
Abstract
Paeoniflorin (PF), a bioactive monoterpene glucoside, has shown a variety of pharmacological effects such as anti-inflammation and autophagy modulation etc. In this study, we investigated whether and how PF exerted a protective effect against ischemic brain injury in vivo and in vitro. Primary rat cortical neurons underwent oxygen/glucose deprivation/reperfusion (OGD/R) for 90 min. We showed that after OGD/R, a short fragment of histone deacetylase 4 (HDAC4) produced by caspase3-mediated degradation was markedly accumulated in the nucleus and the activity of caspase3 was increased. Treatment with PF (100 nM, 1 μM) significantly improved the viability of cortical neurons after OGD/R. Furthermore, PF treatment could maintain HDAC4 intrinsic subcellular localization and reduce the caspase3 activity without changing the HDAC4 at the transcriptional level. PF treatment significantly reduced OGD/R-caused inhibition of transcriptional factor MEF2 expression and increased the expression of downstream proteins such as GDNF, BDNF, and Bcl-xl, thus exerting a great anti-apoptosis effect as revealed by TUNEL staining. The beneficial effects of PF were almost canceled in HDAC4 (D289E)-transfected PC12 cells after OGD/R. In addition, PF treatment reduced the caspase9 activity, rescued the release of cytochrome c from mitochondria, and maintained the integrity of mitochondria membrane. We conducted in vivo experiments in 90-min-middle cerebral artery occlusion (MCAO) rat model. The rats were administered PF (20, 40 mg/kg, ip, 3 times at the reperfusion, 24 h and 48 h after the surgery). We showed that PF administration dose-dependently reduced infarction area, improved neurological symptoms, and maintained HDAC4 localization in rats after MCAO. These results demonstrate that PF is effective in protecting against ischemic brain injury and inhibit apoptosis through inhibiting the cytochrome c/caspase3/HDAC4 pathway.
Similar content being viewed by others


Introduction
Stroke is the third most common cause of death in most Western countries, and it has been the leading cause of death in China in recent years [1]. A large majority of strokes are ischemic strokes, which are induced by the interruption of blood supply [2]. In the focal ischemic core, neurons are unable to maintain basic function and homeostasis because of deprivation of oxygen and glucose after cell membrane depolarization, oxidative stress, and excitotoxicity [3]. In the penumbra, neurons undergo delayed apoptosis, which is becoming a potential therapeutic target. Current therapeutic interventions aim to achieve thrombolysis, anti-inflammation and neuroprotection. Tissue plasminogen activator (tPA) is an effective and preferred clinical treatment for ischemic stroke patients and is also the only medical treatment approved by the United States Food and Drug Administration for ischemic stroke [3]. However, the application of tPA is limited by its short therapeutic window of 0–4.5 h [4]. Edaravone is an antioxidant drug targeting free radicals that was approved in Japan to treat acute cerebral infarction in 2015 [5]; however because the pathogenesis of ischemic stroke is very complex, a significant therapeutic effect cannot be achieved by targeting oxidative stress alone. Therefore, holistic neuroprotective strategies may be new methods for decreasing the degree of cerebral infarction ischemia injury and promoting functional recovery.
Paeoniflorin (PF), extracted from a traditional Chinese herb named Paeonia lactiflora, is a monoterpene glycoside [6]. Numerous experiments have found that PF has marked anti-inflammatory, antioxidant, and neuroprotective effects. In the cardiovascular system, PF can alleviate myocardial damage through protecting against apoptosis by regulating Bax/Bcl-2 expression [7] and lower serum total cholesterol levels in hyperholesterolemic rats [8]. PF administration can alleviate chronic pain by reducing the response of astrocytes, decreasing the phosphorylation of p38 [9], and reducing arthritis-induced injury via the PI3K-Akt-mTOR pathway [10]. To ameliorate intestinal ischemic injury, PF pretreatment can activate the LKB1/AMPK signaling pathway to improve ischemia-induced impairment of autophagic flux [11]. An increasing number of studies have consistently emphasized how epigenetic modification affects the pathological process of stroke [12, 13]. HDAC4, an important member of the class IIa family, has been reported to be a mediator of many crucial physiological and pathological processes [14]. A previous study showed that stroke can induce the nuclear shuttling of HDAC4 between neurons across cortical layers in neurons in the peri-infarct cortex, but that stroke does not increase nuclear HDAC5 levels in neurons [15]. However, the role of this unique shuttling in the process of ischemic stroke is still unknown.
The aim of this paper was to determine whether and how PF can regulate HDAC4 to exert neuroprotective effects in the process of ischemic stroke. First, we found that PF could exert apparent protective effects in vitro. Then, we found that after oxygen/glucose deprivation, PF could influence HDAC4 subcellular localization driven by caspase3-induced degradation, decrease the binding of HDAC4 and MEF2A, reverse the expression of many downstream proteins, including anti-inflammatory proteins and trophic factors, and inhibit the activity of caspase3. Furthermore, we found that PF blocked the release of cytochrome c and maintained the integrity of the mitochondrial membrane, which resulted in inhibition of caspase3 activation and intrinsic apoptosis. We found that PF could maintain HDAC4 subcellular localization in vivo and that the infarct volume was reduced in PF-treated MCAO model rats compared with the vehicle-treated MCAO model rats. Overall, these findings demonstrated that PF exerted a strong anti-apoptotic and neuroprotective effect through the cytochrome c/caspase3/HDAC4 pathway in the process of ischemic stroke.
Materials and methods
Animals
Adult male Sprague-Dawley rats weighing 250–70 g were purchased from SLAC Laboratory Animals Co., Ltd (Shanghai, China) and used in this study. The rats were randomly assigned to groups and housed in a controlled environment (12 h light/12 h dark cycle, temperature of 20–24 °C). The experiments were reviewed and approved by the Institutional Animal Care and Use Committee of Shanghai Institute of Material Medica. All experimental procedures were performed in accordance with the guidelines of the Association of Assessment and Accreditation of Laboratory Animal Care International.
Models of MCAO
All animals were acclimatized for one week before surgery. Transient focal cerebral ischemia was induced as previously described [16]. The animals were anesthetized with 4% isoflurane in 66%:30% N2O/O2 and then a nose cone that delivered 2% isoflurane was used for anesthesia maintenance (RWD Life Science, Shenzhen, China). A 4–0 monofilament nylon surgical suture (Sunbio Biotech Co. Ltd, Beijing, China) with a rounded tip was introduced through the external carotid artery into the internal carotid artery to block the middle cerebral artery. After 90 min of occlusion, the suture was withdrawn to induce reperfusion. Cerebral blood flow (CBF) was monitored to ensure that CBF was reduced by at least 75% by using laser Doppler (moorVMS-LDF2, Axminster, UK). Core body temperature was maintained at 37 ± 0.5 °C with a feedback-controlled heat pad (Hugo Sachs Elektronik, March-Hugstetten, Germany). The animals were transferred to their cages after they awoke from anesthesia. The sham group underwent the same surgical procedures without insertion of a suture.
Experimental design
The rats were randomly divided (n = 10–13) into five groups. All groups were given saline or compounds by intraperitoneal injection based on weight three times, i.e., at the time of reperfusion and 24 and 48 h after surgery. The sham group and model group were given saline. PF and edaravone were freshly prepared separately and dissolved in normal saline. The PF-treated animals were divided into two groups: the 20 mg/kg PF group and 40 mg/kg PF group. The dose of edaravone administered to the edaravone group was 6 mg/kg. The animals were sacrificed 7 days or 14 days after MCAO. PF was prepared by a collaborator’s research group (Prof Yang Ye’s research group) [17] and dissolved in saline. Edaravone was purchased from a commercial source (Selleck, China). All behavioral experiments were executed as shown in Supplementary Fig. S4a.
Infarct volume quantification
We used TTC (T8877, Sigma-Aldrich) staining to measure the cerebral infarct volume. Rat brains were frozen for 10 min and sectioned into 2-mm-thick coronal sections on the 7th day after MCAO. The brain sections were stained with 1% TTC at 37 °C for 6 min and then fixed in 4% paraformaldehyde (PFA). All sections were imaged and then analyzed using Image-Pro Plus 7.0 (Media Cybernetics, Silver Spring, MD, USA). We calculated the infarct volume (%) with the formula: the volume of the contralateral hemisphere/(the volume of the total brain/2) × 100%. All the investigators were blinded to the treatment groups.
Neurological function scores
Neurological deficit scores were assessed according to a classical scoring system with few modifications [2]. The animals were scored as follows: 0, no observable deficit; 1, unable to fully stretch the right forelimb; 2, circling to the right side but normal posture at rest; 3, unilateral circling to the right side; 4, occasionally leaning to the right side and declined locomotor activity; and 5, unable to sustain normal posture at rest. Neurological deficits were assessed every day, and all investigators were blinded to the treatment groups.
Animal behavior test
Rotarod test
The rats were trained on the rotarod, which gradually accelerated from 4 to 40 rpm over 5 min, 3 days before ischemic stroke induction and tested on the fourth and seventh days after surgery (IITC Life Science, Woodland Hills, CA, USA) [2]. The latency for the rats to fall was recorded. Each rat underwent three trials each day with at least a 30 min interval between trials for three consecutive days. The mean latency on the last day of training was recorded as the baseline control. The mean latency over 3 trails with 30 min intervals was determined on the testing day. All investigators were blinded to the treatment groups.
Open-field test
The rats were placed in the center of a plastic open-field arena in a quiet environment for 7 min to test locomotor behavior on the 5th day and the 12th day after MCAO. The distance traveled, active time, rearing time (time spent making vertical movements), and number of vertical movements were analyzed (Jiliang Software Technology Company, Shanghai, China) in the last 5 min. All the investigators were blinded to the treatment groups.
Morris water maze test
We used the Morris water maze to test the learning and memory abilities of rats after MCAO. The test was performed as described previously [2, 18,19,20]. The water maze was 120 cm in diameter and 32 cm in depth. The water maze was divided into four equal-sized quadrants. In one of the quadrants, there was a 10-cm diameter platform that was 2 cm below the surface of the water, which was opaque. The temperature was maintained at ~22 °C. Above the top of the maze, we installed a camera connected to an analysis system (Jiliang Software Technology Company, Shanghai, China) to track the swimming paths of the rats. The Morris water maze test was performed for 4 days beginning on the 11th day. Each rat underwent three trials per day with at least a 30 min interval between trials for 3 consecutive days, and each trial lasted until the rat found the platform. The rats were released from locations in the three quadrants that did not hold the platform. Each rat was placed into the maze facing the tank wall at the appropriate location and allowed to swim for 90 s to find the platform. The order of three starting locations was random. In the training phase, the latency to find and climb on the platform was observed and recorded if the rat could find the platform. If the rat could not find the platform within 90 s, we guided it to the platform and allowed it to stay on the platform for 10 s. In the test phase, which was performed on the 14th day after MCAO, the platform was removed. The rat was placed into the water opposite the previous location of the platform, where the distance to the previous location of the platform was the longest.
The amount of time that the rats tried to find the invisible platform within 90 s was analyzed. All the investigators were blinded to the treatment groups.
Nissl staining
We used a Nissl staining kit (Beyotime Biotechnology, Shanghai, China) to assess the integrity of pyramidal neurons in the hippocampal CA1 region on day 14 after MCAO following the manufacturer’s protocol. The brain sections were immersed in Nissl staining solution for 10 min at 37 °C. Then, the sections were washed in distilled water twice followed by 95% ethyl alcohol and xylene. We observed the stained brain sections under a microscope (Leica Camera, Germany).
Plasmid construction
An HDAC4 (D289E) plasmid, which contained a point mutation in which Asp-289 was replaced by Glu-289, was generated by polymerase chain reaction (PCR) with a full-length human HDAC4 plasmid as the template, ligated into the expression vector HA-pcDNA3.0 and sequenced. A full-length human HDAC4 plasmid was used in an earlier study by our laboratory [14].
Cell culture and OGD/R
Primary cortical neurons were prepared from embryonic Sprague-Dawley rats on E17 and cultured in neurobasal medium with B27 supplement (Thermo Fisher Scientific, USA) and L-glutamine as previously described [2]. The neurons were cultured at 37 °C in 5% CO2 atmosphere. The medium was changed every 2 days. On the 7th day, the neurons were used for oxygen–glucose deprivation/reperfusion (OGD/R) experiments. The normal medium was replaced with glucose-free Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific, USA), and the cells were incubated in a chamber containing 95% N2/5% CO2/0% O2 (Whitley H35 Hypoxystation, Don Whitley Scientific). The neurons were placed in the experimental hypoxia chamber for 90 min, removed, and then cultured under normal conditions for another 24 h. For the PF-treated groups, we added PF at doses of 100 nM and 1 μM to the glucose-free DMEM and neurobasal medium. For the edaravone, Z-VAD-FMK and Mdivi-1 treated groups, we added the appropriate compound to the neurobasal medium. Cell viability was evaluated by the MTT assay.
PC12 cells were cultured in DMEM/5% FBS/10% horse serum and transfected with HDAC4 (D289E) using FuGENE HD Transfection Reagent (Promega, Madison, WI, USA). For the OGD/R experiment, cells were placed in a hypoxic chamber for 3 h and then cultured under normal conditions for 6 h. For the PF-treated groups, we added PF at doses of 100 nM and 1 μM to the glucose-free DMEM and normal medium.
Immunofluorescence staining
Brains were postfixed in PFA for 24 h and dehydrated in 30% (wt/vol) sucrose for 48 h beginning on the 14th day after MCAO. Next, 20 μm coronal slices were obtained with a Leica freezing microtome. The primary antibodies used for immunofluorescence staining were as follows: anti-HDAC4 (ab12172, Abcam), anti-HA tag (3724S, CST), and anti-Tuj1 (ab78078, Abcam) antibodies. The sections were incubated with DAPI (100 ng/mL, Roche, Mannheim, Germany) and Alexa Fluor 488- and Alexa Fluor 555-conjugated secondary antibodies (Thermo Fisher Scientific) overnight at 4 °C. The cells were washed with ice-cold PBS and fixed in 4% polyformaldehyde for 20 min at 4 °C. Then, the cells were treated with 10% goat serum in PBS with 0.1% Triton-X at room temperature for an hour. The cells were incubated with anti-HDAC4 and anti-NeuN (ab177487, Abcam) antibodies overnight at 4 °C. The cells were incubated with Alexa Fluor 405-, Alexa Fluor 488- and Alexa Fluor 555-conjugated secondary antibodies and DAPI for nuclear staining for an hour at room temperature. After washing with PBS three times, the slides were fixed with Dako fluorescent mounting media (Dako Corporation, Carpinteria, CA, USA). All the samples were observed with a confocal microscope (Olympus, Japan).
Cell viability assay
MTT solution (0.5 mg/mL) was added to each well (96-well plates), and the plates were incubated for 4 h at 37 °C. The formazan dye was dissolved in DMSO, and the plates were gently mixed for 15 min on a shaker. The absorbance was read with a microplate reader (NovoStar, BMGLABTECH, Offenburg, Germany) at 595 nm.
Western blot
The cultured cells were washed with precooled PBS and lysed in RIPA buffer with proteinase and phosphatase inhibitor cocktail (Sigma-Aldrich) for 30 min to extract total protein. Cytoplasmic and nuclear proteins were separated according to the protocol of the SC-003 MinuteTM Fractionation Kit (Invent Biotechnologies, Beijing, China). The concentrations of all protein samples were measured using a BCA assay Kit (Thermo Fisher Scientific). The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to the PVDF membranes (Merck Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk and incubated with the primary antibody overnight at 4 °C. After incubation with HRP-conjugated secondary antibodies (Cell Signaling Technology, Beverly, MA, USA), the protein bands were detected with an ECL kit (Merck Millipore, Billerica, MA, USA). The blots were probed with the following specific antibodies: anti-HDAC4 (ab12172, Abcam); anti-GDNF (ab176564, Abcam), anti-BDNF (ab108319, Abcam), anti-Bcl-xl (ab32370, Abcam), anti-cytochrome c (ab133504, Abcam), anti-VDAC1 (ab154856, Abcam), anti-Lamin B1 (ab16048, Abcam), and anti-β-actin (60008-1-Ig, ProteinTech) antibodies.
Separation of cytoplasmic and mitochondrial protein
Cytoplasmic and mitochondrial protein was extracted according to the protocol of the Mitochondria Fractionation Kit (Abcam, London, UK). The samples were analyzed by Western blotting.
Luciferase reporter assay
A MEF2 reporter (QIAGEN) was transiently transfected into PC12 cells with FuGENE HD Transfection Reagent. After OGD, luciferase activity was measured with a Dual-Luciferase Reporter Assay System (Promega), and the absorbance was read with a microplate reader (Novostar, Germany).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
After OGD/R treatment, total RNA was extracted by using a Total RNA Isolation Kit (EZ Bioscience, China). Then, the mRNA was converted to cDNA using HiScript RT SuperMix for qPCR (Vazyme, Nanjing, China). The cDNA was used for real-time PCR with SYBR Green according to the manufacturer’s protocol (Vazyme, Nanjing, China). The expression of the target gene was calculated by the 2−∆∆Ct method. The PCR primers are listed in Supplementary Table S1.
Caspase activity assay
Caspase3 and caspase9 activity was determined by a colorimetric assay following the manufacturer’s protocol (Beyotime Biotechnology, Shanghai, China). Caspase3 and caspase9 can separately catalyze the synthesis of p-nitroaniline, which can be quantified by measuring the absorption at a wavelength of 405 nm (NovoStar, Germany). Primary neurons were lysed with lysis buffer and then centrifuged at 16,000 × g for 15 min at 4 °C. The activity of caspase3 or caspase9 in the supernatant was assayed at 405 nm. The activity was expressed as a ratio of the control.
JC-1 staining
A mitochondrial membrane potential assay kit (Beyotime Biotechnology, Shanghai, China) that uses JC-1 as a fluorescence probe was used to measure the mitochondrial membrane potential. Normally, JC-1 is present in the mitochondrial matrix as aggregates and emits red fluorescence. During apoptosis, when the mitochondrial membrane potential is low, JC-1 exists as monomers, which emit green fluorescence. All the samples subjected to OGD/R were fixed and then observed with a confocal microscope (Olympus, Japan) after JC-1 staining.
Statistical analysis
Quantification and statistical analysis were carried out with GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). All values are presented as the mean ± SEM, and the results from at least three independent experiments were compared by one-way ANOVA followed by Turkey’s multiple comparisons test.
Results
PF maintained HDAC4 localization after OGD/R injury in vitro
In our previous study, we found that PF preconditioning could exert a similar protective effect as ischemic preconditioning in rats after MCAO. In addition, inhibition of HDAC3, an HDAC subtype, mediates ischemic preconditioning [2]. Thus, we wondered whether other HDAC subtypes contribute to the protective effect of PF after ischemic injury. The class II histone deacetylases (HDACs) HDAC4 and HDAC5, which can shuttle between the cytoplasm and the nucleus, are abundantly expressed in the brain and participate in the regulation of neurodegeneration [21,22,23]. Under physiological conditions, HDAC4 is mainly found in the cytoplasm. After MCAO, the expression of HDAC4, but not HDAC5, markedly increased in the nuclei of neurons in the peri-infarct cortex [15]. Based on these results, we explored whether PF could affect HDAC4 localization. We first used an OGD/R model to mimic the pathological process in neurons during the stroke. Primary neurons were obviously damaged after OGD/R treatment. Cell viability was significantly increased in the 100 nM and 1 μM PF groups compared with the OGD/R group (Fig. 1a) (OGD/R, 0.64 ± 0.05 vs. 100 nM, 0.85 ± 0.04; P < 0.01; OGD/R, 0.64 ± 0.05 vs. 1 μM, 0.83 ± 0.03; P < 0.05). Next, we assessed the expression of HDAC4 in cultured primary neurons and PC12 cells subjected to OGD/R by immunofluorescence. The results showed that HDAC4 was obviously located in the nucleus in primary neurons and PC12 cells after OGD/R treatment, while 100 nM and 1 μM PF prevented the accumulation of HDAC4 in the nucleus (Fig. 1b and Supplementary Fig. S1b) (OGD/R, 74.17 ± 10.57 vs. 100 nM, 18.33 ± 6.87; P < 0.001; OGD/R, 74.17 ± 10.57 vs. 1 μM, 17.5 ± 6.85; P < 0.001). These results indicated that PF exerted a neuroprotective effect which was likely related to HDAC4 localization.

a Primary cortical neurons treated with or without PF were subjected to oxygen/glucose deprivation for 90 min. PF obviously increased cell viability. ###P < 0.001, OGD/R group versus control group; **P < 0.01, PF 100 nM group versus OGD/R group; *P < 0.05, 1 μM PF group versus OGD/R group; one-way ANOVA. b Representative images of HDAC4 subcellular localization and the quantitative statistical analysis of the percentage of HDAC4/NeuN colocalization in cultured cortical neurons following OGD/R treatment. The arrow indicates the accumulation of HDAC4 in primary neurons. Scale bar: 20 μm. ###P < 0.001, OGD/R group versus control group; ***P < 0.001, 100 nM PF group and 1 μM PF group versus OGD/R group; one-way ANOVA. All these results are expressed as the mean ± SEM of four independent experiments.
PF modulated HDAC4 distribution by reducing caspase3-mediated degradation under OGD/R treatment
We analyzed full-length HDAC4 protein levels by immunoblot analysis. The expression of full-length HDAC4 dramatically decreased in nuclear lysates after OGD/R, while PF slightly reversed this decrease (Fig. 2a) (control vs. OGD/R, 0.31 ± 0.07; P < 0.001), indicating that full-length HDAC4 was not transported to the nucleus. Based on other experiments, caspase3-induced degradation can generate a short fragment of HDAC4 (~38 kDa), and co-IP showed that cleaved caspase3 combined with the ~38 kDa HDAC4 fragment (Supplementary Fig. S2a). Western blot analysis of the nuclear lysates of primary neurons revealed that the level of the short HDAC4 fragment (~38 kDa) obviously increased in the nucleus following OGD/R treatment (Fig. 2b) (control vs. OGD/R, 8.19 ± 1.32; P < 0.001). Furthermore, we found that OGD/R-treated primary neurons showed a decrease in the protein expression of full-length HDAC4 in cytoplasmic lysates and in whole-cell lysates, indicating that the change in the subcellular localization of HDAC4 under pathological conditions was caused by degradation of HDAC4 rather than inhibition of its transport (Fig. 2c, d) (control vs. OGD/R, 0.36 ± 0.11; P < 0.05; control vs. OGD/R; P < 0.001). We next assessed the involvement of HDAC4 in the protective effect of PF, and we found that 100 nM and 1 μM PF obviously increased full-length HDAC4 protein levels in whole-cell lysates and reduced the level of the short HDAC4 fragment in nuclear lysates (Fig. 2d) (OGD/R vs. 100 nM, 0.75 ± 0.13; P < 0.01; OGD/R vs. 1 μM, 0.68 ± 0.09; P < 0.05). Considering that HDAC4 subcellular localization can be mediated by many factors such as phosphorylation [25] or degradation mediated by caspase3 [26, 27], we added Z-VAD-FMK, a caspase3 inhibitor, to cultured cortical neurons. We found that it increased full-length HDAC4 protein levels in cultured cortical neurons under OGD treatment (Fig. 2d) (OGD/R vs. Z-VAD-FMK, 0.66 ± 0.07; P < 0.05). Since degradation mediated by caspase3 involves the Asp-289 residue of HDAC4 [21], we transfected a HA-HDAC4 (D289E) plasmid, which contained a point mutation, into PC12 cells (the transfection efficiency was ~50.50 ± 2.31 percentage, data not shown). The results showed that HDAC4 (D289E) was always localized in the cytoplasm after OGD/R whether or not PF was administered (Fig. 2e). However, the protective effect of PF was abolished after transfection of the HDAC4 (D289E) plasmid (Fig. 2f) (control vs. OGD/R, 0.67 ± 0.04; P < 0.001). The qPCR results showed no apparent change in HDAC4 mRNA expression, which proved that neither ischemic injury nor PF could influence the transcript level of HDAC4 (Supplementary Fig. S2b). These results indicated that PF may exert its protective effect through maintaining HDAC4 in the cytoplasm by modulating caspase3-mediated degradation after OGD/R treatment.

a The expression of full-length HDAC4 was assessed by Western blotting in nuclear lysates from cortical neurons. ###P < 0.001, OGD/R group versus control group. b The level of the small fragment of HDAC4 (~38 kDa) was increased in the neuronal nuclei after OGD/R treatment. ###P < 0.01, OGD/R group versus control group. c The expression of full-length HDAC4 was reduced in the cytoplasmic lysates and was increased by PF treatment. #P < 0.05, OGD/R group versus control group. d PF and the caspase3 inhibitor Z-VAD-FMK increased the expression of full-length HDAC4 in whole-cell lysates from primary neurons after OGD/R treatment. ###P < 0.001, OGD/R group versus control group; **P < 0.01, 100 nM PF group versus OGD/R group; *P < 0.05, 1 μM PF group and Z-VAD-FMK group versus OGD/R group. All these results are expressed as the mean ± SEM of five independent experiments. e Representative images of HA-HDAC4 (D289E) subcellular localization following OGD/R treatment in PC12 cells. Scale bar: 20 μm. f The viability of PC12 cells transfected with the HDAC4 (D289E) plasmid and treated with or without PF after OGD/R treatment. ###P < 0.001, OGD/R group versus control group. All these results are expressed as the mean ± SEM of three independent experiments; one-way ANOVA.
PF decreased caspase3 activity by preventing the release of cytochrome c
Our data already showed that PF could reduce caspase3-dependent HDAC4 degradation after ischemic injury, but we wanted to know how PF modulates caspase3 activity. Caspase3 is a critical target of caspase9, which is generated by cleavage of the apoptosome by cytochrome c released from the mitochondria [22, 23]. We found that caspase3 and caspase9 activity increased after OGD/R treatment, and that both doses of PF reversed this increase (Fig. 3a, b) (Caspase3: OGD/R, 1.67 ± 0.12 vs. 100 nM, 1.22 ± 0.14; P < 0.05; OGD/R, 1.67 ± 0.12 vs. 1 μM, 1.22 ± 0.07; P < 0.05; Caspase9: OGD/R, 1.99 ± 0.13 vs. 100 nM, 0.93 ± 0.18; P < 0.01; OGD/R, 1.99 ± 0.13 vs. 1 μM, 0.97 ± 0.22; P < 0.01). We next measured the expression levels of cytochrome c in the cytoplasm and mitochondria. Western blot analysis of mitochondrial lysates, where cytochrome c should be found under physiological conditions, revealed that the protein level of cytochrome c was reduced (Fig. 3c), while the expression of cytochrome c increased in cytoplasmic lysates following OGD treatment (Fig. 3d), indicating that cytochrome c was released from the mitochondria into the cytoplasm during OGD/R. Treatment with 100 nM or 1 μM PF obviously prevented the release of cytochrome c (Fig. 3c, d) (mitochondrial lysates: OGD/R, 0.16 ± 0.06 vs. 100 nM, 0.63 ± 0.14; P < 0.01; OGD/R, 0.16 ± 0.06 vs. 1 μM, 0.57 ± 0.11; P < 0.05; cytoplasmic lysates: OGD/R, 1.94 ± 0.06 vs. 100 nM, 1.20 ± 0.09; P < 0.001; OGD/R, 1.94 ± 0.06 vs. 1 μM, 1.18 ± 0.15; P < 0.001). Furthermore, we used a JC-1 staining assay to assess the integrity of the mitochondria membrane. The integrity of mitochondria was disrupted following OGD/R, and PF protected mitochondria from injury during the pathological process (Fig. 3e). Since mitochondrial fragmentation can be blocked by the inhibition of Drp1 [24], we used Mdivi-1, a Drp1 inhibitor, and Z-VAD-FMK to confirm the effect of mitochondria in the development of ischemic injury. Z-VAD-FMK dramatically decreased the increase in caspase3 activity after OGD/R treatment (Fig. 3a) (caspase3: OGD/R vs. Z-VAD-FMK, 0.94 ± 0.08; P < 0.001). Mdivi-1 and Z-VAD-FMK obviously protected primary neurons after OGD/R treatment (Fig. 3f) (OGD/R, 0.63 ± 0.02 vs. Mdivi-1, 0.79 ± 0.02; P < 0.01; OGD/R, 0.63 ± 0.02 vs. Z-VAD-FMK, 0.76 ± 0.04; P < 0.05). These results indicated that PF can maintain the integrity of mitochondria by preventing the release of cytochrome c, reducing the activity of caspase9, and inhibiting the activation of caspase3.

a PF and Z-VAD-FMK apparently decreased caspase3 activity in cultured cortical neurons after OGD/R treatment. ###P < 0.001, OGD/R group versus control group; *P < 0.05, 100 nM PF and 1 μM PF groups versus OGD/R group; ***P < 0.001, Z-VAD-FMK group versus OGD/R group. b PF significantly decreased caspase9 activity in cultured cortical neurons after OGD/R treatment. #P < 0.05, OGD/R group versus control group; **P < 0.01, 100 nM PF and 1 μM PF groups versus OGD/R group. c, d The protein expression of cytochrome c in mitochondrial lysates and cytoplasmic lysates was assessed by Western blotting. ###P < 0.001, OGD/R group versus control group; **P < 0.01, ***P < 0.001, 100 nM PF group versus OGD/R group; *P < 0.05 and ***P < 0.001, 1 μM PF group versus OGD/R group. e Representative images of JC-1 staining following OGD/R and PF treatment. Green fluorescence indicates disintegration of the mitochondrial membrane. Scar bar: 20 μm. f Primary cortical neurons treated with Mdivi-1 or Z-VAD-FMK were subjected to oxygen/glucose deprivation/reperfusion for 90 min. Mdivi-1 and Z-VAD-FMK obviously increased cell viability. ###P < 0.001, OGD/R group versus control group; **P < 0.01, Mdivi-1 group versus OGD/R group, *P < 0.05, Z-VAD-FMK group versus OGD/R group. All these results are expressed as the mean ± SEM of five independent experiments; one-way ANOVA.
PF exerted an anti-apoptotic effect through modulating the HDAC4 pathway
Class II HDACs play fundamental roles in muscle development and neuronal activity by binding tissue-specific transcription factors such as MEF2 and CREB [25,26,27]. We speculate that during the process of stroke, some transcription factors may be influenced by nuclear HDAC4 accumulation. MEF2A, a transcription factor repressed by direct binding with HDAC4, can modulate neuronal differentiation and cell survival through regulating downstream proteins. We performed luciferase promoter assays to evaluate the degree of transcriptional repression of MEF2. The OGD/R-treated group showed a significant reduction in the transcriptional activity of MEF2A. Both doses of PF reversed the decrease in the transcriptional activity of MEF2A after OGD/R treatment (Fig. 4a) (OGD/R, 10.23 ± 0.94 vs. 100 nM, 21.29 ± 3.40; P < 0.05; OGD/R, 10.23 ± 0.94 vs. 1 μM, 20.82 ± 3.49; P < 0.05). The change in transcriptional activity also altered the mRNA level and protein expression of MEF2-dependent genes such as Bcl-xl, GDNF and BDNF. Under OGD/R conditions, PF increased the expression of Bcl-xl at both the transcriptional and translational levels (Fig. 4b, c) (Bcl-xl qPCR; OGD/R, 0.38 ± 0.08 vs. 100 nM, 1.21 ± 0.19; P < 0.01; OGD/R, 0.38 ± 0.08 vs. 1 μM, 1.29 ± 0.19; P < 0.01; Bcl-xl: OGD/R, 0.52 ± 0.06 vs. 100 nM, 0.94 ± 0.10; P < 0.05; OGD/R, 0.52 ± 0.06 vs. 1 μM, 0.94 ± 0.12; P < 0.05) and rescued the protein expression of the trophic factors GDNF and BDNF (Fig. 4d, e) (GDNF: OGD/R, 0.66 ± 0.05 vs. 100 nM, 1.07 ± 0.07; P < 0.001; OGD/R, 0.66 ± 0.05 vs. 1 μM, 0.97 ± 0.04; P < 0.01; BDNF: OGD/R, 0.33 ± 0.06 vs. 100 nM, 1.00 ± 0.12; P < 0.001; OGD/R, 0.33 ± 0.06 vs. 1 μM, 0.84 ± 0.08; P < 0.01). To further confirm the involvement of this pathway, we measured the expression of these proteins in PC12 cells after transfection of HDAC4 (D289E). The results indicated that after OGD/R injury, the protein expression of Bcl-xl and BDNF was significantly reduced, GDNF expression was slightly reduced (Bcl-xl: control vs. OGD/R, 0.46 ± 0.12; P < 0.05; BDNF: control vs. OGD/R, 0.35 ± 0.07; P < 0.01; GDNF: control vs. OGD/R, 0.69 ± 0.09), and the effects of PF were almost canceled out (Fig. 4f, g, h). Considering that the expression of Bcl-xl, which acts as an anti-apoptotic protein, was affected, we used the TUNEL assay to further investigate whether PF could reduce the number of TUNEL-positive neurons. The results revealed that PF could exert a dramatic anti-apoptotic effect against OGD/R injury (Supplementary Fig. S3a) (OGD/R, 40.60 ± 2.69 vs. 100 nM, 11.26 ± 1.65; P < 0.001; OGD/R, 40.60 ± 2.69 vs. 1 μM, 9.07 ± 1.71; P < 0.001). However, the number of TUNEL+ cells was maintained at a high level after PF treatment following transfection with HDAC4 (D289E) (Supplementary Fig. S3b) (control vs. OGD/R, 0.46 ± 0.12; P < 0.05). Based on these data, we confirmed that the protective effect of PF is dependent on caspase3-induced HDAC4 degradation during OGD/R. When caspase3 cannot degrade HDAC4 at a specific site, the protective effect of PF is abolished.

a Quantification of MEF2 transcript levels is shown. #P < 0.05, OGD/R group versus control group; *P < 0.05, 100 nM PF and 1 μM PF groups versus OGD/R group; one-way ANOVA. b, c PF increased the mRNA level and protein expression level of Bcl-xl, which is downstream of MEF2, in cultured cortical neurons. #P < 0.05, OGD/R group versus control group; *P < 0.05 and **P < 0.01, 100 nM PF and 1 μM PF groups versus OGD/R group; one-way ANOVA. d, e The levels of the trophic factors GDNF and BDNF were assessed by Western blotting in whole-cell lysates. #P < 0.05 and ##P < 0.01, OGD/R group versus control group; **P < 0.01, ***P < 0.001, 100 nM PF and 1 μM PF groups versus OGD/R group; one-way ANOVA. (f, g, h) Bcl-xl, GDNF and BDNF levels were assessed by Western blotting in whole-cell lysates after transfection of HDAC4 (D289E) into PC12 cells. The protective effects of PF were abolished. #P < 0.05 and ##P < 0.01, OGD/R group versus control group; one-way ANOVA. All these results are expressed as the mean ± SEM of three independent experiments.
PF modulated HDAC4 distribution and exerted a protective effect in vivo
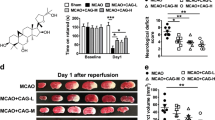
To confirm the results that PF could affect HDAC4 distribution, rats were sacrificed 14 days after MCAO and subjected to double immunohistochemistry with antibodies against HDAC4 and Tuj1. In the cortices of rats in the sham group, HDAC4 was mainly localized in the cytoplasm of Tuj1+ neurons. After MCAO, there was a robust increase in level of HDAC4 in the nucleus, while PF reduced the shuttling of HDAC4 to the nucleus (Fig. 5a). A significant reduction in infarct volume was observed in MCAO rats treated with 20 or 40 mg/kg PF compared with vehicle-treated MCAO rats after reperfusion. The edaravone-treated group also had a lower infarct volume than the vehicle-treated group (Fig. 5b, c) (MCAO, 19.48 ± 4.30 vs. 20 mg/kg PF, 8.61 ± 2.46, P < 0.05; MCAO, 19.48 ± 4.30 vs. 40 mg/kg PF, 5.56 ± 1.09; P < 0.01; MCAO, 19.48 ± 4.30 vs. edaravone, 11.15 ± 2.58).

a Representative images of HDAC4+/Tuj1+ cortical neurons after MCAO and PF treatment. The thick arrow indicates HDAC4 in the nuclei of Tuj1+ neurons. The thin arrow indicates HDAC4 in the cytoplasm of Tuj1+ neurons. Scale bar: 50 μm. b Infarct measurements were performed 7 days (n = 8–10) after MCAO. Representative images of TTC staining. c The infarct volume percentage in each group. **P < 0.01 and *P < 0.05, 40 mg/kg PF and 20 mg/kg PF groups versus the MCAO group; one-way ANOVA. All these results are expressed as the mean ± SEM.
PF improved locomotor ability and cognitive ability in vivo
According to neurological deficit scores, the administration of 40 mg/kg PF apparently alleviated neurological symptoms. On the 7th day, the 20 mg/kg PF group exhibited obvious improvements in neurological deficits. The edaravone group also exhibited lower neurological deficit scores than the MCAO group throughout the 7-day testing period, indicating that PF showed aided neurological recovery after MCAO (Fig. 6a) (day 7: MCAO, 1.56 ± 0.15 vs. 20 mg/kg PF, 0.63 ± 0.21; P < 0.05; MCAO, 1.56 ± 0.15 vs. 40 mg/kg PF, 0.56 ± 0.22; P < 0.05; MCAO, 1.56 ± 0.15 vs. edaravone, 1.13 ± 0.31). Then, we conducted behavioral experiments, which were executed according to the schedule shown in Supplementary Fig. S4a, to examine behavioral recovery after MCAO. After MCAO, rats exhibited obvious locomotor deficits, and when treated with 20 mg/kg or 40 mg/kg PF, they displayed better performance than the MCAO group on the rotarod test on the first testing day (Fig. 6b) (day 4; sham, 121.25 ± 2.88 vs. MCAO, 27.25 ± 4.49; P < 0.001; MCAO, 27.25 ± 4.49 vs. 20 mg/kg PF, 58 ± 8.27; P < 0.05; MCAO, 27.25 ± 4.49 vs. 40 mg/kg PF, 57.13 ± 6.01; P < 0.05), and better performance than the edaravone group. On the second testing day, which was the 7th day after surgery, all groups showed partial recovery of locomotor ability. In the open-field test, which was performed on the 5th day after reperfusion, the sham group displayed normal locomotor activity, while the MCAO group showed dramatic decreases in distance traveled, active time, and time spent making vertical movements, whereas the 40 mg/kg PF group showed significant improvements in all aspects of locomotor activity. The 20 mg/kg PF group exhibited obvious increases in distance traveled and the time spent moving (Fig. 6c, Supplementary Fig. S4b, c, d) (distance traveled: sham, 14,555.03 ± 580.76 vs. MCAO, 6713.62 ± 1105.77; P < 0.001; MCAO, 6713.62 ± 1105.77 vs. 20 mg/kg PF, 13,194.10 ± 1508.11; P < 0.01; MCAO, 6713.62 ± 1105.77 vs. 40 mg/kg PF, 13,997.84 ± 1252.34; P < 0.001; time spent moving: sham, 316.46 ± 2.88 vs. MCAO, 220.28 ± 19.05; P < 0.001; MCAO, 220.28 ± 19.05 vs. 20 mg/kg PF, 290.15 ± 10.69; P < 0.01; MCAO, 220.28 ± 19.05 vs. 40 mg/kg PF, 319.51 ± 13.73; P < 0.001; vertical rearing time: sham, 20.41 ± 2.06 vs. MCAO, 4.13 ± 1.47; P < 0.01; MCAO, 4.13 ± 1.47 vs. 40 mg/kg, 17.33 ± 3.99; P < 0.05; number of vertical rearings: sham, 19.75 ± 1.52 vs. MCAO, 6.38 ± 1.53; P < 0.01; MCAO, 6.38 ± 1.53 vs. 40 mg/kg PF, 17.13 ± 3.10; P < 0.05). Motor function was also improved in the edaravone group compared with the MCAO group. These data illustrated that PF improved locomotor ability in the initial stage after MCAO.

a Functional outcomes were assessed using neurological deficits scores (days 1, 4, and 7 after MCAO) (n = 8). *P < 0.05, **P < 0.01 20 mg/kg PF and 40 mg/kg PF groups versus MCAO group; one-way ANOVA. b The rotarod test (days 4 and 7 after MCAO) was used to test locomotor ability (n = 8). ###P < 0.001, MCAO group versus sham group; *P < 0.05, 20 mg/kg PF and 40 mg/kg PF groups versus MCAO group; one-way ANOVA. c The open-field test (day 5 after MCAO) was used to assess locomotor activity (n = 8). ###P < 0.001, MCAO group versus sham group; **P < 0.01, 20 mg/kg PF group versus MCAO group; ***P < 0.001, 40 mg/kg PF group versus MCAO group; one-way ANOVA. d PF reduced the time required for the rats to find the hidden platform in the training phase (n = 8). #P < 0.05, ##P < 0.01 and ###P < 0.001, MCAO group versus sham group; *P < 0.05, 40 mg/kg PF group versus MCAO group. e The number of times that the rats swam through the location of the platform during the probe phase. ###P < 0.001, MCAO group versus sham group. f Representative swimming tracks of rats in the testing phase are shown. g Representative diagrams of Nissl staining of rat tissues are shown. Scale bar: 1 mm. All these results were expressed as the mean ±SEM.
We measured learning and memory abilities through the Morris water maze task. The escape latency of 40 mg/kg PF group (the time it took to find the hidden platform) was obviously reduced on the last day of the training phase, and the latency was also reduced in the 20 mg/kg PF group (Fig. 6d) (day 13: sham, 22.85 ± 3.99 vs. MCAO, 72.59 ± 7.81; P < 0.001; MCAO, 72.59 ± 7.81 vs. 40 mg/kg PF, 37.31 ± 9.49; P < 0.05). During the testing phase, the number of platform region crossings and the percentage of time spent in the target quadrant were increased in the 20 mg/kg PF and 40 mg/kg PF groups compared with the MCAO group, but not significantly (Fig. 6e and Supplementary Fig. S4f). There was no obvious difference in swimming speed between the groups (Supplementary Fig. S4e), which demonstrated that all the groups had the similar locomotor ability. Rats treated with edaravone did not show obvious improvement in cognitive ability compared with those in the MCAO group in either the training phase or testing phase. The Nissl staining results showed that the integrity of pyramidal neurons in the hippocampal CA1 region was maintained in the 20 mg/kg PF and 40 mg/kg PF groups after MCAO injury, which was consistent with performance in water maze (Fig. 6g). These results indicated that PF obviously alleviates infarction, and improves locomotor and cognitive ability after MCAO.
Discussion
PF, the main active compound extracted from Chinese herbaceous peony, was proven to exhibit anti-inflammatory and protective functions. Previous research supports the claim that PF can inhibit the NF‐κB/MAPK pathway to alleviate inflammatory bowel disease [28]. In an animal model of psoriasis, PF ameliorates psoriatic lesions by suppressing the mRNA expression of IL-6 and IL-22 via the p38/MAPK pathway [29]. In other studies, PF was proven to attenuate neuropathic pain induced by chronic constriction injury by inhibiting the activation of ASK1 and the response of glial cells [30] and decreasing hyperphosphorylated tau levels via the calpain/AKT/GSK-3β-related pathway [31]. Our laboratory found that PF protects cortical neurons after ischemic injury induced by A1R-mediated transactivation of EGFR [32]. In this study, we demonstrated that MCAO rats showed better locomotor ability in the open-field test and rotarod test after PF treatment. In the Morris water maze test, the PF-treated groups showed a significant decrease in latency, which is the most important parameter for evaluating memory and learning, on the last day of the training phase. PF obviously attenuated behavioral and cognitive abnormalities, reduced the infarct volume in an animal model of stroke, and protected the brain from ischemic injury. Furthermore, we explored whether PF could play a protective role following OGD/R, which mimics the pathological process of ischemic stroke in vitro.
HDACs play an important role in the homeostasis of histone acetylation and gene transcription, which is critical for epigenetic modification [33]. HDACs have been grouped into four classes: classical HDACs (I, II, IV), which have similar structures and depend on Zinc ion to exert their enzymatic activity, and class III sirtuins (SIRT1-7), which deacetylate nonhistone proteins and depend on NAD+ to exert their enzymatic activity [33]. Several HDAC inhibitors have been reported to exert protective effects against ischemic stroke. Sodium butyrate, an HDAC inhibitor that alters H3K9ac enrichment and the transcript levels of inflammatory proteins in activated microglia, mediates neuroprotection and anti-inflammatory effects by epigenetic regulation [34]. Sodium valproate, a nonspecific inhibitor of HDAC9, is associated with lower recurrent stroke risk [35]. These HDAC inhibitors mostly target active HDACs, especially class I HDACs, and have limited selectivity. Since HDACs take part in many important epigenetic modulatory processes, nonselective inhibitors may cause unknown side effects. Confirming the specific roles of different HDAC subtypes in the process of stroke is meaningful. Previous studies have demonstrated that class IIa HDACs have low intrinsic HDAC activity against acetyl-lysine substrates, although they are structurally similar to highly active class I HDACs [36]. Unlike class I HDACs, which are localized in the nucleus, class IIa HDACs, led by HDAC4, undergo characteristic nuclear-cytoplasmic trafficking in response to certain cellular signals. The N-terminus and C-terminus of HDAC4 contain a nuclear localization sequence and a nuclear export sequence, respectively [37]. Shutting of HDAC4, a transcription repressor, between the nucleus and cytoplasm has been reported to regulate a transcriptional network that is important for synaptic transmission in the brain [38]. HDAC4 subcellular localization is driven by phosphorylation-dependent shuttling [39] or degradation due to caspase-mediated cleavage [40]. Numerous studies have proven that the nuclear accumulation of HDAC4 is related to the promotion of cell apoptosis [21] and modulation of the NF‐κB-mediated inflammatory pathway [41]. Overactivation of calcium improves the binding between HDAC4 and 14-3-3, resulting in an increase in the transport of HDAC4 to the cytoplasm [42]. In the early stage of myocyte proliferation and apoptosis, HDAC4 can form a complex with MEF2 in the nucleus, inhibiting the expression of and regulatory effect of MEF2 on downstream genes, such as Bcl-2 family members. It has been proven that nuclear HDAC4 influences cell viability by repressing the transcriptional activity of MEF under oxidative stress [14].
According to the immunofluorescence results shown in Fig. 1b, we verified that the subcellular localization of HDAC4 remained largely unchanged in the PF-treated groups. However, the results shown in Fig. 2a indicate that HDAC4 accumulation was induced by degradation of the protein rather than the modulation of protein transport. Then, we detected an increase in the level of a short fragment of HDAC4 (approximately 38 kDa) in the nuclear lysates of primary neurons after OGD/R treatment; this fragment is a degradation product created by caspase3-mediated degradation, as proven by co-IP (Supplemental Fig. S2a), the level of which can be reduced by PF treatment. Fig. 2c, d show that PF reversed the decrease in the protein expression of full-length HDAC4 without changing the mRNA level of HDAC4, indicating that nuclear accumulation of HDAC4 was induced by caspase3-mediated degradation during stroke, not by transcriptional modulation. When we transfected a plasmid carrying a point mutation that could not be cleaved by caspase3, the HDAC4 was maintained in the cytoplasm, which indicated that this phenomenon was indeed caused by caspase3-mediated degradation. However, PF did not exert a protective effect after HDAC4 mutant plasmid transfection, which means caspase3-induced HDAC4 degradation is one of the key contributors to the effect of PF. HDAC4 subcellular localization was maintained in the PF-treated groups through inhibition of the degradation and activity of caspase3. The expression of Bcl-xl, GDNF, and BDNF, downstream proteins of HDAC4, decreased under OGD/R treatment, which was the result of repression of MEF2A caused by HDAC4 that had accumulated in the nucleus. Furthermore, PF exerted an obvious anti-apoptotic effect by augmenting MEF2A transcript levels and increasing the expression of downstream effector proteins, and these effects were abolished by transfection of the HDAC4 (D289E) plasmid, which could not be degraded by caspase3.
Bcl-xl is an important anti-apoptotic protein that is required for neurite outgrowth and potentially inhibits programmed cell death by changing the mitochondrial membrane [43]. Since PF modulated caspase3 activity and affected the expression of Bcl-xl under OGD/R treatment, which are both connected with mitochondria, we wondered whether mitochondria participated in the regulatory effect of PF. Apoptosis, or programmed cell death, plays an essential role in tissue development and the homeostasis of multicellular organisms [44] and is implicated in the pathophysiology of various neurodegenerative disorders, such as stroke [45]. The apoptotic signaling cascade consists of two main parts: the specific apoptosis initiation pathway and the common execution pathway [22]. The intrinsic pathway is activated initially by the formation of a caspase-activating complex called the apoptosome [46], which is a cytochrome c-initiated process. Once the apoptosome is formed, procaspase-9 is recruited to the complex, becomes activated through autocatalysis, and activates effector caspases such as caspase3, caspase-6, and caspase-7 [20, 22, 23]. In our study, PF ameliorated the enhancing effects of caspase9 after OGD treatment. The release of cytochrome c from mitochondria into the cytoplasm was apparently prevented by PF treatment, as reflected by the levels of different proteins. JC-1 staining, which allows evaluation of the mitochondrial membrane potential via analysis of fluorescence, further proved that PF maintained the integrity of the mitochondrial membrane and inhibited OGD/R-induced apoptosis.
Conclusion
In conclusion, PF obviously ameliorated locomotor and cognitive deficits, reduced the infarct volume in vivo, exerted protective effects in vitro, and exerted a notable anti-apoptotic effect through the cytochrome c/caspase3/HDAC4 pathway during the pathological process of stroke. PF, which exerted a marked anti-apoptotic effect, decreased HDAC4 degradation induced by caspase3, which resulted in the nuclear accumulation of a small amount of HDAC4, increased MEF2A transcript levels, and promoted the expression of downstream proteins such as Bcl-xl under OGD/R treatment. The release of cytochrome c from mitochondria into the cytoplasm, which activated caspase3, was also inhibited by PF treatment under OGD/R conditions, and the integrity of the mitochondria membrane was maintained. This study illustrates the unique mechanism under the anti-apoptotic effect of PF and proposes new connections between PF and specific HDAC subtypes in ischemic stroke.
References
Wang W, Jiang B, Sun H, Ru X, Sun D, Wang L, et al. Prevalence, incidence, and mortality of stroke in China: results from a nationwide population-based survey of 480 687 adults. Circulation. 2017;135:759–71.
Yang XY, Wu QM, Zhang L, Feng LY. Inhibition of histone deacetylase 3 (HDAC3) mediates ischemic preconditioning and protects cortical neurons against ischemia in rats. Front Mol Neurosci. 2016;9:131.
Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–9.
Kurzepa J, Kurzepa J, Golab P, Czerska S, Bielewicz J. The significance of matrix metalloproteinase (MMP)−2 and MMP-9 in the ischemic stroke. Int J Neurosci. 2014;124:707–16.
Watanabe K, Tanaka M, Yuki S, Hirai M, Yamamoto Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J Clin Biochem Nutr. 2018;62:20–38.
Xin QQ, Yuan R, Shi WL, Zhu ZC, Wang Y, Cong WH. A review for the anti-inflammatory effects of paeoniflorin in inflammatory disorders. Life Sci. 2019;237:116925.
Nizamutdinova IT, Jin YC, Kim JS, Yean MH, Kang SS, Kim YS, et al. Paeonol and paeoniflorin, the main active principles of paeonia albiflora, protect the heart from myocardial ischemia/reperfusion injury in rats. Planta Med. 2008;74:14–8.
Yang HO, Ko WK, Kim JY, Ro HS. Paeoniflorin: an antihyperlipidemic agent from paeonia lactiflora. Fitoterapia. 2004;75:45–9.
Zhou JY, Wang LY, Wang JX, Wang C, Yang ZH, Wang CL, et al. Paeoniflorin and albiflorin attenuate neuropathic pain via MAPK pathway in chronic constriction injury rats. Evid Based Complement Alternat Med 2016;2016:8082753.
Li PP, Liu DD, Liu YJ, Song SS, Wang QT, Chang Y, et al. BAFF/BAFF-R involved in antibodies production of rats with collagen-induced arthritis via PI3K-Akt-mTOR signaling and the regulation of paeoniflorin. J Ethnopharmacol. 2012;141:290–300.
Wen J, Xu B, Sun YC, Lian MQ, Li YL, Lin Y, et al. Paeoniflorin protects against intestinal ischemia/reperfusion by activating LKB1/AMPK and promoting autophagy. Pharmacol Res. 2019;146:104308.
Aune SE, Herr DJ, Kutz CJ, Menick DR. Histone deacetylases exert class-specific roles in conditioning the brain and heart against acute ischemic injury. Front Neurol. 2015;6:145.
Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 2009;40:2899–905.
Wu QM, Yang XY, Zhang L, Zhang Y, Feng LY. Nuclear accumulation of histone deacetylase 4 (HDAC4) exerts neurotoxicity in models of Parkinson’s disease. Mol Neurobiol. 2017;54:6970–83.
Kassis H, Shehadah A, Chopp M, Roberts C, Zhang ZG. Stroke induces nuclear shuttling of histone deacetylase 4. Stroke. 2015;46:1909–15.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral-artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91.
Jin L, Zhang LM, Xie KQ, Ye Y, Feng LY. Paeoniflorin suppresses the expression of intercellular adhesion molecule-1 (ICAM-1) in endotoxin-treated human monocytic cells. Br J Pharmacol. 2011;164:694–703.
Lu H, Cheng G, Hong F, Zhang L, Hu YH, Feng LY. A novel 2-phenylamino-quinazoline-based compound expands the neural stem cell pool and promotes the hippocampal neurogenesis and the cognitive ability of adult mice. Stem Cells. 2018;36:1273–85.
Wang WZ, Liu LX, Jiang P, Chen C, Zhang T. Levodopa improves learning and memory ability on global cerebral ischemia-reperfusion injured rats in the Morris water maze test. Neurosci Lett. 2017;636:233–40.
Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Duke M, Fujimura M, et al. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci. 2002;22:209–17.
Liu F, Dowling M, Yang XJ, Kao GD. Caspase-mediated specific cleavage of human histone deacetylase 4. J Biol Chem. 2004;279:34537–46.
Cao GD, Xiao M, Sun FY, Xiao X, Pei W, Li J, et al. Cloning of a novel Apaf-1-interacting protein: a potent suppressor of apoptosis and ischemic neuronal cell death. J Neurosci. 2004;24:6189–201.
Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, et al. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci. 2004;24:2750–9.
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25.
Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. Embo J. 1999;18:5099–107.
McKinsey TA, Zhang CL, Lu JR, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–11.
Miska EA, Langley E, Wolf D, Karlsson C, Pines J, Kouzarides T. Differential localization of HDAC4 orchestrates muscle differentiation. Nucleic Acids Res. 2001;29:3439–47.
Zhang JJ, Dou W, Zhang EY, Sun A, Ding LL, Wei XH, et al. Paeoniflorin abrogates DSS-induced colitis via a TLR4-dependent pathway. Am J Physiol-Gastrointest Liver Physiol. 2014;306:G27–G36.
Yu JH, Xiao ZC, Zhao RZ, Lu CJ, Zhang YM. Paeoniflorin suppressed IL-22 via p38 MAPK pathway and exerts anti-psoriatic effect. Life Sci. 2017;180:17–22.
Zhou DL, Zhang SQ, Hu L, Gu YF, Cai YM, Wu DQ, et al. Inhibition of apoptosis signal-regulating kinase by paeoniflorin attenuates neuroinflammation and ameliorates neuropathic pain. J Neuroinflammation. 2019;16:83.
Ma XH, Duan WJ, Mo YS, Chen JL, Li S, Zhao W, et al. Neuroprotective effect of paeoniflorin on okadaic acid-induced tau hyperphosphorylation via calpain/Akt/GSK-3beta pathway in SH-SY5Y cells. Brain Res. 2018;1690:1–11.
Zhong M, Song WL, Xu YC, Ye Y, Feng LY. Paeoniflorin ameliorates ischemic neuronal damage in vitro via adenosine A1 receptor-mediated transactivation of epidermal growth factor receptor. Acta Pharmacol Sin. 2015;36:298–310.
He M, Zhang B, Wei XB, Wang ZY, Fan BX, Du PC, et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J Cell Mol Med. 2013;17:531–42.
Patnala R, VArumugam T, Gupta N, Dheen ST. HDAC inhibitor sodium butyrate-mediated epigenetic regulation enhances neuroprotective function of microglia during ischemic stroke. Mol Neurobiol. 2017;54:6391–411.
Brookes RL, Crichton S, Wolfe CDA, Yi QL, Li LX, Hankey GJ, et al. Sodium valproate, a histone deacetylase inhibitor, is associated with reduced stroke risk after previous ischemic stroke or transient ischemic attack. Stroke. 2018;49:54–61.
Gaur V, Connor T, Sanigorski A, Martin SD, Bruce CR, Henstridge DC, et al. Disruption of the class IIa HDAC corepressor complex increases energy expenditure and lipid oxidation. Cell Rep. 2016;16:2802–10.
Hou F, Wei W, Qin X, Liang J, Han S, Han AZ, et al. The posttranslational modification of HDAC4 in cell biology: mechanisms and potential targets. J Cell Biochem. 2020;121:930–7.
Sando R 3rd, Gounko N, Pieraut S, Liao L, Yates J 3rd, Maximov A. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell. 2012;151:821–34.
Paroni G, Fontanini A, Cernotta N, Foti C, Gupta MP, Yang XJ, et al. Dephosphorylation and caspase processing generate distinct nuclear pools of histone deacetylase 4. Mol Cell Biol. 2007;27:6718–32.
Paroni G, Mizzau M, Henderson C, Del Sal G, Schneider C, Brancolini C. Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol Biol Cell. 2004;15:2804–18.
Usui T, Okada M, Mizuno W, Oda M, Ide N, Morita T, et al. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am J Physiol-Heart Circulatory Physiol. 2012;302:H1894–H1904.
McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol. 2001;21:6312–21.
Park HA, Licznerski P, Alavian KN, Shanabrough M, Jonas EA. Bcl-xL is necessary for neurite outgrowth in hippocampal neurons. Antioxid Redox Signal. 2015;22:93–108.
Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–9.
Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415.
Hu YM, Benedict MA, Ding LY, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. Embo J. 1999;18:3586–95.
Acknowledgements
We thank Prof Yang Ye for providing the compound paeoniflorin. This study was supported by grants from the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (2018ZX09711002-003-013), and the Scientific Innovation Project of the Chinese Academy of Sciences (XDA12040304 and XDA12040216). This project was supported by the Shanghai Committee of Science and Technology, China (18DZ2290200).
Author information
Authors and Affiliations
Contributions
LYF conceived and designed the experiments. YFL performed the experiments. LZ contributed to the animal behavior studies. YFL and QW analyzed the data. YFL and LYF wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Liu, Yf., Zhang, L., Wu, Q. et al. Paeoniflorin ameliorates ischemic injury in rat brain via inhibiting cytochrome c/caspase3/HDAC4 pathway. Acta Pharmacol Sin 43, 273–284 (2022). https://doi.org/10.1038/s41401-021-00671-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00671-y
Keywords
This article is cited by
-
Salvianolic acid C attenuates cerebral ischemic injury through inhibiting neuroinflammation via the TLR4-TREM1-NF-κB pathway
Chinese Medicine (2024)
-
Exercise Alleviates Fluoride-Induced Learning and Memory Impairment in Mice: Role of miR-206-3p and PREG
Biological Trace Element Research (2024)
