
Abstract
Xanthine oxidoreductase (XOR) is a critical, rate-limiting enzyme that controls the last two steps of purine catabolism by converting hypoxanthine to xanthine and xanthine to uric acid. It also produces reactive oxygen species (ROS) during the catalytic process. The enzyme is generally recognized as a drug target for the therapy of gout and hyperuricemia. The catalytic products uric acid and ROS act as antioxidants or oxidants, respectively, and are involved in pro/anti-inflammatory actions, which are associated with various disease manifestations, including metabolic syndrome, ischemia reperfusion injury, cardiovascular disorders, and cancer. Recently, extensive efforts have been devoted to understanding the paradoxical roles of XOR in tumor promotion. Here, we summarize the expression of XOR in different types of cancer and decipher the dual roles of XOR in cancer by its enzymatic or nonenzymatic activity to provide an updated understanding of the mechanistic function of XOR in cancer. We also discuss the potential to modulate XOR in cancer therapy.
Similar content being viewed by others
Introduction
Xanthine oxidoreductase (XOR) is a pivotal enzyme that acts as a catalyst for the oxidation of hypoxanthine to xanthine or xanthine to uric acid in the last two steps of purine catabolism in the highest uricotelic primates (Fig. 1). XOR belongs to the family of metal flavin enzymes, which is a homodimer with a molecular mass of approximately 150 kDa for each subunit. Each subunit comprised four redox-active centers: two iron–sulfur redox clusters (Fe2–S2, 20 kDa), one flavin adenine dinucleotide (FAD, 40 kDa), and one molybdopterin (Mo–Co, 85 kDa) [1]. During catalysis, the oxidation of xanthine takes place at the molybdenum center, and the electrons are rapidly transferred to FAD via the Fe/S center [2] (Fig. 2). XOR exists in two interconvertible forms as xanthine dehydrogenase (XDH, EC 1.17.1.4) and xanthine oxidase (XO, EC 1.17.3.2) in mammals. XO reduces oxygen to produce superoxide and H2O2, whereas XDH is able to reduce either oxygen or NAD+ with a greater affinity for the latter. XDH is the predominant form of XOR found in normal cells and tissues, yet XO appears to have an important role during stress or upon immune activation [3, 4]. For example, XDH is converted to XO via proteolytic modification or reversible sulfhydryl oxidation under conditions of ischemia/hypoxia or other forms of stimuli [5]. XOR is also able to oxidase nicotinamide adenine dinucleotide (NADH) or convert nitrites into nitric oxide, especially in environments with low pH or hypoxia, thus generating ROS or reactive nitrogen species (RNS), respectively [6]. Activity as XDH, XO, or nitrite reductase often takes place in the Mo-co domain of XOR, while the FAD domain contains the substrate pocket for the oxidation of NADH [1] (Fig. 2).

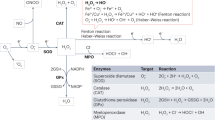
Xanthine oxidoreductase (XOR) is the rate-limiting enzyme that controls the last two steps of purine catabolism by converting hypoxanthine to xanthine and xanthine to uric acid. Inhibitors of XOR such as allopurinol, febuxostat, and topiroxostat are able to inhibit the process. IMPDH inosine monophosphate dehydrogenase, ADSL adenylosuccinate lyase, ADSS adenylosuccinate synthase, GMPS guanine monophosphate, cN-I cytosolic 5ʹ-nucleotidase I, cN-II cytosolic 5’-nucleotidase II, ADA adenosine deaminase, GDA guanine deaminase, PNP purine nucleoside phosphorylase.

XOR is a homodimer and each subunit composes of two non-identical iron-sulfur clusters (2Fe/S), a flavin adenine dinucleotide (FAD) cofactor, and a molybdopterin cofactor (Mo-co) domain. XOR acts as xanthine dehydrogenase (XDH), xanthine oxidase (XO), nicotinamide adenine dinucleotide (NADH) oxidase, and nitrate reductase under different circumstances, participating in purine metabolism, detoxification, and redox homeostasis.
XOR is implicated in functions beyond purine metabolism (Fig. 2). XOR constitutes a major component of the milk-fat globule membrane and colocalizes with the lipid-binding protein adipophilin on the milk-fat globule, exerting an essential role in milk fat droplet secretion from the lactating mammary gland [7]. XOR produces uric acid, as well as ROS and RNS, which are involved in antioxidant or antibacterial defenses, making it a versatile protective housekeeping enzyme and an important component of the innate immune system [8]. Additionally, XOR is involved in the metabolism of a number of endogenous and exogenous substances, including numerous toxic compounds,anticancer and antimetabolic drugs, which is attributed to its low specificity for substrates [9]. Thus, XOR also acts as a detoxifying and drug-metabolizing enzyme.
The activity of XOR is regulated by a variety of factors, including hormones, growth factors, lipopolysaccharide [4, 10], and various inflammatory cytokines, such as interferon-γ (IFN-γ), IFN-α, tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1) and IL-3 [2, 11]. Hypoxia is the most studied factor that regulates the expression and activity of XOR. In general, XOR enzymatic activity tends to be repressed under hyperoxic conditions, while hypoxia was shown to induce a gradual increase in XOR expression in endothelial cells at both pre- and posttranslational levels [12, 13]. p38 MAPK may play a critical role in the activation of XOR, as it has been reported to activate XOR under stress or hypoxia [14].
Under pathological conditions, the excessive activity of XOR results in a high level of uric acid in the plasma, which leads to hyperuricemia and gout [8]. XOR and its byproducts are also considered to be involved in other pathological processes, including tumor lysis syndrome, ischemia reperfusion injury, circulatory shock, vascular disorders in diabetes, adipogenesis, cardiovascular disease, and cancer [15].
The involvement of XOR in the pathogenesis of cancer has been established for ages. Upregulated XOR activity and its byproducts have been observed in cancer cachexia and play important roles in cancer tissue wasting [16, 17]. Inhibition of XOR-induced oxidative stress and subsequent inflammation significantly improved survival in animal models of cancer cachexia [18]. Moreover, the ROS/RNS and uric acid produced by XOR are involved in the development of hyperuricemia, hypertension, dyslipidemia, and insulin resistance, revealing the important role of XOR in metabolic syndrome [19]. Additionally, patients with metabolic syndrome have an increased risk of cancer [20]. Recently, the understanding of XOR in cancer has greatly advanced with progress in cancer genomics and metabolomics [21, 22]. The level of XOR expression is often found to be altered in cancer tissues compared with the corresponding normal tissues [22]. Abnormal XOR expression and activity are observed in various types of cancer and closely associated with the clinical outcome. XOR is implicated both in the initiation and promotion of cancer by the action of its catalytic products or nonenzymatic activity [23, 24]. ROS and uric acid may exert multiple activities, including mutagenesis, pro-angiogenesis, cytotoxicity, pro/anti-apoptosis and pro/antioxidative stress, depending on the circumstances, which also play a vital role in regulating the immune response and cancer metabolism. Furthermore, the influence of XOR on cancer treatment has also been increasingly recognized.
In this review, we elaborate the double-faced role of XOR in different types of cancer based on its enzymatic or nonenzymatic activity, as well the potential opportunities in cancer prevention and therapy by modulating XOR.
Expression and activity of XOR in human cancer
XOR expression widely varies across different tissues according to the data recruited from the Human Protein Atlas (https://www.proteinatlas.org/) (Supplemental Fig. 1). Likewise, the alteration of XOR expression is diversified in cancers originating from different tissue types [25]. As shown in Fig. 3, the mRNA data of XOR in tumor tissue and its paired normal tissue were recruited from TCGA (The Cancer Genome Atlas, https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). The expression and activity of XOR are reduced in tumors derived from tissues with high XOR expression because of the low differentiation of malignant cells [22], such as liver, breast and colon cancers (Fig. 3). In contrast, the expression of XOR significantly increases in tumors derived from low XOR-expressing tissues (Fig. 3), which might be due to elevated demand for purine metabolism and the activation of the inflammatory response [22, 26]. The altered expression and activity of XOR in cancer tissues are usually associated with the prognosis of cancer [27,28,29,30].

The expression of XOR at mRNA level was recruited from TCGA database and plotted using the TIMER (https://cistrome.shinyapps.io/timer/). *P < 0.05; **P < 0.01; ***P < 0.001.
Downregulation of XOR in cancer
XOR expression and activity are much lower in gastrointestinal, colorectal, breast or liver tumor tissues than in their normal counterparts [22] (Fig. 3). Low XOR level has been linked to aggressive phenotypes and unfavorable clinical outcomes in multiple cancers.
In contrast to high level of XOR in normal liver tissue, decreased expression and activity are observed in hepatocellular carcinoma (HCC), and lower XOR confers selective advantages to hepatic cancer cells [31, 32]. Downregulation of XOR is significantly correlated with recurrence and poor prognosis in HCC [31,32,33]. Mechanistically, knocking down or inhibiting XDH promoted the expression of epithelial–mesenchymal transition (EMT) markers as well as cell migration and invasion dependent on the transforming growth factor-β (TGFβ) signaling pathway. In addition, decreased XDH expression is associated with increased gene expression related to the signature of cancer stem cells (CSCs), such as CD44 or CD133, in HCC cells and leads to cancer development [33]. A recent study revealed that loss of XOR potentiated the propagation of HCC stem cells by decreasing ubiquitin-specific peptidase 15 (USP15)-mediated nuclear factor erythroid 2-related factor 2 (NRF2)-Kelch-like ECH associated protein 1 (KEAP1) signaling and ROS accumulation [32]. The results may help understand the role of XOR in HCC, as well as other cancers with reduced XOR expression or activity.
XOR exerts an important role in modulating the pathogenesis of breast cancer. High levels of XOR expression and activity are observed in normal breast epithelium, while XOR expression is markedly reduced in neoplastic breast epithelium [34]. XOR has been recognized as an important functional component of differentiation, and diminished expression indicates poor differentiation [35]. Indeed, decreased XOR expression is associated with parameters of aggressive breast cancer, including poor histologic grade of differentiation, ductal and lobular histologic types, large tumor size, and a high number of positive axillary lymph nodes [29]. Therefore, decreased XOR in mammary epithelial cells was identified as a marker of aggressive breast cancer and predictor of poor clinical outcome [29, 36].
Decreased expression of XOR was also found in colon cancer according to TCGA data (Fig. 3). Similarly, analyses of XOR expression in specimens from 478 patients with colorectal cancer (CRC) showed that XOR was decreased in 62% and undetectable in 22% of the tumors compared to normal tissues [28]. Low expression of XOR was associated with increased CRC risk and poor differentiation as well as the grade of disease in CRC patients [28, 37]. XOR is identified as one of the metabolic markers related to CRC risk and provides significant prognostic information independent of established factors.
Enhanced expression and activity of XOR in cancer
In contrast to the aforementioned cancers, increased levels of XOR are often observed in cancers originating from low XOR-expressing tissues (Fig. 3). For example, XOR expression and activity are significantly higher in prostate cancer [38], laryngeal well-differentiated squamous cell carcinomas [39], and bladder cancer [40] than in the corresponding adjacent normal tissues. Among these cancers, XOR is more often studied in lung cancer and brain tumors.
The expression and activity of XOR is low in normal lung tissue, while an enhanced level of XOR is exhibited in cancerous tissue [26, 41, 42]. Consistently, the levels of xanthine, uric acid and other metabolites produced during purine catabolism are also elevated in lung adenocarcinoma [43, 44], suggesting upregulation of purine metabolism in lung cancer. Meanwhile, the 5-year overall survival rate among patients with lung adenocarcinoma strongly expressing XDH is significantly poorer than those expressing lower levels of XDH in cancer tissue [30]. Non-small-cell lung cancer (NSCLC) with low expression of programmed death ligand 1 (PD-L1) and wild-type epidermal growth factor receptor (EGFR) is often refractory to the available molecularly targeted therapies, and therapeutic options are very limited. Combined analysis of the methylome and transcriptome revealed that XDH was one of the potential therapeutic targets for this subgroup of NSCLC [45]. Nevertheless, the role of XOR in lung cancer remains largely unknown.
Similarly, significantly higher levels of XOR were observed in meningioma and astrocytoma compared with normal brain tissue [46]. Likewise, the rs207444 polymorphism of the XDH gene was associated with the risk of meningioma [47]. Moreover, compared with primary brain tumors or nonbrain metastatic papillary thyroid carcinomas (PTCs), XOR expression was upregulated in brain metastatic PTCs [48]. Metabolomic analysis of the brain of the cachexia model showed that purine metabolism was activated and XO activity was increased. Targeting XO in the brain may be a promising strategy for the treatment of cancer cachexia [49].
Bipolar nature of XOR in cancer
As XOR expression and activity are differentially altered in various tumor types, it is not surprising that XOR plays complex and paradoxical roles in modulating cancer. XOR is an important cellular source of ROS. The dual roles of ROS in cancer prevention and promotion have been extensively reviewed [50] (Fig. 4). On the other hand, XOR is usually considered the rate-limiting step for the production of uric acid. Uric acid is a major water-soluble antioxidant in human plasma but may also possess intracellular pro-oxidant activity (Fig. 4). Moreover, XOR may also regulate cancer development independent of its enzymatic activity.

XOR may exert protumoral or tumor suppressive role via modulating tumor cells as well tumor microenviroment by its catalytic products uric acid and ROS.
Tumor-promoting role of XOR
ROS
XOR is a physiological source of ROS, including superoxide ions, hydrogen peroxide, and nitric oxide, which function as second messengers in the activation of various pathways involved in tumorigenesis [23]. XOR-derived ROS have also been shown to promote tumorigenesis by inducing mutagenesis, cell proliferation, angiogenesis, and reshaping the tumor microenvironment (Fig. 4).
ROS are thought to be oncogenic, causing damage to DNA, proteins, and lipids, promoting genetic instability and activating various signaling cascades related to tumorigenesis [50]. XOR is involved in the metabolism of alcohol and has been linked to the increased risk of breast cancer in alcoholics because ROS generated from XOR can induce carcinogenic mutations and DNA damage [51, 52]. ROS produced by XOR also cause lipid peroxidation and protein oxidation, which results in damage to the function and structure of the cell membrane and protein dysfunction, thus triggering pro-carcinogenic processes in prostate cancer [38].
XOR-derived ROS could act as second messengers promoting vascular growth. ROS stimulate the activation of various transcription factors, including hypoxia-inducible factor-1 (HIF-1) and nuclear factor κB (NF-κB), followed by the upregulation of angiogenic molecules, such as vascular endothelial growth factor (VEGF), matrix metalloproteinase (MMP), and urokinase-type plasminogen activator (uPA), that stimulate endothelial cell proliferation and migration [53]. NF-κB induced by ROS could also promote cancer-associated inflammatory signaling, including the production of cytokines and the expression of a set of antiapoptotic proteins [54, 55]. XO-derived ROS have been identified as a novel and critical component in the regulation of HIF-1α [56]. Upregulation of HIF-1α plays a central role in tumor progression by activating various hallmarks of cancer, such as glucose metabolism, angiogenesis, migration, and invasion [57]. Indeed, chemically induced hypoxia increased XOR activity and the subsequent ROS in human glioma cells, which further induced HIF-1α expression and contributed to both tumor development and progression [58]. Moreover, overexpression of XOR was found to upregulate the expression of HIF-1 and glycolysis in hepatoma cells [59].
XO has also been shown to regulate tumor growth by modulating the tumor microenvironment. The knock-in XO-locked form of XOR in mice strongly increased transplanted tumor growth, which was due to the increased regulatory T cells (Tregs) in the tumor tissue as a result of the high level of ROS produced by macrophages [60]. Moreover, XO-derived ROS in macrophages mediated the activation of the NLRP3 inflammasome and IL-1β release [61], which may exacerbate the inflammatory response and promote tumorigenesis.
These findings suggest that XOR-derived ROS contribute to cancer-associated inflammatory signaling as well as to tumor progression by activating angiogenesis, cell migration, and invasion in tumor cells. Hence, scavenger of ROS and XOR inhibitors may be considered effective strategies for cancer treatment.
Uric acid
Uric acid is the final product of XOR. It is becoming clear that a high level of uric acid may indicate higher cancer incidence and tumor stage and is negatively associated with survival in cancer patients [62, 63]. Evidence has shown that preoperative serum uric acid (SUA) is an independent and significant prognostic predictor in esophageal squamous cell carcinoma patients who undergo R0 esophagectomy and patients with pancreatic cancer [64, 65]. The SUA level was revealed to gradually increase from stage I to stage IV in CRC, suggesting that the SUA level reflected the progression of CRC [66]. The SUA level also correlates with the incidence of urological cancer, including bladder and renal cancer [67]. Therefore, management of uric acid levels may be relevant for the improvement of treatment in patients where hyperuricemia disorders are associated with tumors.
Increasing evidence supports that high levels of uric acid can be responsible for oxidant and proinflammatory activity with consequent cell transformation by reacting with ROS, NO, and RNS [19]. Although uric acid is generally regarded as a potent antioxidant in the extracellular environment, an excessive intracellular concentration of uric acid induces an oxidative burst in many types of cells [68, 69]. Upregulated uric acid could increase the production of ROS by reducing nitric oxide availability or disrupting cellular redox homeostasis [70, 71]. In addition, an excessive intracellular concentration of uric acid may result in feedback on the downregulation of XOR expression, thus increasing the purine salvage pathway and supporting cell proliferation in transformed cells [24].
Uric acid also plays an important role in regulating fructose metabolism, which has been recognized as a key driving force in metabolic syndrome and cancers [72]. In the process of fructose metabolism, generated AMP is further degraded by AMP deaminase and XOR, resulting in the production of uric acid [73]. Elevated uric acid was found to prevent fructose metabolites from channeling mitochondrial oxidation in human HCC HepG2 cells by decreasing aconitase activity in the mitochondria, resulting in the inhibition of the TCA cycle, mitochondrial dysfunction and enhanced glycolysis [72]. Meanwhile, uric acid is able to promote lipid synthesis by increasing the accumulation of citrate and subsequent translocation from mitochondria to the cytosol [74]. These effects may converge to promote cell proliferation and cancer growth.
Moreover, a high level of uric acid is able to induce inflammatory stress, which may also contribute to tumorigenesis. When uric acid is saturated in body fluids, it nucleates into crystals of monosodium urate (MSU) [75], which are ingested by phagocytes and stimulate NLRP3 inflammasomes and produce the proinflammatory form of IL-1β by activating caspase-1-mediated cleavage of pro-IL-1β [76]. Soluble uric acid is also able to promote IL-1β production dependent on NLRP33 and Myd88 [71]. IL-1β is a key proinflammatory cytokine and plays a key role in carcinogenesis and tumor growth. IL-1β upregulation was shown to exert protumoral activity via the immunosuppressive network of myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and Tregs [77]. IL-1β promotes tumor development by driving chronic nonresolved inflammation and endothelial cell activation, influencing angiogenesis and metastasis [78]. Increased levels of IL-1β are correlated with poor prognosis and invasiveness in experimental tumor models and in various types of cancers, including melanoma, colon, lung, breast, or head and neck cancers.
Nonenzymatic activity
XOR can also confer a cancer-promoting action through nonenzymatic activities (Fig. 4). It has been reported that XOR can interact with butyrophilin1A1 (BTN1A1) by binding to the PRY/SPRY/B30.2 domain [79]. BTN1A1 and XOR are highly expressed in the lactating mammary gland and are secreted into milk associated with the milk fat globule membrane. BTN1A1 belongs to the B7-like T cell coregulatory molecule family, which is of increasing interest in cancer immunotherapy, as it may represent a novel subset of immune checkpoint regulators [80]. Consistently, BTN1A1 plays a vital role in the immune system, which has been described to inhibit T cell proliferation in vitro and reduce the expression of cytokines associated with T cell activation, such as IL-2 and IFN-γ [81, 82]. Thus, XOR may modulate the immune response by binding BTN1A1, which could affect cancer development and treatment.
Tumor-suppressive role of XOR
ROS
XOR-derived ROS could be antitumorigenic and reduce the growth of cancer [50, 83]. Excessive ROS production confers cytotoxic effects and leads to apoptosis in cancer cells [84]. XOR-derived ROS may trigger both apoptosis and necrosis in proliferating human lymphocytes [85]. XOR activation was found in prostate cancer cells after alternol treatment, which resulted in ROS accumulation and apoptotic cell death [86]. It has also been revealed that autophagy induced by cathepsin S induced XO-generated ROS, leading to oxidative DNA damage and subsequently cancer cell death [87]. XOR-derived ROS decreased the expression of COX-2 in breast cancer cells, which in turn significantly reduced the expression of MMP-1 and MMP-3 as well as cell migration [88].
ROS are an important component of the innate immune system. ROS derived from XOR participate in the innate immune response of phagocytotic cells, mediating the effect of phagocytotic killing of tumor cells [8]. ROS produced by XOR have also been implicated in the activation of the complement system, an important feature of the systemic innate immune system [89]. Complement activation could eradicate tumor cells through complement-dependent cytotoxicity [90].
Uric acid
Increasing studies have indicated that elevated serum uric acid (normal SUA: 3-6.8 mg/dl) was associated with low cancer risk and cancer mortality, such as prostate cancer, laryngeal squamous cell cancer, and breast cancer [91,92,93,94]. Uric acid is known to act as an important antioxidant. Uric acid donates a single electron to ROS and is converted to the relatively stable urate radical, which then decays to allantoin [95]. Uric acid also exhibits a secondary antioxidant effect by chelating iron [96]. The antioxidative activity of uric in the circulation may be involved in the prevention of cancer [97]. SLC2A9 acts as a uric acid transporter, which is a direct p53 targeted gene and a key downstream effector in the reduction of ROS by transporting uric acid as a source of antioxidants [98]. Consistently, overexpression of SLC2A9 inhibited the proliferation of HCC cells by reducing intracellular ROS levels [99]. Uric acid may also promote the immune response against tumor cells. Dendritic cells are activated by uric acid and enhance the T cell response to foreign antigens [100, 101]. It has been reported that uric acid enhanced the antitumor immunity of dendritic cell-based vaccines by increasing the accumulation of activated CD8+ T cells and increasing the production of IFN-γ [102].
Maintaining the balance of nucleotide metabolism
XOR plays a vital role in maintaining the balance of nucleotide metabolism. Decreased XOR activity in tumor cells leads to a shift in the purine anabolic-catabolic balance, which may confer selective advantages to malignant cells [25]. Inhibition of XOR has been found to promote reutilization of hypoxanthine and ATP production through the salvage pathway [103], which was necessary to provide purine nucleotides and maintain energy for tumor cells [104, 105].
Targeting XOR for tumor therapy
Effective treatment for cancer is considered a great unmet medical need. XOR has been shown to play oncogenic or suppressive roles in cancer, and the potential of targeting XOR in tumor therapy has been explored in recent years.
Inhibition of XOR
XOR inhibitors, including allopurinol, febuxostat, and topiroxostat, were initially approved for the treatment of gout and conditions associated with hyperuricemia [106] (Fig. 1). Allopurinol is the first approved XOR inhibitor and has been used worldwide since 1966 [15]. Allopurinol and its active metabolite oxypurinol are structural analogs of purines that inhibit the enzymatic activity of XOR by competing with substrates [107]. Febuxostat is a selective XOR inhibitor approved in 2009 for the treatment of hyperuricemia with improved tolerability compared to allopurinol [108]. Febuxostat noncompetitively blocks the activity of the two forms of XOR by binding to their active site [109]. Topiroxostat (FYX-051) is a hybrid-type XOR inhibitor exhibiting both structure- and mechanism-based inhibition [110] and was first approved in Japan in 2013 [111]. There is a continuous and intensive effort to develop new XOR inhibitors to improve the efficacy and safety profiles [112].
Pharmacological inhibition of XOR for the treatment of cancer cachexia
Cancer cachexia is a multifactorial metabolic syndrome characterized by weight loss, anorexia, muscle loss, and systemic inflammation [113]. Cancer cachexia not only impairs the quality of life of cancer patients but also reduces the efficacy and increases the toxicity of chemotherapy, thereby highlighting the need for more effective treatment [114]. Cancer cachexia is associated with muscle wasting and atrophy, while ROS play a key role in the development of muscle atrophy in response to inflammation related to cancer cachexia [17, 115]. Atrophic factors can activate XO and increase the generation of ROS. ROS trigger multiple processes, including mitochondrial dysfunction, the ubiquitin-proteasome system, and myonuclear apoptosis, which further promote skeletal muscle atrophy and wasting in cancer patients, finally leading to cancer cachexia [16]. In this regard, XO is an attractive target for cancer cachexia therapy.
Upregulated uric acid levels and an approximately 52-fold induction of XO-produced ROS were observed in the Yoshida hepatoma cancer cachexia model [18]. XO inhibition by allopurinol or oxypurinol improved survival and reduced wasting in this model [18] (Table 1). Compared to allopurinol, febuxostat also demonstrated significant efficacy in the same model with fewer adverse reactions, indicating a promising candidate for cancer cachexia treatment [116] (Table 1).
Pharmacological inhibition of XOR for the treatment of tumor lysis syndrome
Tumor lysis syndrome (TLS) is one of the most common complications related to cancer therapy and is a life-threatening condition caused by an abrupt release of intracellular metabolites after the lysis of tumor cells during chemotherapy [117, 118]. During TLS, the rapid accumulation of uric acid derived from the breakdown of nucleic acids may lead to kidney failure [119]. Accordingly, management of the level of serum uric acid is important to prevent TLS during chemotherapy. Since the production of uric acid is dependent on XOR, it has been validated as a therapeutic target for TLS. Allopurinol can be preferentially used in patients with low or intermediate risk for TLS [120]. Allopurinol has been applied to decrease uric acid levels in patients with lymphoma and acute lymphoblastic leukemia during chemotherapy, which could prevent TLS and reduce other toxic effects [121, 122] (Table 1). Febuxostat represents an attractive alternative to allopurinol in patients with renal insufficiency or hypersensitivity to allopurinol. The efficacy and safety of febuxostat have been proven in patients with hematological malignancies and solid tumors who received chemotherapy in clinical trials [123, 124] (Table 1).
Direct targeting cancer by inhibiting XOR
With the increasing understanding of the role of XOR in cancer development, inhibition of XOR has been explored to target cancer directly. Preclinical studies of XOR inhibitors for cancer treatment are summarized in Table 1. It has been shown that long-term allopurinol use decreased the risk of prostate cancer in patients with gout [125]. Consistently, allopurinol was found to drastically induce apoptosis in human hormone-refractory prostate cancer cells in combination with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [126]. Moreover, allopurinol alone was able to trigger apoptosis in allopurinol-sensitive NSCLC cells, while allopurinol combined with the JAK2 inhibitor CEP-33779 induced cell death in resistant cells [127]. Several studies have indicated that febuxostat possessed anticancer activity. Febuxostat showed cytotoxic effects in NSCLC A549 cells by inducing apoptosis mediated by caspase 3 [128]. Formulation of febuxostat by emulsomes strongly enhanced its cytotoxic activity in human colorectal carcinoma cells by improving its cellular penetration [129]. Inhibition of XOR by febuxostat mitigated breast cancer cell migration and pulmonary metastasis under hyperlipidemic conditions, which were associated with decreased ROS generation and MAPK phosphorylation [130]. These studies indicated the potential of XOR inhibitors in directly targeting cancer. However, the application of XOR inhibitors should be avoided in treating cancers originating from tissues with high expression of XOR due to the dual role of XOR in tumorigenesis. For example, it has been reported that pharmacological inhibition of XOR increased breast cancer tumor burden in a mouse ErbB2 breast cancer model [35].
Upregulation of XOR
Consistent with the antitumor role of XOR, it has been demonstrated that XOR overexpression increased the chemosensitivity of HCC cells. Compared with single-drug treatment, the combination of lentivirus-induced XOR and doxorubicin or 5-fluorouracil significantly inhibited tumor growth in nude mice [32]. Moreover, XOR was required for the antitumor activity of gemcitabine in a murine breast cancer model [131]. These findings suggest that induction of XOR expression or possibly activation in cancer may enhance the therapeutic efficacy of chemotherapy in cancers with reduced expression of XDH.
Conclusions and perspectives
Purines are the most abundant metabolic substrates and provide necessary components for DNA and RNA to support the rapid proliferation of cancer cells. Enhanced purine biosynthesis, including the complementary salvage pathway and de novo biosynthetic pathway, is tightly associated with the progression of cancer [132,133,134]. Recent studies proved that alterations in the activity and expression of metabolic enzymes involved in purine degradation, such as cytosolic 5’-nucleotidase II (cN-II), purine nucleoside phosphorylase (PNP), and adenosine deaminase (ADA), contributed to tumor progression [25, 135, 136]. As the rate-limiting enzyme in purine catabolism, the association between XOR and cancer has been increasingly recognized. Mounting evidence suggests that alterations in XOR expression and activity are often found in various types of cancers and are closely related to the prognosis of the disease. Increased or decreased XOR may be found in cancers dependent on the originated tissue types. XOR expression and activity are usually attenuated in HCC, breast cancer, and CRC but enhanced in lung cancer and brain tumors. ROS and uric acid, the major products during XOR-mediated catabolism, may exert protumoral or antitumor activity by various double-faced effects on the oxidative response, apoptosis, metabolism, and immune response, highlighting a dual role of XOR in the initiation and progression of human cancer. While XOR inhibitors are promising in the treatment of cancer cachexia and TLS, direct targeting of cancer by modulating XOR is much less studied. Nevertheless, the current findings shed light on the potential for further development of XOR modulators for the treatment of cancer.
To further exploit the role of XOR in cancer, a few challenges need to be addressed. First, the mechanism of XOR in the development of cancer remains elusive. Metabolomics will help to further elucidate the function of XOR-mediated purine metabolism and the enzymatic products in cancer. Second, the potential distinct role of the two forms of XOR (XDH or XO) in tumor-related pathways needs to be further clarified, which will help lay the foundation to discover isoform-selective modulators for precise therapy. Finally, although pharmacological inhibition of XOR has shown antitumor activity, the efficacy and safety of XOR inhibitors as a constituent of the therapeutic regimen warrants further investigation.
References
Bortolotti M, Polito L, Battelli MG, Bolognesi A. Xanthine oxidoreductase: one enzyme for multiple physiological tasks. Redox Biol. 2021;41:101882.
Schmidt HM, Kelley EE, Straub AC. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019;21:101072.
Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med. 2002;33:774–97.
Giulia BM, Andrea B, Letizia P. Pathophysiology of circulating xanthine oxidoreductase: New emerging roles for a multi-tasking enzyme. Biochim Biophys Acta. 2014;1842:1502–17.
Nishino T, Okamoto K, Eger BT, Pai EF, Nishino T. Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008;275:3278–89.
Kelley EE. A new paradigm for XOR-catalyzed reactive species generation in the endothelium. Pharmacol Rep. 2015;67:669–74.
McManaman JL, Palmer CA, Wright RM, Neville MC. Functional regulation of xanthine oxidoreductase expression and localization in the mouse mammary gland: evidence of a role in lipid secretion. J Physiol. 2002;545:567–79.
Ojha R, Singh J, Ojha A, Singh H, Sharma S, Nepali K. An updated patent review: xanthine oxidase inhibitors for the treatment of hyperuricemia and gout (2011-5). Expert Opin Ther Pat. 2017;27:311–45.
Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase in drug metabolism: beyond a role as a detoxifying enzyme. Curr Med Chem. 2016;23:4027–36.
Roberts LE, Fini MA, Derkash N, Wright RM. PD98059 enhanced insulin, cytokine, and growth factor activation of xanthine oxidoreductase in epithelial cells involves STAT3 and the glucocorticoid receptor. J Cell Biochem. 2007;101:1567–87.
Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606.
Kelley EE, Hock T, Khoo NK, Richardson GR, Johnson KK, Powell PC, et al. Moderate hypoxia induces xanthine oxidoreductase activity in arterial endothelial cells. Free Radic Biol Med. 2006;40:952–9.
Terada LS, Piermattei D, Shibao GN, McManaman JL, Wright RM. Hypoxia regulates xanthine dehydrogenase activity at pre- and posttranslational levels. Arch Biochem. Biophysics. 1997;348:163–8.
Seymour KJ, Roberts LE, Fini MA, Parmley LA, Oustitch TL, Wright RM. Stress activation of mammary epithelial cell xanthine oxidoreductase is mediated by p38 MAPK and CCAAT/enhancer-binding protein-beta. J Biol Chem. 2006;281:8545–58.
Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114.
Abrigo J, Elorza AA, Riedel CA, Vilos C, Simon F, Cabrera D, et al. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid Med Cell Longev. 2018;2018:2063179.
Assi M, Rebillard A. The Janus-faced role of antioxidants in cancer cachexia: new insights on the established concepts. Oxid Med Cell Longev. 2016;2016:9579868.
Springer J, Tschirner A, Hartman K, Palus S, Wirth EK, Ruis SB, et al. Inhibition of xanthine oxidase reduces wasting and improves outcome in a rat model of cancer cachexia. Int J Cancer. 2012;131:2187–96.
Battelli MG, Bortolotti M, Polito L, Bolognesi A. Metabolic syndrome and cancer risk: the role of xanthine oxidoreductase. Redox Biol. 2019;21:101070.
Battelli MG, Bortolotti M, Polito L, Bolognesi A. The role of xanthine oxidoreductase and uric acid in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2557–65.
Agarwal A, Banerjee A, Banerjee UC. Xanthine oxidoreductase: a journey from purine metabolism to cardiovascular excitation-contraction coupling. Crit Rev Biotechnol. 2011;31:264–80.
Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med. 2016;5:546–57.
Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase-derived reactive species: physiological and pathological effects. Oxid Med Cell Longev. 2016;2016:3527579.
Fini MA, Elias A, Johnson RJ, Wright RM. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin and Transl Med. 2012;1:16.
Garcia-Gil M, Camici M, Allegrini S, Pesi R, Petrotto E, Tozzi MG. Emerging role of purine metabolizing enzymes in brain function and tumors. Int J Mol Sci. 2018;19:3598.
You L, Fan Y, Liu X, Shao S, Guo L, Noreldeen HAA, et al. Liquid chromatography-mass spectrometry-based tissue metabolic profiling reveals major metabolic pathway alterations and potential biomarkers of lung cancer. J Proteome Res. 2020;19:3750–60.
Linder N, Haglund C, Lundin M, Nordling S, Ristimaki A, Kokkola A, et al. Decreased xanthine oxidoreductase is a predictor of poor prognosis in early-stage gastric cancer. J Clin Pathol. 2006;59:965–71.
Linder N, Martelin E, Lundin M, Louhimo J, Nordling S, Haglund C, et al. Xanthine oxidoreductase - clinical significance in colorectal cancer and in vitro expression of the protein in human colon cancer cells. Eur J Cancer. 2009;45:648–55.
Nina Linder JL, Jorma I, Mikael L, Kari O. Raivio, Heikki J. Down-regulated xanthine oxidoreductase is a feature of aggressive breast cancer. Clin Cancer Res. 2005;11:4372–81.
Konno H, Minamiya Y, Saito H, Imai K, Kawaharada Y, Motoyama S, et al. Acquired xanthine dehydrogenase expression shortens survival in patients with resected adenocarcinoma of lung. Tumour Biol. 2012;33:1727–32.
Stirpe F, Ravaioli M, Battelli MG, Musiani S, Grazi GL. Xanthine oxidoreductase activity in human liver disease. Am J Gastroenterol. 2002;97:2079–85.
Sun Q, Zhang Z, Lu Y, Liu Q, Xu X, Xu J, et al. Loss of xanthine oxidoreductase potentiates propagation of hepatocellular carcinoma stem cells. Hepatology. 2020;71:2033–49.
Chen GL, Ye T, Chen HL, Zhao ZY, Tang WQ, Wang LS, et al. Xanthine dehydrogenase downregulation promotes TGFbeta signaling and cancer stem cell-related gene expression in hepatocellular carcinoma. Oncogenesis. 2017;6:e382.
Cook W, Chu R, Saksela M, Raivio K, Yeldandi A. Differential immunohistochemical localization of xanthine oxidase in normal and neoplastic human breast epithelium. Int J Oncol. 1997;11:1013–7.
Fini MA, Monks J, Farabaugh SM, Wright RM. Contribution of xanthine oxidoreductase to mammary epithelial and breast cancer cell differentiation in part modulates inhibitor of differentiation-1. Mol Cancer Res. 2011;9:1242–54.
Shan L, He M, Yu M, Qiu C, Lee NH, Liu ET, et al. cDNA microarray profiling of rat mammary gland carcinomas induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and 7,12-dimethylbenz[a]anthracene. Carcinogenesis. 2002;23:1561–8.
Miao Y, Li Q, Wang J, Quan W, Li C, Yang Y, et al. Prognostic implications of metabolism-associated gene signatures in colorectal cancer. PeerJ. 2020;8:e9847.
Veljkovic A, Hadzi-Dokic J, Sokolovic D, Basic D, Velickovic-Jankovic L, Stojanovic M, et al. Xanthine oxidase/dehydrogenase activity as a source of oxidative stress in prostate cancer tissue. Diagnostics (Basel). 2020;10:668.
Durak İ, Işik CÜ, Canbolat O, Akyol Ö, Kavutçu M. Adenosine deaminase, 5′ nucleotidase, xanthine oxidase, superoxide dismutase, and catalase activities in cancerous and noncancerous human laryngeal tissues. Free Radic Biol Med. 1993;15:681–4.
Metwally NS, Ali SA, Mohamed AM, Khaled HM, Ahmed SA. Levels of certain tumor markers as differential factors between bilharzial and non-biharzial bladder cancer among Egyptian patients. Cancer Cell Int. 2011;11:8.
Kaynar H, Meral M, Turhan H, Keles M, Celik G, Akcay F. Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu-Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer Lett. 2005;227:133–9.
Tsao SM, Yin MC, Liu WH. Oxidant stress and B vitamins status in patients with non-small cell lung cancer. Nutr Cancer. 2007;59:8–13.
Wikoff WR, Grapov D, Fahrmann JF, DeFelice B, Rom WN, Pass HI, et al. Metabolomic markers of altered nucleotide metabolism in early stage adenocarcinoma. Cancer Prev Res (Philos). 2015;8:410–8.
Moreno P, Jimenez-Jimenez C, Garrido-Rodriguez M, Calderon-Santiago M, Molina S, Lara-Chica M, et al. Metabolomic profiling of human lung tumor tissues - nucleotide metabolism as a candidate for therapeutic interventions and biomarkers. Mol Oncol. 2018;12:1778–96.
Hu W, Wang G, Yarmus LB, Wan Y. Combined methylome and transcriptome analyses reveals potential therapeutic targets for EGFR wild type lung cancers with low PD-L1 expression. Cancers (Basel). 2020;12:2496.
Kijkog WE, BeIce A, Ozyyurt E, Tepeler Z. Xanthine oxidase levels in human brain tumors. Cancer Letters. 1990;50:179–81.
Rajaraman P, Brenner AV, Neta G, Pfeiffer R, Wang SS, Yeager M, et al. Risk of meningioma and common variation in genes related to innate immunity. Cancer Epidemiol Biomark Prev. 2010;19:1356–61.
Schulten H-J, Hussein D, Al-Adwani F, Karim S, Al-Maghrabi J, Al-Sharif M. Microarray expression profiling identifies genes, including cytokines, and biofunctions, as diapedesis, associated with a brain metastasis from a papillary thyroid carcinoma. Am J Cancer Res. 2016;6:2140–61.
Uzu M, Nonaka M, Miyano K, Sato H, Kurebayashi N, Yanagihara K, et al. A novel strategy for treatment of cancer cachexia targeting xanthine oxidase in the brain. J Pharmacol Sci. 2019;140:109–12.
Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64.
Dumitrescu RG, Shields PG. The etiology of alcohol-induced breast cancer. Alcohol. 2005;35:213–25.
Wright RM, Mcmanaman JL, Repine JE. Alcohol-induced breast cancer:a proposed mechanism. Free Radic Biol Med. 1999;26:348–54.
Bir SC, Kolluru GK, Fang K, Kevil CG. Redox balance dynamically regulates vascular growth and remodeling. Semin Cell Dev Biol. 2012;23:745–57.
Liu J, Wang C, Liu F, Lu Y, Cheng J. Metabonomics revealed xanthine oxidase-induced oxidative stress and inflammation in the pathogenesis of diabetic nephropathy. Anal Bioanal Chem. 2015;407:2569–79.
Lugrin J, Rosenblatt-Velin N, Parapanov R, Liaudet L. The role of oxidative stress during inflammatory processes. Biol Chem. 2014;395:203–30.
Nanduri J, Vaddi DR, Khan SA, Wang N, Makarenko V, Semenza GL, et al. HIF-1alpha activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS One. 2015;10:e0119762.
Balamurugan K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer. 2016;138:1058–66.
Griguer CE, Oliva CR, Kelley EE, Giles GI, Lancaster JR Jr, Gillespie GY. Xanthine oxidase-dependent regulation of hypoxia-inducible factor in cancer cells. Cancer Res. 2006;66:2257–63.
Shi DY, Xie FZ, Zhai C, Stern JS, Liu Y, Liu SL. The role of cellular oxidative stress in regulating glycolysis energy metabolism in hepatoma cells. Mol Cancer. 2009;8:32.
Kusano T, Ehirchiou D, Matsumura T, Chobaz V, Nasi S, Castelblanco M, et al. Targeted knock-in mice expressing the oxidase-fixed form of xanthine oxidoreductase favor tumor growth. Nat Commun. 2019;10:4904.
Ives A, Nomura J, Martinon F, Roger T, LeRoy D, Miner JN, et al. Xanthine oxidoreductase regulates macrophage IL1beta secretion upon NLRP3 inflammasome activation. Nat Commun. 2015;6:6555.
Yim K, Bindayi A, McKay R, Mehrazin R, Raheem OA, Field C, et al. Rising serum uric acid level is negatively associated with survival in renal cell carcinoma. Cancers (Basel). 2019;11:536.
Yang S, He X, Liu Y, Ding X, Jiang H, Tan Y, et al. Prognostic significance of serum uric acid and gamma-glutamyltransferase in patients with advanced gastric cancer. Dis Markers. 2019;2019:1415421.
Chen YF, Li Q, Chen DT, Pan JH, Chen YH, Wen ZS, et al. Prognostic value of pre-operative serum uric acid levels in esophageal squamous cell carcinoma patients who undergo R0 esophagectomy. Cancer Biomark. 2016;17:89–96.
Stotz M, Szkandera J, Seidel J, Stojakovic T, Samonigg H, Reitz D, et al. Evaluation of uric acid as a prognostic blood-based marker in a large cohort of pancreatic cancer patients. PLoS One. 2014;9:e104730.
Mao L, Guo C, Zheng S. Elevated urinary 8-oxo-7,8-dihydro-2’-deoxyguanosine and serum uric acid are associated with progression and are prognostic factors of colorectal cancer. Onco Targets Ther. 2018;11:5895–902.
Chen CJ, Yen JH, Chang SJ. Gout patients have an increased risk of developing most cancers, especially urological cancers. Scand J Rheumatol. 2014;43:385–90.
Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol. 2007;293:C584–96.
Yu M-A, Sánchez-Lozada LG, Johnson RJ, Kang D-H. Oxidative stress with an activation of the renin–angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hyperts. 2010;28:1234–42.
So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120:1791–9.
Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep. 2017;7:39884.
Nakagawa T, Lanaspa MA, Millan IS, Fini M, Rivard CJ, Sanchez-Lozada LG, et al. Fructose contributes to the Warburg effect for cancer growth. Cancer Metab. 2020;8:16.
Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–15.
Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem. 2012;287:40732–44.
Rock KL, Kataoka H, Lai JJ. Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol. 2013;9:13–23.
Shi Y, Mucsi AD, Ng G. Monosodium urate crystals in inflammation and immunity. Immunol Rev. 2010;233:203–17.
Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta-a friend or foe in malignancies? Int J Mol Sci. 2018;19:2155.
Rébé C, Ghiringhelli F. Interleukin-1β and cancer. Cancers. 2020;12:1791.
Jeong J, Rao AU, Xu J, Ogg SL, Hathout Y, Fenselau C, et al. The PRY/SPRY/B30.2 Domain of Butyrophilin 1A1 (BTN1A1) Binds to Xanthine Oxidoreductase: IMPLICATIONS FOR THE FUNCTION OF BTN1A1 IN THE MAMMARY GLAND AND OTHER TISSUES*. J Biol Chem. 2009;284:22444–56.
Arnett HA, Viney JL. Immune modulation by butyrophilins. Nat Rev Immunol. 2014;14:559–69.
Smith IA, Knezevic BR, Ammann JU, Rhodes DA, Aw D, Palmer DB, et al. BTN1A1, the mammary gland butyrophilin, and BTN2A2 are both inhibitors of T cell activation. J Immunol. 2010;184:3514–25.
LaRocca J, Pietruska J, Hixon M. Akt1 is essential for postnatal mammary gland development, function, and the expression of Btn1a1. PLoS One. 2011;6:e24432.
Haddow A, Lamirande GDE, Bergel F, Bray RC, Gilbert DA. Anti-tumour and biochemical effects of purified bovine xanthine oxidase in C3H and C mice. Nature. 1958;182:1144–6.
Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, et al. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed Pharmacother. 2015;74:101–10.
Battelli MG, Musiani S, Tazzari PL, Stirpe F. Oxidative stress to human lymphocytes by xanthine oxidoreductase activity. Free Radic Res. 2001;35:665–79.
Xu H, Li C, Mozziconacci O, Zhu R, Xu Y, Tang Y, et al. Xanthine oxidase-mediated oxidative stress promotes cancer cell-specific apoptosis. Free Radic Biol Med. 2019;139:70–9.
Huang CC, Chen KL, Cheung CHA, Chang JY. Autophagy induced by cathepsin S inhibition induces early ROS production, oxidative DNA damage, and cell death via xanthine oxidase. Free Radic Biol Med. 2013;65:1473–86.
Fini MA, Orchard-Webb D, Kosmider B, Amon JD, Kelland R, Shibao G, et al. Migratory activity of human breast cancer cells is modulated by differential expression of xanthine oxidoreductase. J Cell Biochem. 2008;105:1008–26.
Tanhehco EJ, Yasojima K, Fau - McGeer PL, McGeer Pl, Fau - Washington RA, et al. Free radicals upregulate complement expression in rabbit isolated heart. Am J Physiol Heart Circ Physiol. 2000;279:H195–H201.
Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest. 2017;127:780–9.
Taghizadeh N, Vonk JM, Boezen HM. Serum uric acid levels and cancer mortality risk among males in a large general population-based cohort study. Cancer Causes Control. 2014;25:1075–80.
Kuhn T, Sookthai D, Graf ME, Schubel R, Freisling H, Johnson T, et al. Albumin, bilirubin, uric acid and cancer risk: results from a prospective population-based study. Br J Cancer. 2017;117:1572–9.
Benli E, Cirakoglu A, Ayyildiz SN, Yuce A. Comparison of serum uric acid levels between prostate cancer patients and a control group. Cent Eur J Urol. 2018;71:242–7.
Hsueh CY, Shao M, Cao W, Li S, Zhou L. Pretreatment serum uric acid as an efficient predictor of prognosis in men with laryngeal squamous cell cancer: a retrospective cohort study. Oxid Med Cell Longev. 2019;2019:1821969.
Yu R, Schellhorn HE. Recent applications of engineered animal antioxidant deficiency models in human nutrition and chronic disease. The J Nutr. 2013;143:1–11.
Barros MP, Ganini D, Lorenço-Lima L, Soares CO, Pereira B, Bechara EJH, et al. Effects of acute creatine supplementation on iron homeostasis and uric acid-based antioxidant capacity of plasma after wingate test. J Int Soc Sports Nutr. 2012;9:25.
Ames BN, Cathcart R, Schwiers E, Hochsteint P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci USA. 1981;78:6858–62.
Itahana Y, Han R, Barbier S, Lei Z, Rozen S, Itahana K. The uric acid transporter SLC2A9 is a direct target gene of the tumor suppressor p53 contributing to antioxidant defense. Oncogene. 2014;34:1799–810.
Han X, Yang J, Li D, Guo Z. Overexpression of uric acid transporter SLC2A9 inhibits proliferation of hepatocellular carcinoma cells. Oncol Res. 2019;27:533–40.
Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–21.
Jerome KR, Corey L. The danger within. N Engl J Med. 2004;350:411–2.
Wang Y, Ma X, Su C, Peng B, Du J, Jia H, et al. Uric acid enhances the antitumor immunity of dendritic cell-based vaccine. Sci Rep. 2015;5:16427.
Furuhashi M. New insights into purine metabolism in metabolic diseases: role of xanthine oxidoreductase activity. Am J Physiol Endocrinol Metab. 2020;319:E827–E34.
Yin J, Ren W, Huang X, Deng J, Li T, Yin Y. Potential mechanisms connecting purine metabolism and cancer therapy. Front Immunol. 2018;9:1697.
Townsend MH, Robison RA, O’Neill KL. A review of HPRT and its emerging role in cancer. Med Oncol. 2018;35:89.
Vickneson K, George J. Xanthine oxidoreductase inhibitors. Handb Exp Pharmacol. 2021;264:205–28.
Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM. Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin Pharmacokinet. 2007;46:623–44.
Bardin T, Richette P. The role of febuxostat in gout. Curr Opin Rheumatol. 2019;31:152–8.
Frampton JE. Febuxostat: a review of its use in the treatment of hyperuricaemia in patients with gout. Drugs. 2015;75:427–38.
Matsumoto K, Okamoto K, Ashizawa N, Nishino T. FYX-051: a novel and potent hybrid-type inhibitor of xanthine oxidoreductase. J Pharmacol Exp Ther. 2011;336:95–103.
Luo Z, Yu G, Han X, Yang T, Ji Y, Huang H, et al. Prediction of the pharmacokinetics and pharmacodynamics of topiroxostat in humans by integrating the physiologically based pharmacokinetic model with the drug-target residence time model. Biomed Pharmacother. 2020;121:109660.
Smelcerovic A, Tomovic K, Smelcerovic Z, Petronijevic Z, Kocic G, Tomasic T, et al. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. Eur J Med Chem. 2017;135:491–516.
Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Prim. 2018;4:17105.
Schmidt SF, Rohm M, Herzig S, Berriel, Diaz M. Cancer cachexia: more than skeletal muscle wasting. Trends Cancer. 2018;4:849–60.
Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. 2014;14:754–62.
Konishi M, Pelgrim L, Tschirner A, Baumgarten A, von Haehling S, Palus S, et al. Febuxostat improves outcome in a rat model of cancer cachexia. J Cachexia Sarcopenia Muscle. 2015;6:174–80.
Alakel N, Middeke JM, Schetelig J, Bornhauser M. Prevention and treatment of tumor lysis syndrome, and the efficacy and role of rasburicase. Onco Targets Ther. 2017;10:597–605.
Nicholaou T, Wong R, Davis ID. Tumour lysis syndrome in a patient with renal-cell carcinoma treated with sunitinib malate. Lancet. 2007;369:1923–4.
Wilson FP, Berns JS. Tumor lysis syndrome: new challenges and recent advances. Adv Chronic Kidney Dis. 2014;21:18–26.
Sood AR, Burry LD. FCCP, Cheng DoretKF. Clarifying the role of Rasburicase in tumor lysis syndrome. Pharmacotherapy. 2007;27:111–21.
Stuckert AJ, Schafer ES, Bernhardt MB, Baxter P, Brackett J. Use of allopurinol to reduce hepatotoxicity from 6-mercaptopurine (6-MP) in patients with acute lymphoblastic leukemia (ALL). Leuk Lymphoma. 2020;61:1246–9.
Cohen G, Cooper S, Sison EA, Annesley C, Bhuiyan M, Brown P. Allopurinol use during pediatric acute lymphoblastic leukemia maintenance therapy safely corrects skewed 6-mercaptopurine metabolism, improving inadequate myelosuppression and reducing gastrointestinal toxicity. Pediatr Blood Cancer. 2020;67:e28360.
Tamura K, Kawai Y, Kiguchi T, Okamoto M, Kaneko M, Maemondo M, et al. Efficacy and safety of febuxostat for prevention of tumor lysis syndrome in patients with malignant tumors receiving chemotherapy: a phase III, randomized, multi-center trial comparing febuxostat and allopurinol. Int J Clin Oncol. 2016;21:996–1003.
Takai M, Yamauchi T, Ookura M, Matsuda Y, Tai K, Kishi S, et al. Febuxostat for management of tumor lysis syndrome including its effects on levels of purine metabolites in patients with hematological malignancies - a single institutio’s, pharmacokinetic and pilot prospective study. Anticancer Res. 2014;34:7287.
Shih HJ, Kao MC, Tsai PS, Fan YC, Huang CJ. Long-term allopurinol use decreases the risk of prostate cancer in patients with gout: a population-based study. Prostate Cancer Prostatic Dis. 2017;20:328–33.
Yasuda T, Yoshida T, Goda AE, Horinaka M, Yano K, Shiraishi T, et al. Anti-gout agent allopurinol exerts cytotoxicity to human hormone-refractory prostate cancer cells in combination with tumor necrosis factor-related apoptosis-inducing ligand. Mol Cancer Res. 2008;6:1852–60.
Tavassoly I, Hu Y, Zhao S, Mariottini C, Boran A, Chen Y, et al. Genomic signatures defining responsiveness to allopurinol and combination therapy for lung cancer identified by systems therapeutics analyses. Mol Oncol. 2019;13:1725–43.
Alfaifi MY, Shati AA, Elbehairi SEI, Fahmy UA, Alhakamy NA, Md S. Anti-tumor effect of PEG-coated PLGA nanoparticles of febuxostat on A549 non-small cell lung cancer cells. 3 Biotech. 2020;10:133.
Fahmy UA, Aldawsari HM, Badr-Eldin SM, Ahmed OAA, Alhakamy NA, Alsulimani HH, et al. The encapsulation of febuxostat into emulsomes strongly enhances the cytotoxic potential of the drug on HCT 116 colon cancer cells. Pharmaceutics. 2020;12:956.
Oh SH, Choi SY, Choi HJ, Ryu HM, Kim YJ, Jung HY, et al. The emerging role of xanthine oxidase inhibition for suppression of breast cancer cell migration and metastasis associated with hypercholesterolemia. FASEB J. 2019;33:7301–14.
Xu X, Rao G, Li Y. Xanthine oxidoreductase is required for genotoxic stressinduced NKG2D ligand expression and gemcitabine-mediated antitumor activity. Oncotarget. 2016;7:59220–35.
Zhou W, Yao Y, Scott AJ, Wilder-Romans K, Dresser JJ, Werner CK, et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun. 2020;11:3811.
Ma F, Zhu Y, Liu X, Zhou Q, Hong X, Qu C, et al. Dual-specificity tyrosine phosphorylation-regulated kinase 3 loss activates purine metabolism and promotes hepatocellular carcinoma progression. Hepatology. 2019;70:1785–803.
Zhou Q, Lin M, Feng X, Ma F, Zhu Y, Liu X, et al. Targeting CLK3 inhibits the progression of cholangiocarcinoma by reprogramming nucleotide metabolism. J Exp Med. 2020;217:e20191779.
Lars Petter J, Laurent C. Therapeutic perspectives for cN-II in cancer. Curr Med Chem. 2013;20:4292–303.
Furman RR, Hoelzer D. Purine nucleoside phosphorylase inhibition as a novel therapeutic approach for B-cell lymphoid malignancies. Semin Oncol. 2007;34:S29–34.
Acknowledgements
This work was supported by National Natural Science Foundation of China [81773760 & 81973345].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, Mm., Meng, Lh. The double faced role of xanthine oxidoreductase in cancer. Acta Pharmacol Sin 43, 1623–1632 (2022). https://doi.org/10.1038/s41401-021-00800-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00800-7
