentration of NADPH likely determines the 11β-HSD1 activity [218]. Similarly, aldosterone and 18-hydroxycorticosterone synthesis by adrenal mitochondria of the bullfrog, sheep, or rat requires the presence of NADPH [219]. In bovine adrenal homogenates, Kahnt and Neher [220] showed that both corticosterone and 18-hydroxycorticosterone were converted to aldosterone utilizing NAD, NADP, or NADPH. In contrary to this, Raman et al. [221] using ovine adrenal homogenates, found that the transformation of 18-hydroxycorticosterone to aldosterone required added or generated NADPH. Excessive dietary carbohydrate intake induced adipose tissue expansion is attributed to NADPH overproduction. Such a pattern is indispensable for de novo fatty acid synthesis in developing adipocytes or for upgrading the triacylglycerol content in adipocytes. Contradictory to it, it is reported that the decline in G6PD activity might restrain the NADPH level leading to redox homeostasis and lipid deposition in 3T3-L1 cells [222].
Cancer
As discussed before, NADPH provides the major reducing power for guarding cells from oxidative damage, biosynthesis of DNA, RNA, fatty acids, and cholesterol, and also acts as a substrate to produce ROS. Rapidly multiplying cells accordingly need NADPH for their regular function to proliferate and survive. It is reported that the suppression of NADPH reduces glutathione levels leading to ROS-induced hindered apoptosis/necrosis of multiple myeloma cells [223]. In one of the studies, downregulation of NADPH by suppressing tyrosine is proposed to have therapeutic applicability in cancer treatment [224]. In preclinical trials, inhibition of G6PD by 6-AN has proven anti-tumor effects in leukemia, glioblastoma, and lung cancer [225].
Similarly, cisplastin, one of the chemotherapeutic agents, induces kidney cell death by impairing IDH2 function in mitochondria. It is found that cisplastin decreases mitochondrial NADPH and GSH levels, increases oxidative stress, and regulates apoptosis. This suggests that the mitochondrial IDH2-NADPH-GSH antioxidant pathway is a target for inhibiting cisplastin-mediated kidney cell death [226]. IDH2 knockdown efficiently boosts the apoptosis of mouse melanoma B16F10 cells through the modulation of ROS generation. The mutual activity of a ROS generator and the dominance of antioxidant enzyme activity alter the intracellular redox status and aid in cancer management [227]. Moreover, as a result of augmented metabolism, i.e., simultaneous generation of high levels of nucleotides for DNA synthesis and repair, cancer cells typically exhibit higher intracellular ROS levels than normal cells. Pike et al. showed that cancer cells can oxidize both endogenous and exogenous fatty acids, providing NADPH for protection against oxidative stress and preventing ATP loss and cell death. Hence, any signaling pathways regulating the NADPH might help control cancer cell survival [228]. Likewise, diminished NADPH production and a subsequent high ROS level were observed in mitochondrial malic enzyme deficient cells. Hence, mitochondrial ME deficiency resulting in compromised NADPH production provides a critical “collateral lethality” therapeutic strategy for treating pancreatic ductal adenocarcinoma patients [229]. Under high oxidative conditions, the PPP consumes more glucose to compensate for the depleted GSH; in fact, a high oxidative stress drives G6P to the PPP, generating NADPH for antioxidant defenses [176]. Activation of the PPP has also been demonstrated in cancer cells, probably to produce more NADPH to combat oxidative stress [230]. The evidence that PPP flux is mostly higher in some human cancer cells indicates that the PPP might play a vital role in cancer cell proliferation [231] and generate a relatively high level of NADPH to combat ROS [230]. Hence, inhibiting NADPH production in cancer cells would tentatively decelerate macromolecular biosynthesis and render the transformed cells to free radical-mediated damage [232].
Chronic fatigue syndrome (CFS)
Chronic fatigue syndrome is a long-term disorder described by extreme fatigue and a wide range of symptoms. The fatigue may deteriorate with the physical or mental activity that does not recover with rest. A person showing any of its symptoms, including lethargy, muscle aches, and weakness, and headaches, when treated with NADH (5–15 mg) or NADPH (1–5 mg) or their derivatives exhibited significant improvement in physical strength and performance with time without any adverse effects [233]. Different dosage forms such as tablets, capsules, solution, suspension, spray, emulsions, suppositories for administration are available [233]. When the patient diagnosed with CFS was administered 2.5 mg of NADPH intravenously, three times a week for 4 weeks, the patient showed distinct improvement in the CFS (from 142 to 90; a score of 200 is the worst case of chronic fatigue syndrome, a score of 50 is normal for a healthy individual). The patient was able to perform the physical exercise to a greater extent and with less problems afterward [233]. It is assumed that the mechanism of action in treating CFS patients depends on the ability of NADH/NADPH to stimulate cellular ATP generation [233].
Based on the above discussion, we can say the NADPH plays major role in the biosynthesis of nucleic acids, fatty acid acyl chains, cholesterol, and steroid hormones. It is also a central player in cellular redox balance, being required for the regeneration of unconjugated free glutathione and thus, playing the main role in the control of ROS and eventually protecting cells against oxidative stress. NADPH has been shown to reduce the risk of several types of cancers, CVDs, hypertension, Alzheimer’s disease, and Parkinson’s disease. Likewise, in inflammatory pathways, NADPH acts as a substrate for NADPH oxidase in neutrophils and phagocytes, which use these enzymes to kill pathogens by generating superoxides. On the other hand, NADPH fuels cellular ROS production via its role as a substrate for the NOX family proteins (NOX1–7) and of reactive nitrogen species by way of nitric oxide synthases. All pathological conditions such as atherosclerosis, cardiovascular disease, neurodegeneration, cancer, and fibrotic remodeling of multiple organs, etc., instigate the involvement of high levels of ROS. The demand for NADPH is particularly high in proliferating cancer cells due to the two major functions: first, NADPH acts as a cofactor for the synthesis of nucleotides, proteins, and fatty acids; and second, NADPH is required for decrease in the high levels of ROS in cancer cells which results from increased metabolic activity.
ROS are double-edged swords with protective and destructive capabilities. Both these facts are widely asserted and acknowledged, but the mystery regarding the level of ROS at which there is a change from physiological action to pathological action is still unclear. Together, these data suggest that cellular redox homeostasis results from a delicate balance between NADPH‐dependent protection against oxidant stress and NADPH‐dependent reductive stress. However, the results of numerous studies imply that the NADPH might possess greater beneficial effects than their detrimental effects. Given that the NADPH deficiency induces such pathological conditions to high complications, it is not surprising that NADPH plays a beneficial role in health. Indeed, the evidence that NADPH reduces risk factors/ mortality has been widely accepted for the past several decades. Although the mechanism responsible for NADPH-induced protection against chronic diseases remains a topic of debate, the undeniable evidence that NADPH protects against all-cause mortality supports the conclusion that NADPH is more a “friend” and not a “foe”. However, these conclusions come from limited data. More population studies in humans are needed to better elucidate the effects of NADPH to clarify the controversial role of NADPH. Sensitive, dynamic indicators of NAD(P)(H) that can be efficiently targeted to specific subcellular compartments, that can be readily visualized, and that has demonstrable utility in vivo should be developed. A better definition of the localization, functioning, and regulation of redox couples should then permit targeted manipulation for specific effects.
Conclusions
In this review, we demonstrate the integral and powerful role the NADPH plays in maintaining cellular homeostasis and how targeting or exploiting key functions of the NADPH can revert this coenzyme/cofactor from supportive friend to lethal foe. Nevertheless, many questions about the modulation of NADPH in different compartments in normal conditions and in response to stressed conditions remain obscure. Newly‐recognized biosynthetic enzymes such as eNAMPT and MNADK and newly‐developed genetically encoded fluorescent biosensors have improved our understanding of how compartmentalized NAD(H)/NADP(H) pools inter‐relate and how these two cofactors collectively maintain cellular redox metabolism and cellular energy metabolism under normal and pathological circumstances. Moreover, additional study of the pathways and regulatory mechanisms of NADPH generation in different compartments, mechanisms by which NADPH plays a role in various diseases will provide innovative insights into its roles in human health and diseases and may find a value of NADPH for treatment of certain diseases.
References
Scheibe R, Dietz KJ. Reduction–oxidation network for flexible adjustment of cellular metabolism in photoautotrophic cells. Plant Cell Environ. 2012;35:202–16.
Pollak N, Dölle C, Ziegler M. The power to reduce: pyridine nucleotides–small molecules with a multitude of functions. Biochem J. 2007;402:205–18.
Jeon S-M, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–6.
Stanton RC. Glucose‐6‐phosphate dehydrogenase, NADPH, and cell survival. IUBMB life. 2012;64:362–9.
Adler L, Chen C, Koutalos Y. Mitochondria contribute to NADPH generation in mouse rod photoreceptors. J Biol Chem. 2014;289:1519–28.
Hothersall JS, Gordge M, Noronha-Dutra AA. Inhibition of NADPH supply by 6‐aminonicotinamide: effect on glutathione, nitric oxide and superoxide in J774 cells. FEBS Lett. 1998;434:97–100.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
Lowry OH, Passonneau JV, Schulz DW, Rock MK. The measurement of pyridine nucleotides by enzymatic cycling. J Biol Chem. 1961;236:2746–55.
Xie W, Xu A, Yeung ES. Determination of NAD+ and NADH in a single cell under hydrogen peroxide stress by capillary electrophoresis. Anal Chem. 2009;81:1280–4.
Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302.
Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell. 2014;55:253–63.
Fischer E, Zamboni N, Sauer U. High-throughput metabolic flux analysis based on gas chromatography–mass spectrometry derived 13C constraints. Anal Biochem. 2004;325:308–16.
Jones DP. Determination of pyridine dinucleotides in cell extracts by high-performance liquid chromatography. J Chromatogr. 1981;225:446–9.
Mayevsky A, Rogatsky GG. Mitochondrial function in vivo evaluated by NADH fluorescence: from animal models to human studies. Am J Physiol Cell Physiol. 2007;292:615–40.
Zhao Y, Yang Y. Real-time and high-throughput analysis of mitochondrial metabolic states in living cells using genetically encoded NAD+/NADH sensors. Free Radic Biol Med. 2016;100:43–52.
Blacker TS, Duchen MR. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med. 2016;100:53–65.
Bilan DS, Matlashov ME, Gorokhovatsky AY, Schultz C, Enikolopov G, Belousov VV. Genetically encoded fluorescent indicator for imaging NAD+/NADH ratio changes in different cellular compartments. Biochim Biophys Acta. 2014;1840:951–7.
Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, et al. Biosensor reveals multiple sources for mitochondrial NAD+. Science. 2016;352:1474–7.
Cameron WD, Bui CV, Hutchinson A, Loppnau P, Gräslund S, Rocheleau JV. Apollo-NADP+: a spectrally tunable family of genetically encoded sensors for NADP+. Nat Methods. 2016;13:352–8.
Hung YP, Albeck JG, Tantama M, Yellen G. Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab. 2011;14:545–54.
Zhao Y, Jin J, Hu Q, Zhou H-M, Yi J, Yu Z, et al. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab. 2011;14:555–66.
Zhao FL, Zhang C, Zhang C, Tang Y, Ye BC. A genetically encoded biosensor for in vitro and in vivo detection of NADP+. Biosens Bioelectron. 2016;77:901–6.
Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, et al. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab. 2015;21:777–89.
Tao R, Zhao Y, Chu H, Wang A, Zhu J, Chen X, et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat Methods. 2017;14:720–8.
Zou Y, Wang A, Huang L, Zhu X, Hu Q, Zhang Y, et al. Illuminating NAD+ metabolism in live cells and in vivo using a genetically encoded fluorescent sensor. Dev Cell. 2020;53:240–52.
Vanengelenburg SB, Palmer AE. Fluorescent biosensors of protein function. Curr Opin Chem Biol. 2008;12:60–5.
Pouvreau S. Genetically encoded reactive oxygen species (ROS) and redox indicators. Biotechnol J. 2014;9:282–93.
Lukyanov KA, Belousov VV. Genetically encoded fluorescent redox sensors. Biochim Biophys Acta. 2014;1840:745–56.
Sallin O, Reymond L, Gondrand C, Raith F, Koch B, Johnsson K. Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. Elife 2018;7:e32638.
Yu Q, Xue L, Hiblot J, Griss R, Fabritz S, Roux C, et al. Semisynthetic sensor proteins enable metabolic assays at the point of care. Science. 2018;361:1122–6.
Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001;15:10–2.
Xiao W, Wang R-S, Handy DE, Loscalzo J. NAD (H) and NADP (H) redox couples and cellular energy metabolism. Antioxid Redox Signal. 2018;28:251–72.
Fessel JP, Oldham WM. Pyridine dinucleotides from molecules to man. Antioxid Redox Signal. 2018;28:180–212.
Palmieri F, Rieder B, Ventrella A, Blanco E, Do PT, Nunes-Nesi A, et al. Molecular identification and functional characterization of Arabidopsis thaliana mitochondrial and chloroplastic NAD+ carrier proteins. J Biol Chem. 2009;284:31249–59.
Todisco S, Agrimi G, Castegna A, Palmieri F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J Biol Chem. 2006;281:1524–31.
Alano CC, Ying W, Swanson RA. Poly (ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–902.
Ying W, Garnier P, Swanson RA. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem Biophys Res Commun. 2003;308:809–13.
Li H, Liu T-F, Lazrak A, Peracchia C, Goldberg GS, Lampe PD, et al. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J Cell Biol. 1996;134:1019–30.
Bevans CG, Kordel M, Rhee SK, Harris AL. Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J Biol Chem. 1998;273:2808–16.
Luongo TS, Eller JM, Lu M-J, Niere M, Raith F, Perry C, et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature. 2020;588:174–9.
Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223.
Yang Y, Sauve AA. NAD+ metabolism:bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016;1864:1787–800.
Friebe D, Neef M, Kratzsch J, Erbs S, Dittrich K, Garten A, et al. Leucocytes are a major source of circulating nicotinamide phosphoribosyltransferase (NAMPT)/pre-B cell colony (PBEF)/visfatin linking obesity and inflammation in humans. Diabetologia. 2011;54:1200–11.
Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–7.
Romacho T, Valencia I, Ramos-González M, Vallejo S, López-Esteban M, Lorenzo O, et al. Visfatin/eNampt induces endothelial dysfunction in vivo: a role for toll-like receptor 4 and NLRP3 inflammasome. Sci Rep. 2020;10:1–13.
Garten A, Schuster S, Penke M, Gorski T, De Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11:535–46.
Revollo JR, Körner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–75.
Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, et al. SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. 2015;21:706–17.
Yoshida M, Satoh A, Lin JB, Mills KF, Sasaki Y, Rensing N, et al. Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab. 2019;30:329–42.
Chang YH, Chang DM, Lin KC, Shin SJ, Lee YJ. Visfatin in overweight/obesity, type 2 diabetes mellitus, insulin resistance, metabolic syndrome and cardiovascular diseases: a meta‐analysis and systemic review. Diabetes Metab Res Rev. 2011;27:515–27.
Romacho T, Sánchez-Ferrer CF, Peiró C. Visfatin/Nampt: an adipokine with cardiovascular impact. Mediators Inflamm. 2013;2013:1–15.
Chen X, Zhao S, Song Y, Shi Y, K Leak R, Cao G. The role of nicotinamide phosphoribosyltransferase in cerebral ischemia. Curr Top Med Chem. 2015;15:2211–21.
Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun. 2016;7:13103.
Ohashi K, Kawai S, Murata K. Identification and characterization of a human mitochondrial NAD kinase. Nat Commun. 2012;3:1248.
Lerner F, Niere M, Ludwig A, Ziegler M. Structural and functional characterization of human NAD kinase. Biochem Biophys Res Commun. 2001;288:69–74.
Zhang R. MNADK, a novel liver-enriched mitochondrion-localized NAD kinase. Biol Open. 2013;2:432–8.
Zhang R. MNADK, a long‐awaited human mitochondrion‐localized NAD kinase. J Cell Physiol. 2015;230:1697–701.
Zhang K, Kim H, Fu Z, Qiu Y, Yang Z, Wang J, et al. Deficiency of the mitochondrial NAD kinase causes stress-induced hepatic steatosis in mice. Gastroenterology. 2018;154:224–37.
Pollak N, Niere M, Ziegler M. NAD kinase levels control the NADPH concentration in human cells. J Biol Chem. 2007;282:33562–71.
Bradshaw PC. Cytoplasmic and mitochondrial NADPH-coupled redox systems in the cegulation of aging. Nutrients. 2019;11:504.
Agledal L, Niere M, Ziegler M. The phosphate makes a difference: cellular functions of NADP. Redox Rep. 2010;15:2–10.
Houten SM, Denis S, te Brinke H, Jongejan A, van Kampen AH, Bradley EJ, et al. Mitochondrial NADP (H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia. Hum Mol Genet. 2014;23:5009–16.
Liu L, Shah S, Fan J, Park JO, Wellen KE, Rabinowitz JD. Malic enzyme tracers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nat Chem Biol. 2016;12:345–55.
Minich T, Yokota S, Dringen R. Cytosolic and mitochondrial isoforms of NADP+‐dependent isocitrate dehydrogenases are expressed in cultured rat neurons, astrocytes, oligodendrocytes and microglial cells. J Neurochem. 2003;86:605–14.
Rydström J. Mitochondrial NADPH, transhydrogenase and disease. J Biochim Biophys Acta. 2006;1757:721–6.
Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206.
Jin L, Li D, Alesi GN, Fan J, Kang H-B, Lu Z, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27:257–70.
Hecker PA, Leopold JA, Gupte SA, Recchia FA, Stanley WC. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am J Physiol Heart Circ Physiol. 2012;304:H491–H500.
Frederiks WM, Kümmerlin IP, Bosch KS, Vreeling-Sindelarova H, Jonker A, Noorden CJV. NADPH production by the pentose phosphate pathway in the zona fasciculata of rat adrenal gland. J Histochem Cytochem. 2007;55:975–80.
Jain M, Brenner DA, Cui L, Lim CC, Wang B, Pimentel DR, et al. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res. 2003;93:e9–e16.
Tian W-N, Braunstein LD, Apse K, Pang J, Rose M, Tian X, et al. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol. 1999;276:C1121–C31.
Tian W-N, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem. 1998;273:10609–17.
Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–H90.
Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci. 1996;93:6770–4.
Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301:H1723–H41.
Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang Y-Y, Loscalzo AJ, et al. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J Biol Chem. 2003;278:32100–6.
Guzik TJ, Griendling KK. NADPH oxidases: molecular understanding finally reaching the clinical level? Antioxid Redox Signal. 2009;11:2365–70.
Lee JH, Park JW. Attenuated mitochondrial NADP+-dependent isocitrate dehydrogenase activity induces apoptosis and hypertrophy of H9c2 cardiomyocytes. Biochimie. 2014;99:110–8.
Haraguchi CM, Mabuchi T, Yokota S. Localization of a mitochondrial type of NADP-dependent isocitrate dehydrogenase in kidney and heart of rat: an immunocytochemical and biochemical study. J Histochem Cytochem. 2003;51:215–26.
Maeng O, Kim YC, Shin HJ, Lee JO, Huh TL, Kang KI, et al. Cytosolic NADP+-dependent isocitrate dehydrogenase protects macrophages from LPS-induced nitric oxide and reactive oxygen species. Biochem Biophys Res Commun. 2004;317:558–64.
Veech R, Eggleston L, Krebs H. The redox state of free nicotinamide–adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem J. 1969;115:609.
Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 2001;276:16168–76.
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci. 2011;108:19611–6.
Yang Z, Zhang H, Hung HC, Kuo CC, Tsai LC, Yuan HS, et al. Structural studies of the pigeon cytosolic NADP+‐dependent malic enzyme. Protein Sci. 2002;11:332–41.
Sanz N, Díez-Fernández C, Valverde A, Lorenzo M, Benito M, Cascales M. Malic enzyme and glucose 6-phosphate dehydrogenase gene expression increases in rat liver cirrhogenesis. Br J Cancer. 1997;75:487.
Simpson E, Estabrook R. The “malate shuttle” and control of steroid hydroxylation in the adrenal cortex. Adv Enzym Regul. 1969;7:259–79.
Santos MM, Raghevendran V, Kötter P, Olsson L, Nielsen J. Manipulation of malic enzyme in Saccharomyces cerevisiae for increasing NADPH production capacity aerobically in different cellular compartments. Metab Eng. 2004;6:352–63.
Kampjut D, Sazanov LA. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature. 2019;573:291–5.
Jackson JB. Proton translocation by transhydrogenase. FEBS Lett. 2003;545:18–24.
Earle SR, Anderson WM, Fisher RR. Evidence that reconstituted bovine heart mitochondrial transhydrogenase functions as a proton pump. FEBS Lett. 1978;91:21–4.
Kern SE, Price-Whelan A, Newman DK. Extraction and measurement of NAD (P)+ and NAD (P) H. In: Methods in Molecular Biology. Springer: 2014. p 311–23.
Nickel AG, Von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–84.
Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet. 2012;44:740.
Yin F, Sancheti H, Cadenas E. Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim Biophys Acta. 2012;1817:401–9.
Ronchi JA, Francisco A, Passos LAC, Figueira TR, Castilho RF. The contribution of nicotinamide nucleotide transhydrogenase to peroxide detoxification is dependent on the respiratory state and counterbalanced by other sources of NADPH in liver mitochondria. J Biol Chem. 2016;291:20173–87.
Smith HQ, Li C, Stanley CA, Smith TJ. Glutamate dehydrogenase, a complex enzyme at a crucial metabolic branch point. Neurochem Res. 2019;44:117–32.
Dimovasili C, Fadouloglou VE, Kefala A, Providaki M, Kotsifaki D, Kanavouras K, et al. Crystal structure of glutamate dehydrogenase 2, a positively selected novel human enzyme involved in brain biology and cancer pathophysiology. J Neurochem. 2021;157:802–15.
Ciccarese F, Ciminale V. Escaping death: mitochondrial redox homeostasis in cancer cells. Front Oncol. 2017;7:1–16.
Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology. 2017;6:11–37.
Engel PC. Glutamate dehydrogenases: the why and how of coenzyme specificity. Neurochem Res. 2014;39:426–32.
Spanaki C, Zaganas I, Kleopa KA, Plaitakis A. Human GLUD2 glutamate dehydrogenase is expressed in neural and testicular supporting cells. J Biol Chem. 2010;285:16748–56.
Spanaki C, Kotzamani D, Petraki Z, Drakos E, Plaitakis A. Expression of human GLUD1 and GLUD2 glutamate dehydrogenases in steroid producing tissues. Mol Cell Endocrinol. 2015;415:1–11.
DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–24.
Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci USA. 2014;111:14217–22.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5.
Hong C, Zheng J, Li X. Inhibition of GOT1 sensitizes colorectal cancer cells to 5-fluorouracil. Cancer Chemother Pharmacol. 2017;79:835.
Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc Natl Acad Sci USA. 2011;108:15163–8.
Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25:27–42.
Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr. 2010;30:57–81.
Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–83.
Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi S, et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013;4:e877–e89.
Ducker GS, Chen L, Morscher RJ, Ghergurovich JM, Esposito M, Teng X, et al. Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 2016;23:1140–53.
Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–91.
Zong W-X, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. 2016;61:667–76.
Slavov N, Budnik BA, Schwab D, Airoldi EM, van Oudenaarden A. Constant growth rate can be supported by decreasing energy flux and increasing aerobic glycolysis. Cell Rep. 2014;7:705–14.
Shin M, Momb J, Appling DR. Human mitochondrial MTHFD2 is a dual redox cofactor-specific methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase. Cancer Metab. 2017;5:11.
Newman AC, Maddocks OD. One-carbon metabolism in cancer. Br J Cancer. 2017;116:1499–504.
Goodman RP, Calvo SE, Mootha VK. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J Biol Chem. 2018;293:7508–16.
Nikiforov A, Dölle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem. 2011;286:21767–78.
LaNoue K. Metabolite transport in mitochondria. Annu Rev Biochem. 1979;48:871–922.
Pardo B, Contreras L. Redox shuttles in the brain. In: Neural metabolism in vivo. (Springer, 2012), p 841–83.
Macdonald MJ. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets further implication of cytosolic NADPH in insulin secretion. J Biol Chem. 1995;270:20051–8.
Bobyleva V, Kneer N, Bellei M, Battelli D, Lardy HA. Concerning the mechanism of increased thermogenesis in rats treated with dehydroepiandrosterone. J Bioenerg Biomembr. 1993;25:313–21.
Nielsen TT, Støttrup NB, Løfgren B, Bøtker HE. Metabolic fingerprint of ischaemic cardioprotection: importance of the malate–aspartate shuttle. Cardiovasc Res. 2011;91:382–91.
Lane M, Gardner DK. Mitochondrial malate-aspartate shuttle regulates mouse embryo nutrient consumption. J Biol Chem. 2005;280:18361–7.
Macdonald MJ. Evidence for the malate aspartate shuttle in pancreatic islets. Arch Biochem Biophys. 1982;213:643–9.
Martin B, Denton R. Intracellular localization of enzymes in white-adipose-tissue fat-cells and permeability properties of fat-cell mitochondria. Transfer of acetyl units and reducing power between mitochondria and cytoplasm. Biochem J. 1970;117:861–77.
Iyanagi T. Molecular mechanism of phase I and phase II drug‐metabolizing enzymes: implications for detoxification. Int Rev Cytol. 2007;260:35–112.
Porter TD. Electron transfer pathways in cholesterol synthesis. Lipids. 2015;50:927–36.
Iyanagi T. Structure and function of NADPH-cytochrome P450 reductase and nitric oxide synthase reductase domain. Biochem Biophys Res Commun. 2005;338:520–8.
Fico A, Paglialunga F, Cigliano L, Abrescia P, Verde P, Martini G, et al. Glucose-6-phosphate dehydrogenase plays a crucial role in protection from redox-stress-induced apoptosis. Cell Death Differ. 2004;11:823.
Koju N, Taleb A, Zhou J, Lv G, Yang J, Cao X, et al. Pharmacological strategies to lower crosstalk between nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria. Biomed Pharmacother. 2019;111:1478–98.
Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–92.
Meldrum NU, Tarr HLA. The reduction of glutathione by the Warburg-Christian system. Biochem J. 1935;29:108.
Ost M, Keipert S, van Schothorst EM, Donner V, van der Stelt I, Kipp AP, et al. Muscle mitohormesis promotes cellular survival via serine/glycine pathway flux. FASEB J. 2015;29:1314–28.
Kirsch M, De Groot H. NAD (P) H, a directly operating antioxidant? FASEB J. 2001;15:1569–74.
Nakamura H. Thioredoxin and its related molecules: update 2005. Antioxid Redox Signal. 2005;7:823–8.
Bodega G, Alique M, Bohórquez L, Morán M, Magro L, Puebla L, et al. Young and especially senescent endothelial microvesicles produce NADPH: The fuel for their antioxidant machinery. Oxid Med Cell Longev. 2018;2018:3183794.
Qin Y-Y, Li M, Feng X, Wang J, Cao L, Shen X-K, et al. Combined NADPH and the NOX inhibitor apocynin provides greater anti-inflammatory and neuroprotective effects in a mouse model of stroke. Free Radic Biol Med. 2017;104:333–45.
Takac I, Schröder K, Brandes RP. The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr Hypertens Rep. 2012;14:70–8.
Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal. 2006;8:1549–61.
Phillips DC, Woollard KJ, Griffiths HR. The anti‐inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br J Pharmacol. 2003;138:501–11.
Griffiths HR, Gao D, Pararasa C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017;12:50–7.
Linderholm AL, Onitsuka J, Xu C, Chiu M, Lee W-M, Harper RW. All-trans retinoic acid mediates DUOX2 expression and function in respiratory tract epithelium. Am J Physiol Lung Cell Mol Physiol. 2010;299:L215–L21.
Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. In: Trends in innate immunity. (Karger Publishers, 2008), vol 15, p 164-87.
Green SJ, Crawford R, Hockmeyer J, Meltzer M, Nacy C. Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha. J Immunol. 1990;145:4290–7.
Seguin MC, Klotz FW, Schneider I, Weir JP, Goodbary M, Slayter M, et al. Induction of nitric oxide synthase protects against malaria in mice exposed to irradiated Plasmodium berghei infected mosquitoes: involvement of interferon gamma and CD8+ T cells. J Exp Med. 1994;180:353–8.
Francisco A, Engel DF, Figueira TR, Rogério F, Andreza F, Castilho RF. Mitochondrial NAD (P)+ transhydrogenase is unevenly distributed in different brain regions, and its loss causes depressive-like behavior and motor dysfunction in mice. Neuroscience. 2020;440:210–29.
Gotti S, Sica M, Viglietti‐Panzica C, Panzica G. Distribution of nitric oxide synthase immunoreactivity in the mouse brain. Microsc Res Technol. 2005;68:13–35.
Guzik TJ, West NE, Pillai R, Taggart DP, Channon KM. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension. 2002;39:1088–94.
Guzik T, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation. J Physiol Pharmacol. 2003;54:469–87.
Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci USA. 2005;102:9936–41.
Borutaite V, Hope H, Brown GC. Arachidonate and NADPH oxidase synergise with iNOS to induce death in macrophages: mechanisms of inflammatory degeneration. Pharmacol Rep. 2006;58:96.
Li H, Poulos TL. Structure–function studies on nitric oxide synthases. J Inorg Biochem. 2005;99:293–305.
Yoshii K, Tajima F, Ishijima S, Sagami I. Changes in pH and NADPH regulate the DNA binding activity of neuronal PAS domain protein 2, a mammalian circadian transcription factor. Biochemistry. 2015;54:250–9.
Yoshii K, Ishijima S, Sagami I. Effects of NAD (P) H and its derivatives on the DNA-binding activity of NPAS2, a mammalian circadian transcription factor. Biochem Biophys Res Commun. 2013;437:386–91.
Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15:R271–R7.
Liu AC, Lewis WG, Kay SA. Mammalian circadian signaling networks and therapeutic targets. Nat Chem Biol. 2007;3:630–9.
Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. In: Circadian clocks. (Springer, 2013), p 3–27.
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–9.
Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205.
Stangherlin A, Reddy AB. Regulation of circadian clocks by redox homeostasis. J Biol Chem. 2013;288:26505–11.
Rutter J, Reick M, Wu LC, McKnight SL. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science. 2001;293:510–4.
O’Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature. 2011;469:498–503.
Rey G, Valekunja UK, Feeney KA, Wulund L, Milev NB, Stangherlin A, et al. The pentose phosphate pathway regulates the circadian clock. Cell Metab. 2016;24:462–73.
Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radic Biol Med. 2012;52:539–55.
Go YM, Jones DP. Redox theory of aging: implications for health and disease. Clin Sci. 2017;131:1669–88.
Son MJ, Kwon Y, Son T, Cho YS. Restoration of mitochondrial NAD+ levels delays stem cell senescence and facilitates reprogramming of aged somatic cells. Stem Cells. 2016;34:2840–51.
Tischler ME, Friedrichs D, Coll K, Williamson JR. Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch Biochem Biophys. 1977;184:222–36.
Jongkind JF, Verkerk A, Poot M. Glucose flux through the hexose monophosphate shunt and NADP (H) levels during in vitro ageing of human skin fibroblasts. Gerontology. 1987;33:281–6.
Zhu XH, Lu M, Lee B-Y, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci USA. 2015;112:2876–81.
Aw TY. Postnatal changes in pyridine nucleotides in rat hepatocytes: composition and O2 dependence. Pediatr Res. 1991;30:112.
Lenaz G, D’Aurelio M, Pich MM, Genova M, Ventura B, Bovina C, et al. Mitochondrial bioenergetics in aging. Biochim Biophys Acta. 2000;1459:397–404.
Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–52.
Ma C, Pi C, Yang Y, Lin L, Shi Y, Li Y, et al. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting Sirt1. PLoS One. 2017;12:e0170930.
Nóbrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, et al. G6PD protects from oxidative damage and improves healthspan in mice. Nat Commun. 2016;7:1–9.
Ghosh D, Levault KR, Brewer GJ. Relative importance of redox buffers GSH and NAD (P) H in age‐related neurodegeneration and Alzheimer disease‐like mouse neurons. Aging Cell. 2014;13:631–40.
Benzi G, Moretti A. Age-and peroxidative stress-related modifications of the cerebral enzymatic activities linked to mitochondria and the glutathione system. Free Radic Biol Med. 1995;19:77–101.
Schwartz AG, Pashko LL. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Res. Rev. 2004;3:171–87.
Ulusu NN. Glucose-6-phosphate dehydrogenase deficiency and Alzheimer’s disease: partners in crime? The hypothesis. Med Hypotheses. 2015;85:219–23.
Krishnan S, Rani P. Evaluation of selenium, redox status and their association with plasma amyloid/tau in Alzheimer’s disease. Biol Trace Elem Res. 2014;158:158–65.
Vural H, Demirin H, Kara Y, Eren I, Delibas N. Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer’s disease. J Trace Elem Med Biol. 2010;24:169–73.
Verdile G, Keane KN, Cruzat VF, Medic S, Sabale M, Rowles J, et al. Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm. 2015;2015:1–17.
Birkmayer W, Birkmayer J, Horowski R, Wachtel H, Loschmann PA, inventors. Agent for treatment of Parkinson’s disease. United States patent 4,970,200. Nov. 13 1990.
Birkmayer J, inventors. Treatment of Parkinson’s disease with NADPH. United States patent 5,019,561. May 28 1991.
Herrero Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat Cell Biol. 2009;11:747–52.
Zhou Y, Wu J, Sheng R, Li M, Wang Y, Han R, et al. Reduced nicotinamide adenine dinucleotide phosphate inhibits MPTP-induced neuroinflammation and neurotoxicity. Neuroscience. 2018;391:140–53.
Dunn L, Allen GF, Mamais A, Ling H, Li A, Duberley KE, et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson’s disease. Neurobiol Aging. 2014;35:1111–5.
Birkmayer JG, inventors. NADH and NADPH pharmaceuticals for treating hypertension. United States patent 5,668,114. Sep. 16 1997.
Dong LH, Li L, Song Y, Duan ZL, Sun SG, Lin YL, et al. TRAF6-mediated SM22α K21 ubiquitination promotes G6PD activation and NADPH production, contributing to GSH homeostasis and VSMC survival in vitro and in vivo. Circ Res. 2015;117:684–94.
Jain M, Cui L, Brenner DA, Wang B, Handy DE, Leopold JA, et al. Increased myocardial dysfunction after ischemia-reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation. 2004;109:898–903.
Ku HJ, Ahn Y, Lee JH, Park KM, Park JW. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic Biol Med. 2015;80:84–92.
Fazal L, Laudette M, Paula-Gomes S, Pons S, Conte C, Tortosa F, et al. Multifunctional mitochondrial Epac1 controls myocardial cell death. Circ Res. 2017;120:645–57.
Boslett J, Hemann C, Zhao YJ, Lee HC, Zweier JL. Luteolinidin protects the postischemic heart through CD38 inhibition with preservation of NAD (P)(H). J Pharmacol Exp Ther. 2017;361:99–108.
Reyes LA, Boslett J, Varadharaj S, De Pascali F, Hemann C, Druhan LJ, et al. Depletion of NADP (H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc Natl Acad Sci USA. 2015;112:11648–53.
Huang YS, Wang LX, Huang GR. Plasma redox state and its clinical significance in elderly patients with acute myocardial infarction. Chin J Geriatric Heart Brain Vessel Dis. 2013;15:132–4.
Zhu J, Wang YF, Chai XM, Qian K, Zhang LW, Peng P, et al. Exogenous NADPH ameliorates myocardial ischemia–reperfusion injury in rats through activating AMPK/mTOR pathway. Acta Pharmacol Sin. 2020; 41:535–45.
Matsui R, Xu S, Maitland KA, Hayes A, Leopold JA, Handy DE, et al. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation. 2005;112:257–63.
Pei H, Song X, Peng C, Tan Y, Li Y, Li X, et al. TNF-α inhibitor protects against myocardial ischemia/reperfusion injury via Notch1-mediated suppression of oxidative/nitrative stress. Free Radic Biol Med. 2015;82:114–21.
Uysal A, Sahna E, Ozguler I, Burma O, Ilhan N. Effects of apocynin, an NADPH oxidase inhibitor, on levels of ADMA, MPO, iNOS and TLR4 induced by myocardial ischemia reperfusion. Perfusion. 2015;30:472–7.
Cortassa S, O’Rourke B, Aon MA. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim Biophys Acta. 2014;1837:287–95.
Handy DE, Loscalzo J. Responses to reductive stress in the cardiovascular system. Free Radic Biol Med. 2017;109:114–24.
Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G–αB-crystallin causes aberrant desmin and αB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91.
Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, et al. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol. 2006;41:340–9.
Zuurbier C, Eerbeek O, Goedhart P, Struys E, Verhoeven N, Jakobs C, et al. Inhibition of the pentose phosphate pathway decreases ischemia–reperfusion-induced creatine kinase release in the heart. Cardiovasc Res. 2004;62:145–53.
Hecker PA, Lionetti V, Ribeiro RF Jr, Rastogi S, Brown BH, O’Connell KA, et al. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ Heart Fail. 2013;6:118–26.
Li M, Zhou ZP, Sun M, Cao L, Chen J, Qin YY, et al. Reduced nicotinamide adenine dinucleotide phosphate, a pentose phosphate pathway product, might be a novel drug candidate for ischemic stroke. Stroke. 2016;47:187–95.
Zhao G, Zhao Y, Wang X, Xu Y. Knockdown of glucose-6-phosphate dehydrogenase (G6PD) following cerebral ischemic reperfusion: the pros and cons. Neurochem Int. 2012;61:146–55.
Huang Q, Sun M, Li M, Zhang D, Han F, Wu JC, et al. Combination of NAD+ and NADPH offers greater neuroprotection in ischemic stroke models by relieving metabolic stress. Mol Neurobiol. 2018;55:6063–75.
Díaz Flores M, Ibáñez Hernández MA, Galván RE, Gutiérrez M, Durán Reyes G, Medina Navarro R, et al. Glucose-6-phosphate dehydrogenase activity and NADPH/NADP+ ratio in liver and pancreas are dependent on the severity of hyperglycemia in rat. Life Sci. 2006;78:2601–7.
Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, Stevens RD, et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem. 2006;281:30593–602.
Ammon HP, Patel TN, Steinke J. The role of the pentose phosphate shunt in glucose-induced insulin release: in vitro studies with 6-aminonicotinamide, methylene blue, NAD+, NADH, NADP+, NADPH and nicotinamide on isolated pancreatic rat islets. Biochim Biophys Acta. 1973;297:352–67.
Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J, et al. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and β-cell apoptosis. FASEB J. 2010;24:1497–505.
Xu Y, Osborne BW, Stanton RC. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am J Physiol Ren Physiol. 2005;289:F1040–F7.
Perner A, Nielsen S, Rask-Madsen J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med. 2003;29:642–5.
Gupte RS, Floyd BC, Kozicky M, George S, Ungvari ZI, Neito V, et al. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD (P) H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med. 2009;47:219–28.
Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. 2016;6:26933.
Agarwal AK. Cortisol metabolism and visceral obesity: role of 11β‐hydroxysteroid dehydrogenase type I enzyme and reduced co‐factor NADPH. Endocr Res. 2003;29:411–8.
Fraser R, Lantos C. 18-Hydroxycorticosterone: a review. J Steroid Biochem. 1978;9:273–86.
Kahnt F, Neher R. Über die adrenale Steroid‐Biosynthese in vitro I. Umwandlung endogener und exogener Vorstufen im Nebennieren‐Homogenat des Rindes. Helvetica Chim Acta. 1965;48:1457–76.
Raman PB, Sharma D, Dorfman R. Studies on aldosterone biosynthesis in vitro. Biochemistry. 1966;5:1795–804.
Torres-Ramírez N, Baiza-Gutman LA, García-Macedo R, Ortega-Camarillo C, Contreras-Ramos A, Medina-Navarro R, et al. Nicotinamide, a glucose-6-phosphate dehydrogenase non-competitive mixed inhibitor, modifies redox balance and lipid accumulation in 3T3-L1 cells. Life Sci. 2013;93:975–85.
Yin L, Kosugi M, Kufe D. Inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death by down-regulating TIGAR expression and depleting NADPH. Blood. 2012;119:810–6.
Lui VWY, Wong EYL, Ho K, Ng PKS, Lau CPY, Tsui SKW, et al. Inhibition of c-Met downregulates TIGAR expression and reduces NADPH production leading to cell death. Oncogene. 2011;30:1127.
Budihardjo II, Walker DL, Svingen PA, Buckwalter CA, Desnoyers S, Eckdahl S, et al. 6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin. Clin Cancer Res. 1998;4:117–30.
Kong MJ, Han SJ, Kim JI, Park JW, Park KM. Mitochondrial NADP+-dependent isocitrate dehydrogenase deficiency increases cisplatin-induced oxidative damage in the kidney tubule cells. Cell Death Dis. 2018;9:488.
Ku HJ, Kwon OS, Kang BS, Lee DS, Lee HS, Park JW. IDH2 knockdown sensitizes tumor cells to emodin cytotoxicity in vitro and in vivo. Free Radic Res. 2016;50:1089–97.
Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta. 2011;1807:726–34.
Dey P, Baddour J, Muller F, Wu CC, Wang H, Liao WT, et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature. 2017;542:119–23.
Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–54.
Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. 2014;5:592–602.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95.
Birkmayer JG, inventors. NADH and NADPH pharmaceuticals for treating chronic fatigue syndrome. United States patent 5,712,259. Jan. 27 1998.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81973315, 82173811, 81730092), Natural Science Foundation of Jiangsu Higher Education (20KJA310008), Jiangsu Key Laboratory of Neuropsychiatric Diseases (BM2013003), and the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Koju, N., Qin, Zh. & Sheng, R. Reduced nicotinamide adenine dinucleotide phosphate in redox balance and diseases: a friend or foe?. Acta Pharmacol Sin 43, 1889–1904 (2022). https://doi.org/10.1038/s41401-021-00838-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00838-7
