
Abstract
Systemic sclerosis (SSc) is a life-threatening chronic connective tissue disease with the characteristics of skin fibrosis, vascular injury, and inflammatory infiltrations. Though inhibition of phosphodiesterase 4 (PDE4) has been turned out to be an effective strategy in suppressing inflammation through promoting the accumulation of intracellular cyclic adenosine monophosphate (cAMP), little is known about the functional modes of inhibiting PDE4 by apremilast on the process of SSc. The present research aimed to investigate the therapeutic effects and underlying mechanism of apremilast on SSc. Herein, we found that apremilast could markedly ameliorate the pathological manifestations of SSc, including skin dermal thickness, deposition of collagens, and increased expression of α-SMA. Further study demonstrated that apremilast suppressed the recruitment and activation of macrophages and T cells, along with the secretion of inflammatory cytokines, which accounted for the effects of apremilast on modulating the pro-fibrotic processes. Interestingly, apremilast could dose-dependently inhibit the activation of M1 and T cells in vitro through promoting the phosphorylation of CREB. In summary, our research suggested that inhibiting PDE4 by apremilast might provide a novel therapeutic option for clinical treatment of SSc patients.
Similar content being viewed by others
Introduction
Systemic sclerosis (SSc), with global disproportionate but increasing morbidity and disability, is an autoimmune disease, which threatens multiple tissues with the manifestations of finger swelling, subcutaneous tissue hardening, and muscle atrophy [1]. The pathological triggers for SSc remained partially defined and extensively involved the crosstalk and interaction among genetic factors, epigenetics, environmental deterioration, and disturbance of immune systems [2]. Fibrosis with progressive accumulation of extracellular matrix (ECM) proteins, including collagens, elastin, glycosaminoglycans, and fibronectin, represents a prominent pathological feature and distinguishing hallmark of SSc patients [3]. Currently, no single therapeutic approach for connective tissue diseases has been proved uniformly effective. Therefore, there exists a great requirement for developing novel therapeutic strategies for SSc treatment.
Mounting evidences indicated that alternations of innate and adaptive immune responses, such as infiltration of immune cells and secretion of pro-inflammatory cytokines, played a prominent role in the initiation and development of SSc [4, 5]. Upon exposure of multiple pathological mediators, skin-resident and newly recruited macrophages could differentiate into classically inflammatory macrophages (M1) and immune-regulating macrophages (M2), which have been intriguingly implicated in the pathogenesis of skin fibrosis [6]. In addition to the intrinsic specialty of macrophages, cytokines derived from activated macrophages could further lead to the differentiation of T cells and abnormal activation of skin fibroblasts [7]. Therein, TGF-β derived from macrophages and T-helper cells is considered as the main pathogenic triggers of tissue fibrosis. The inactive precursor of TGF-β could be converted to its biologically active form via integrins-mediated activation [8].
Phosphodiesterase 4 (PDE4) belongs to the PDEs superfamily and specifically hydrolyzes the cyclic adenosine monophosphate (cAMP) to regulate intracellular signaling transduction [9]. With an extensive distribution in neurons, immune cells, and epithelial cells, PDE4 might be incorporated into the specific signalosomes and specially regulate cAMP-predominant signaling events [10]. In terms of the transduction of G-protein-coupled receptors, adenylate cyclase could be activated and subsequently lead to the accumulation of intracellular cAMP and further activation of protein kinase A (PKA) [11]. Previous reports have demonstrated that inhibition of PDE4 displayed multiple pharmacological effects in psoriasis, inflammatory airway diseases, and ulcerative colitis through modulating the PKA-dominant phosphorylation of cAMP-response element binding protein (CREB) and antagonism of nuclear translocation of NF-κB [12]. Apremilast is a patent PDE4 inhibitor bearing with the distinct chemical skeleton and specifically regulates the expression of pro-inflammatory and anti-inflammatory mediators. Due to the therapeutic capacities, apremilast was approved by FDA in 2014 for the treatment of moderate-to-severe chronic plaque psoriasis and active psoriatic arthritis in adults [13].
Over the past 10 years, apremilast, with a well-defined tolerance and oral availability, has been considered as a breakthrough in the development of PDE4 inhibitors [14]. In the previous research by Maier et al., the efficacies of specific inhibition of PDE4 by rolipram and apremilast were investigated in bleomycin (BLM)-induced skin fibrosis [15]. Whereas, the underlying mechanism of apremilast on the function of macrophages and T cells remained ill-defined in skin fibrosis. Herein, the present research was designed to uncover the modulatory effects of apremilast on BLM-induced murine SSc model and to provide new insights toward the development of clinical therapeutic strategies for SSc patients.
Materials and methods
BLM-induced murine model of SSc and drug treatment
All animals’ feeding and laboratory programs were implemented according to the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Bioethics Committee of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Wild-type female C57BL/6J mice (8 weeks old, 20–22 g, IACUC protocol# 2018-10-TW-12) were obtained from Shanghai Laboratory Animal Center of the Chinese Academy of Sciences. The mice were reared under specific pathogen-free conditions with the period of 12 h of light/12 h of darkness, suitable temperature, and humidity. All mice were randomly allotted with standard laboratory fodder and drinking water, and acclimated in our facility for 1 week before the experiments began.
Mice were randomly divided into four groups (normal, vehicle receiving BLM and 0.5% carboxymethylcellulose sodium (Sigma-Aldrich, St. Louis, MO, USA) plus 0.25% Tween 80 (Sigma-Aldrich), drug treatment group receiving BLM and 25 mg/kg of apremilast, drug treatment group receiving BLM and 5 mg/kg of apremilast with 10 mice per group. BLM (TargetMol, Boston, USA) was dissolved in phosphate buffer saline (PBS), prepared into a stock solution of 300 μg/ml, and then disinfected by filtration. According to the experimental instructions, BLM (1.5 mg/kg) was injected subcutaneously with an insulin needle into the settled position on the shaved back skin of C57BL/6J mice, which was lasting for 4 weeks as previously described [16, 17]. Apremilast (Selleck, Shanghai, China) was dissolved in 0.5% carboxymethylcellulose sodium plus 0.25% Tween 80, and then mice were taken orally once a day for consecutive 4 weeks. In the end of treatment, all mice were euthanized and skin biopsies were collected for further analysis.
Histopathological assessment and immunohistochemistry
All skin biopsies were obtained from BLM-induced murine SSc model and fixed in 10% formaldehyde buffered in PBS solution. Immobilized skin slices were dewaxed and stained with hematoxylin and eosin (H&E) or Masson’s trichrome reagents to visualize the histopathological manifestations and deposition of collagens, respectively. Subsequently, the histopathological features were observed and scored under the light microscope (Olympus, Tokyo, Japan) by three researchers who were double-blind about experimental conditions. For immunohistochemistry, skin slices were incubated with anti-α-SMA antibodies (Cell Signaling Technology, Beverly, MA, USA) overnight at 4 °C and then stained with the secondary antibodies (R&D systems, Minneapolis, MN, USA) according to the instructions. The positive areas of α-SMA were observed under the light microscope. The images were analyzed and quantified using the Image-Pro Plus software (Media Cybernetics, Silver Springs, MD, USA).
Cell cultures and treatments
Bone-marrow-derived macrophages (BMDMs) were differentiated in the presence of macrophage colony-stimulating factor (M-CSF, PeproTech, Rocky Hill, NJ, USA) as previous description [18]. Briefly, the 6-week-old C57BL/6J mice were euthanized and the tibia and femur were disinfected to obtain bone marrow cells. The cells were dispersed in the Iscove’s Modified Dulbecco’s Medium (IMDM, Gibco, Grand Island, NY, USA), and then the Red Blood Cell Lysis Buffer (Beyotime, Shanghai, China) was added to deplete red blood cells. Subsequently, cells were cultured in IMDM containing 10% fetal bovine serum (FBS, Gibco), 10-ng/ml M-CSF, 100-U/ml penicillin, and 100-mg/ml streptomycin for 7 days. Non-adherent cells were removed and new growth media were added on day 3. To induce the polarization of macrophages in vitro, BMDMs were incubated with 10-ng/ml IFN-γ (PeproTech) plus 1-μg/ml lipopolysaccharide (LPS, Sigma-Aldrich), 10-ng/ml IL-4 (PeproTech) plus 10-ng/ml IL-13 (PeproTech) for M1 and M2 induction, respectively. Apremilast was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 50 mM. The stock solution of apremilast was diluted into the required concentration with the culture media, in which the final concentration of DMSO did not exceed 0.1% (v/v).
Polyclonal CD4+ T cells were separated from murine spleens by using EasySepTM mouse CD4+ T-Cell isolation kit (Stemcell, Vancouver, BC, Canada). In order to obtain the pure CD4+ T cells, immunomagnetic negative selection was performed according to the instructions. Purified CD4+ T cells were cultured with anti‐CD3 antibodies (5 μg/ml) plus anti‐CD28 antibodies (2 μg/ml, Thermo Fisher Scientific, Pittsburgh, PA, USA) in the presence or absence of apremilast at the indicated concentrations for 48 h. The cytokine levels in the supernatants were determined by ELISA.
Preparation of skin cell suspensions
The depilated back skins were snipped into small pieces and then digested in RPMI-1640 media (Gibco) containing 10% FBS, 2-mg/ml collagenase IV (Sigma-Aldrich), 1.5-mg/ml hyaluronidase (Sigma-Aldrich), and 0.03-mg/ml DNase I (Roche Applied Science) at 37 °C for 2 h [19]. After grinding, the skin cells were filtered through the 70-μm Falcon cell strainers (BD Biosciences, San Jose, CA, USA). Single cell suspensions were centrifugated at 1200 rpm for 5 min, resuspended in RPMI-1640 media, and used for the following flow cytometric analysis.
Flow cytometric analysis
Skin single cell suspensions were washed with cold PBS and stained with fixable viability dye eFluor™ 780 (eBioscience, San Diego, CA, USA) at 4 °C for 30 min to identify the viable cells from the dead cells. For staining the cell surface proteins, cell suspensions were incubated with FITC‐conjugated anti‐CD4 (BD Biosciences Cat# 553651), FITC‐conjugated anti‐Gr‐1 (Thermo Fisher Scientific Cat# 11‐5931‐81), FITC‐conjugated anti‐CD86 (BD Biosciences Cat# 561962), FITC-conjugated anti-CCR5 (abcam Cat# 258785–9), phycoerythrin (PE)‐conjugated anti‐CD25 (BD Biosciences Cat# 553866), PE‐conjugated anti‐F4/80 (BD Biosciences Cat# 565410), PE-conjugated anti-CD11c (BD Biosciences Cat# 553802), PE-conjugated anti-CXCR3 (BD Biosciences Cat# 562152), PE-conjugated anti-CD301 (Biolegend Cat# 145704), Percp‐Cy5.5‐conjugated anti‐CD11b (BD Biosciences Cat# 550993), Percp‐Cy5.5‐conjugated anti‐CD4 (BD Biosciences Cat# 550954), allophycocyanin (APC)‐conjugated anti‐CD4 (BD Biosciences Cat# 553051), APC‐conjugated anti-Ly6C (BD Biosciences Cat# 560595), APC‐conjugated anti-CD11b (Biolegend Cat# 101212), APC‐conjugated anti-CD206 (BD Biosciences Cat# 17-2061-82), brilliant violet 421 (BV421)‐conjugated anti‐CD3 (BD Biosciences Cat# 564008), BV421‐conjugated anti-CD11b (BD Biosciences Cat# 562605), or BV421‐conjugated anti-CX3CR1 (Biolegend Cat# 149023). For intracellular staining, cells were fixed, permeabilized, and then labeled with PE‐conjugated anti‐IL‐17A (BD Biosciences Cat# 559502), Percp‐Cy5.5‐conjugated anti-IL-4 (BD Biosciences Cat# 560700), Alexa Fluor 647‐conjugated anti-p-CREB (Cell Signaling Technology Cat# 14001S), or Percp‐Cy5.5‐conjugated anti‐Foxp3 antibodies (Thermo Fisher Scientific Cat# 45‐5773‐82). The data were further analyzed by the FlowJo software (Tree Star, Ashland, OR, USA).
SiRNA interference
To decrease the expression of CREB in BMDMs, siRNA sequences targeting CREB (RiboBio, Shanghai, China) were designed and transfected according to the instructions. Briefly, siRNA was mixed with Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific) in serum-free Opti-MEM media and then transfected into the BMDMs. Cells were collected for further validation after 72-h transfection.
Immunofluorescence analysis
BMDMs were grown on the coverslips, fixed with 4% paraformaldehyde for 30 min, and permeabilized with 1% Triton X-100 for 10 min. After washing with PBS for three times, blocking buffer (Beyotime) was incubated for 1 h to block the nonspecific bindings. Subsequently, the coverslips were stained with anti-p-CREB antibody or anti-p-NF-κB antibody (Cell Signaling Technology) overnight at 4 °C. Primary labellings for unconjugated fluorescein were detected by FITC-conjugated secondary antibodies and then counterstained with DAPI. The representative immunofluorescent images were captured under the Leica TCS SPS microscope (Wetzlar, Germany).
Cytokines analysis by ELISA
Skin samples were cut from the frozen back skins in each group, homogenized, and then centrifuged at 12,000 rpm for 10 min. The supernatants were collected, and BCA Protein Assay Kits (Thermo Fisher Scientific) were used to determine the concentration of total proteins. The levels of TNF-α, IL-6, IL-12, and IL-17 in skin homogenates and cell culture media were quantified by the ELISA kits (BD Pharmingen) according to the instructions.
Western blot analysis
Cells and skin samples were lysed in SDS Lysis Buffer (Beyotime) in the presence of protease inhibitors. Total proteins were uniformed by using BCA Protein Assay Kits (Thermo Fisher Scientific). The protein samples were mixed with the loading buffers, subjected to the SDS-PAGE gels, and then transferred onto the nitrocellulose membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK). Nonspecific bindings were blocked with SuperBlockTM T20 Blocking Buffer (Thermo Fisher Scientific). Subsequently, the membranes were incubated with the primary antibodies, including anti-α-SMA, anti-Smad4, anti-p-Smad3, anti-CREB, anti-p-CREB, anti-NF-κB, anti-p-NF-κB, anti-ERK, anti-p-ERK, anti-MAPK, anti-p-MAPK, anti-PI3K, anti-p-PI3K, anti-GAPDH, or anti-Tubulin antibodies (Cell Signaling Technology) overnight at 4 °C. The HRP-conjugated secondary antibodies were added and incubated for 1 h at room temperature. The positive signals were detected by ChemiDoc™ MP Imaging System (Bio‐Rad, Hercules, CA, USA).
RNA extraction, cDNA synthesis, and real-time PCR
Total RNA in skin tissues and BMDMs was extracted by the RNAsimple total RNA kits (Tiangen, Beijing, China) and then reversely transcribed by cDNA Synthesis SuperMix (Biotool, Houston, TX, USA). For performing real-time PCR experiments, SYBR Green Real-time PCR Master Mix (TOYOBO, Osaka, Japan) was used on an Applied Biosystems 7500 Fast Real‐Time PCR System (Applied Biosystems, Foster city, CA, USA). β-actin acted as the housekeeping gene and fold changes of the gene expression were normalized to β-actin using the ΔΔCt method.
cAMP determination
Frozen skin tissues were homogenized in 10 volumes of 0.1-M HCl and centrifuged at 600 × g for 10 min to pellet the debris. The supernatants were collected for further analysis. Enzo Direct cAMP ELISA kits (Enzolifesciences, New York, USA) were used to determine the content of cAMP in skin lesions according to the instructions.
Statistical analysis
All data were presented as mean ± SEM. One-way ANOVA followed by Dunnett’s multiple comparison test was applied to determine the significant difference by using the GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered to represent a significant difference.
Results
Inhibition of PDE4 by apremilast attenuated the pathological manifestations in BLM-induced murine skin fibrosis
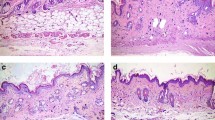
To uncover the therapeutic capacity of inhibition of PDE4, oral administration of apremilast was conducted in BLM-induced SSc. As illustrated in Fig. 1a, mice from the vehicle group generally displayed more black patches around the injection locations than the normal individuals, which were proven to be associated with the disease severity and hair follicle damage in the inflamed sections [20]. Consistently, apremilast could dramatically attenuate the pathological manifestations in a dose-dependent manner (Fig. 1a). In the histopathological assessment by H&E and Masson staining, compared with the vehicle mice, the histological scores, dermal thickening, and deposition of collagens were significantly reversed upon apremilast treatment (Fig. 1b–e). Moreover, the immunohistochemistry results revealed that activated fibroblasts, indicated by α-SMA positive staining, were largely scattered in the dermal layers, and decreased in the apremilast-treated group (Fig. 1c, e). Collectively, our findings demonstrated that inhibition of PDE4 by apremilast exerted a curative effect on the BLM-induced skin fibrosis. To fully illustrate the underlying mechanism of apremilast in skin fibrosis, mice treated with 25 mg/kg of apremilast were selected in the following investigations.

a Representative back skin phenotypes of BLM-induced SSc. b Histological scores. c Representative skin sections with H&E staining (top, ×100 magnification), Masson’s trichrome staining (middle, ×100 magnification), and immunohistochemical staining of α-SMA (bottom, ×100 magnification). d Dermal thickness from each group. e Quantification of collagen volume fraction and α-SMA positive cells. Data were shown as mean ± SEM; n = 10 mice per group. ***P < 0.001, significantly different from vehicle (BLM only) group.
Inhibition of PDE4 by apremilast suppressed the expression of pro-fibrotic factors in BLM-induced murine skin fibrosis
Given that apremilast could retard the process of skin fibrosis, we further explored whether apremilast directly exhibited modulatory effects on the expression of pro-fibrotic factors. Previous reports have suggested that multiple signaling pathways were involved in the development and deterioration of tissue fibrosis, in which TGF-β/Smads signaling pathway functioned as the dominant element [21]. In line with the pathological phenotypes, apremilast could obviously decrease the mRNA level of TGF-β (Fig. 2a) and modulate the expression and phosphorylation of Smad proteins, as evidenced by decreasing the expression of Smad4 and phospho-Smad3 and increasing the expression of Smad7 (Fig. 2b). Consequently, apremilast downregulated the expression of several types of collagens, including Col1a1, Col13a1, and Col4a1 (Fig. 2c). Moreover, increased expression of α-SMA, fibroblast activation protein (FAP), fibronectin, platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and Vimentin were observed in vehicle mice and obviously suppressed following apremilast treatment (Fig. 2d, e). Taken together, we found that inhibition of PDE4 suppressed the deposition of ECMs and collagens through regulating the classic TGF-β/Smads signaling pathway.

a The mRNA level of TGF-β in skin samples. b Western blotting assay and quantification analysis of α-SMA, Smad4, Smad7, and p-Smad3. c The mRNA levels of collagen subtypes, Col1a1, Col3a1, and Col4a1, in skin samples. d The mRNA levels of α-SMA, FAP, and Fibronectin in skin samples. e The mRNA levels of PDGF, VEGF, and Vimentin in skin samples. Data were shown as mean ± SEM; n = 10 mice per group. *P < 0.05, **P < 0.01, and ***P < 0.001, significantly different from vehicle (BLM only) group.
Inhibition of PDE4 by apremilast regulated the expression and downstream signaling transduction of PDE4
Previous studies have suggested that inhibition of PDE4 could modulate the expression of PDE4 in inflammatory bowel diseases and psoriasis [22, 23]. In the present research, we found that in BLM-induced skin fibrosis, apremilast specifically decreased the mRNA expression of PDE4A, PDE4B, and PDE4C subtypes (Fig. 3a), and subsequently promoted the accumulation of cAMP in the inflamed skins (Fig. S1). Consistently, apremilast exerted the capacity on promoting the phosphorylation of CREB in CD3+ T cells and CD11b+ myeloid cells (Fig. 3b). The function of CREB was mediated by the cAMP–PDE4–PKA axis, and phosphorylation of CREB was closely related to the transcriptional regulation of several intracellular events [24]. Herein, protein expression of CREB in BMDMs was knocked down to explore how inhibition of PDE4 affected the activation of macrophages. As illustrated in Fig. 3c, three CREB-targeting siRNAs were designed and transfected into BMDMs and the expression of CREB was largely reduced. In line with our hypothesis, inhibition of PDE4 by apremilast could increase the phosphorylation of CREB, which was nearly impeded upon siCREB transfection (Fig. 3c, bottom). The immunofluorescence further confirmed that increased distribution of phospho-CREB in nuclei was weakened by siCREB intervention (Fig. 3d). In terms of inflammatory mediators’ synthesis, apremilast significantly inhibited the expression level of TNF-α, iNOS, and IL-12; whereas, the effects of apremilast were aborted in siCREB-transfected BMDMs (Fig. 3e). In brief, these findings demonstrated that CREB functioned as the critical element for apremilast in modulating the inflammatory responses in macrophages.

a The mRNA expression level of PDE4 isoforms in skin lesions. b The percentage of p-CREB in T cells (CD3+) and myeloid cells (CD11b+) in skin cell suspensions. c Western blotting analysis of CREB and phosphorylation of CREB in BMDMs. d Representative images of nuclear location of phospho-CREB in BMDMs, assayed by immunofluorescence. e Transfected BMDMs were incubated with apremilast in the presence of LPS for 3 h and the mRNA level of TNF-α, iNOS, and IL-12 was determined by RT-PCR. Data were shown as mean ± SEM; a, b n = 10 mice per group; c–e n = 3. *P < 0.05, **P < 0.01, and ***P < 0.001.
Inhibition of PDE4 by apremilast decreased the infiltration of inflammatory monocytes and the polarization of macrophages
Given the anti-inflammatory effects of apremilast on macrophages, further investigations in the inflamed skin lesions were performed by flow cytometry. Compared with the vehicle group, apremilast decreased the infiltration of myeloid cells (CD11b+), monocytes (CD11b+Ly6C+), dendritic cells (CD11b+CD11c+), macrophages (CD11b+F4/80+), and neutrophils (CD11b+Ly6G+) in the skin lesions (Fig. 4a), as well as the expression of chemokine receptors, CCR2, and CX3CR1 on CD11b+ cells (Fig. 4b). In addition, the percentage of macrophage subtypes M1 (F4/80+CD86+) and M2 (F4/80+CD206+) was also decreased in the skin lesions upon apremilast treatment (Fig. 4c), which were further confirmed by the expression of transcription factors, IRF5, Arg-1, and Ym-1 (Fig. 4d). To verify the outcomes in vitro, BMDMs were differentiated and incubated with LPS plus IFN-γ and IL-4 plus IL-13 to induce the polarization of M1 and M2, respectively. The results in Fig. 4e demonstrated that apremilast displayed a concentration-dependent efficacy on restraining the polarization of M1, while there seemed a negligible effect on the polarization of M2 in vitro. Correspondingly, apremilast could decrease the mRNA and protein level of TNF-α and IL-12p40, the inflammatory cytokines from M1 cells, in a concentration-dependent manner (Fig. 4f). The similar results were observed in the peritoneal-derived macrophages (Fig. 4g). Collectively, we found that inhibition of PDE4 by apremilast could suppress the recruitment of inflammatory monocytes, especially the M1 subtypes.

a The percentage of myeloid cells (CD11b+), monocytes (CD11b+Ly6C+), dendritic cells (CD11b+CD11c+), macrophages (CD11b+F4/80+), neutrophils (CD11b+Gr‐1+), assayed by flow cytometry. b The expression level of CCR2 and CX3CR1 on CD11b+ cells. c The expression level of CD86 and CD206 on F4/80+ cells. d The mRNA levels of IRF5, Arg-1, and Ym-1 in skin lesions. e BMDMs were polarized into M1 or M2 cells in the presence of apremilast and the population of M1 and M2 cells were assayed by flow cytometry. f The mRNA and protein levels of TNF-α and IL-12p40 in BMDMs. g The mRNA and protein levels of TNF-α and IL-12p40 in peritoneal macrophages. Data were shown as mean ± SEM; a–d n = 10 mice per group; e–g n = 3. *P < 0.05, **P < 0.01, and ***P < 0.001.
Inhibition of PDE4 by apremilast inhibited the infiltration and activation of T cells
In addition to macrophages, T cells functioned as the predominant immune cells to produce IFN-γ, IL-2, IL-4, and IL-17, which subsequently aggravated the process of skin fibrosis [25]. Herein, flow cytometry analysis showed that inhibition of PDE4 by apremilast significantly reduced the population of T-helper cells, including Th1, Th2, and Th17 cells (Fig. 5a). Moreover, reductions of activated T cells and Treg cells were also observed in the apremilast-treated mice (Fig. 5b). Conformably, compared to the vehicle group, transcriptional factors associated with T cells differentiation were downregulated upon apremilast treatment (Fig. 5c). Furthermore, the expression of CCR5 and CXCR3 on T cells was also suppressed by apremilast (Fig. 5d). To confirm the fact that inhibition of PDE4 by apremilast exerted modulatory effects on the function of T cells, CD4+ T cells were purified by magnetic beads and cultured in vitro with the anti-CD3/CD28 antibodies. We found that apremilast could inhibit the expression of CD25 and CD69 and production of IFN-γ and IL-2 from activated T cells in a dose-dependent manner (Fig. 5e, f).

a The percentage of Th1, Th2, and Th17 cells in skin lesions was measured by flow cytometry. b The percentage of activated T cells and Treg cells in skin lesions. c The mRNA expression of transcription factors, related with T cells differentiation. d The expression level of CCR5 and CXCR3 on T cells. e CD4+ T cells were purified and incubated with anti‐CD3/CD28 antibodies in the presence of apremilast. The cells were collected for determining the expression of CD25 and CD69; meanwhile, the supernatants were quantified for IFN-γ and IL-2 (f). Data were shown as mean ± SEM; a–d n = 10 mice per group; e, f n = 3. *P < 0.05, **P < 0.01, and ***P < 0.001.
Inhibition of PDE4 by apremilast suppressed the inflammatory responses in BLM-induced murine skin fibrosis
In keeping with the activation of skin fibroblasts, macrophages, and T cells, inflammatory responses occurred in the inflamed locations in BLM-induced skin fibrosis. Increased expression of adhesion molecules, including ICAM-1, MadCAM-1, and E-selectin, were observed in the vehicle group and relieved by apremilast intervention (Fig. 6a). Meanwhile, the mRNA and protein level of certain pro-inflammatory cytokines, TNF-α, IL-6, and IL-17A, were consistently decreased in the apremilast-treated mice (Fig. 6b, c). In line with our previous reports, inhibition of PDE4 by apremilast could directly enhance the phosphorylation of CREB and spontaneously interfere with the activation of pro-inflammatory signaling pathways, such as NF-κB, ERK, MAPK, and PI3K signaling (Fig. 6d, e), which were further confirmed by the immunofluorescent staining of phosphor-NF-κB in the nuclei of BMDMs (Fig. S2). Taken together, inhibition of PDE4 by apremilast exerted protective effects on BLM-induced skin fibrosis, which were mainly due to the suppression of activation of M1 and T cells.

a The mRNA expression of adhesive molecules in skin lesions. b The protein level of inflammatory cytokines in skin lesions, assayed by ELISA. c The mRNA level of inflammatory cytokines in skin lesions. Western blotting analysis of cAMP-dominant phosphorylation of CREB (d) and downstream inflammatory signaling pathways (e). f The mRNA level of IFN-γ, IL-4, and IL-13 in skin lesions. Data were shown as mean ± SEM; n = 10 mice per group. *P < 0.05, **P < 0.01, and ***P < 0.001, significantly different from vehicle (only BLM) group.
Discussion
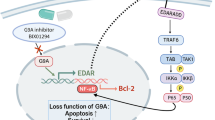
SSc is an autoimmune disease with a relatively high prevalence of disability and characterized by excessive activation of immune responses and pathological exacerbation of tissue fibrosis [26]. Given the stubborn manifestations, there remains a great need for developing novel therapeutic strategies for SSc patients [27]. Accumulating studies have demonstrated that multiple factors, such as genetic susceptibility, environmental pathogens, mechanical stress, and chronic inflammation, were linked with the pathogenesis of SSc [28]. With a wide distribution in various cells, PDE4 acts as an intracellular modulator of cAMP signalosomes, which are involved in modulating the differentiation and function of inflammatory immune cells [29]. During the prolonged development and optimization for PDE4 inhibitors, apremilast, approved for the treatment of psoriasis, attracted the enthusiasm of numerous researchers [30]. Due to the intrinsic expression of PDE4 in immune cells, fibroblasts, and myofibroblasts, PDE4 inhibitors were attempted to be applied in the treatment of SSc [31]. In the previous research, specific inhibition of PDE4 by rolipram was investigated in experimental murine skin fibrosis model, which indicated that rolipram could attenuate the activation of fibroblasts and deposition of collagens [15]. Whereas, the therapeutic effects of apremilast on skin fibrosis and the modulatory mechanism of inhibition of PDE4 on macrophages and T cells remained ill-defined. In the present study, we aimed to uncover the efficacy and the underlying mechanism of apremilast in the process of BLM-induced murine skin fibrosis. Herein, we found that oral administration of apremilast could prevent dermal fibrosis through reducing deposition of collagens and activation of myofibroblasts, which were largely due to the direct suppression of the polarization and function of M1-type macrophages and T cells (Fig. 7).

Inhibition of PDE4 by apremilast exerted protective effects on the experimental skin fibrosis, as evidenced by alleviating the progression of disease deterioration and decreasing the activation of skin fibroblasts by modulating the TGF-β/Smads signaling pathway. Moreover, apremilast could suppress the infiltration and polarization of macrophages and T cells in the inflamed skin tissues, which were largely due to the activation of cAMP-predominant PKA-CREB pathway.
BLM-induced murine SSc model is characterized by obstinate fibrosis and overactivation of immune cells and could extensively simulate the clinical features of human SSc [32]. It has been identified that fibroblasts obtained from the inflamed skin lesions exhibited more potential to produce various types of collagens than healthy individuals [33]. The present research confirmed that skin biopsies from BLM-induced SSc displayed several pathological parameters, such as tightness and stiffness, as well as increased deposition of collagens, activation of fibroblasts, and dermal thickening (Figs. 1 and 2), in which several signaling pathways, including Wnt and TGF-β/Smads, were closely related to the activation and proliferation of fibroblasts in skin fibrosis [34, 35]. Thereinto, TGF-β served a key growth factor directly inducing the destruction of ECM and upregulation of α-SMA through modulating the phosphorylation of downstream proteins, Smad2, and Smad3 [36]. Blockade or intervention of TGF-β signaling has been identified to be effective in attenuating fibrogenesis in liver, kidney, lung, heart, and skin [37]. Consistently, inhibition of PDE4 by apremilast could significantly attenuate the disease severity of SSc (Fig. 1) and reverse the pro-fibrotic process by modulating the signaling transduction of TGF-β/Smads pathway (Fig. 2a–c).
In the skin microenvironments, innate and adaptive immune responses played the vital parts in initiation and deterioration of inflammation and fibrosis in SSc [38], among which activation and polarization of macrophages and T cells have been proven to participate in the key process [39]. The lineage classification and plasticity of macrophages were the key aspects for the function of macrophages [40]. Mounting evidences have implicated that upon exposure of multiple mediators, tissue-resident and newly recruited macrophages could differentiate into classically inflammatory macrophages (M1) and immune-regulating macrophages (M2), which were intriguingly implicated in the pathogenesis of skin fibrosis [40]. On the other hand, there existed a precise communication between macrophages and T cells. In line with the plasticity of macrophages, differentiation and activation of T cells are well-known as the capacity of secreting inflammatory cytokines, IFN-γ, IL-4, and IL-17, and also closely related to the disease severity of SSc [41]. Meanwhile, during the deterioration of SSc, increased level of TGF-β from activated macrophages could subsequently promote the differentiation of Tregs, which were the main source of IL-10 and thought to drive the fibrosis processes [41]. The function and mechanism of Tregs in the process of fibrosis are tissue and context-dependent. In addition to interfering with the Th2 cells, Tregs could also express TGF-β, which not only induced differentiation and maturation of Tregs, but also promoted the activation of fibroblasts [42, 43]. In our research, we found that apremilast could largely impeded the production of TGF-β in the inflamed skin tissues, which indicated that the inducer for Tregs was reduced and might account for the fact that apremilast decreased the population of Tregs. Further investigations revealed that inhibition of PDE4 by apremilast could suppress the infiltration and differentiation of T cells (Fig. 5), which in turn directly inhibited the polarization of M1 cells in a concentration-dependent manner (Figs. 4e–g and 6f). Indeed, many studies have shown that M2 cells played an important role in promoting the activation of fibroblasts. However, compared with the vehicle controls, decreased population of M2 cells were observed in apremilast-treated mice (Fig. 4c, d), which might be attributed to the suppressive effects of apremilast on the function of Th2 cells. Moreover, as demonstrated in Fig. 6f, inhibition of PDE4 could dramatically decrease the production of IL-4 and IL-13, which were the key inducers of M2 cells. Taken together, we speculated that the inhibition of PDE4 by apremilast downregulated the inducers of M2 differentiation without directly interfering with the differentiation of M2 cells.
Numerous clinical trials have designed to expand the therapeutic indications of apremilast for inflammatory and autoimmune diseases [44]. Targeting the regulation of intracellular second messengers has identified to show effects on interrupting the inflammatory cascades at the earlier point [22, 45]. Upregulation of intracellular cAMP could functionally activate PKA, cyclic nucleotide gated ion channels, and exchange protein activated by cAMP [46], which subsequently led to the phosphorylation of cAMP-responsive element binding transcription factors, including CREB and cAMP-responsive element modulator (CREM) [47]. In keeping with previous reports, upon cAMP elevation by inhibiting PDE4 enzymatic activity, phosphorylation of CREB was increased following apremilast treatment (Figs. 3c, d and 6d), which resulted in the suppression of activation of macrophages (Fig. 4e, f). Nevertheless, the effects of apremilast on macrophages could be largely restricted following siCREB transfection (Fig. 3).
This study presented that inhibition of PDE4 by apremilast exerted properties to improve the pathological features in BLM-induced murine skin fibrosis, as evidenced by alleviating the progression of disease deterioration and decreasing the activation of fibroblasts through regulating the TGF-β/Smads signaling pathway. Further study demonstrated that inhibition of PDE4 by apremilast suppressed the infiltration and polarization of macrophages and T cells and inhibited the production of pro-inflammatory cytokines in the inflamed skin lesions. Notably, the effects of apremilast on the differentiation of M1-type macrophages and T-helper cells depended on the phosphorylation of CREB. In brief, the present research provided critical evidence that inhibition of cAMP-dominant PDE4 might be selected for therapeutic intervention in pathological processes of skin fibrosis.
References
Sundaram SM, Chung L. An update on systemic sclerosis-associated pulmonary arterial hypertension: a review of the current literature. Curr Rheumatol Rep. 2018;20:10.
Furue M, Mitoma C, Mitoma H, Tsuji G, Chiba T, Nakahara T, et al. Pathogenesis of systemic sclerosis-current concept and emerging treatments. Immunol Res. 2017;65:790–7.
Rockey DC, Bell PD, Hill JA. Fibrosis-a common pathway to organ injury and failure. N Engl J Med. 2015;372:1138–49.
Toledo DM, Pioli PA. Macrophages in systemic sclerosis: novel insights and therapeutic implications. Curr Rheumatol Rep. 2019;21:31.
Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Prim. 2015;1:15002.
Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425–40.
Phan AT, Goldrath AW, Glass CK. Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity. 2017;46:714–29.
Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Mol Asp Med. 2019;65:2–15.
Raker VK, Becker C, Steinbrink K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front Immunol. 2016;7:123.
Houslay MD, Baillie GS, Maurice DH. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res. 2007;100:950–66.
Burdyga A, Surdo NC, Monterisi S, Di Benedetto G, Grisan F, Penna E, et al. Phosphatases control PKA-dependent functional microdomains at the outer mitochondrial membrane. Proc Natl Acad Sci U S A. 2018;115:E6497–E506.
Li H, Zuo J, Tang W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol. 2018;9:1048.
Keating GM. Apremilast: a review in psoriasis and psoriatic arthritis. Drugs. 2017;77:459–72.
Schafer PH, Parton A, Gandhi AK, Capone L, Adams M, Wu L, et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–55.
Maier C, Ramming A, Bergmann C, Weinkam R, Kittan N, Schett G, et al. Inhibition of phosphodiesterase 4 (PDE4) reduces dermal fibrosis by interfering with the release of interleukin-6 from M2 macrophages. Ann Rheum Dis. 2017;76:1133–41.
Liang M, Lv J, Zou L, Yang W, Xiong Y, Chen X, et al. A modified murine model of systemic sclerosis: bleomycin given by pump infusion induced skin and pulmonary inflammation and fibrosis. Lab Invest. 2015;95:342–50.
Yamamoto T, Takagawa S, Katayama I, Yamazaki K, Hamazaki Y, Shinkai H, et al. Animal model of sclerotic skin. I: Local injections of bleomycin induce sclerotic skin mimicking scleroderma. J Invest Dermatol. 1999;112:456–62.
Sun C, Sun L, Ma H, Peng J, Zhen Y, Duan K, et al. The phenotype and functional alterations of macrophages in mice with hyperglycemia for long term. J Cell Physiol. 2012;227:1670–9.
Huu DL, Matsushita T, Jin G, Hamaguchi Y, Hasegawa M, Takehara K, et al. FTY720 ameliorates murine sclerodermatous chronic graft-versus-host disease by promoting expansion of splenic regulatory cells and inhibiting immune cell infiltration into skin. Arthritis Rheum. 2013;65:1624–35.
Slominski A, Zbytek B, Nikolakis G, Manna PR, Skobowiat C, Zmijewski M, et al. Steroidogenesis in the skin: implications for local immune functions. J Steroid Biochem Mol Biol. 2013;137:107–23.
Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol. 2011;8:42–54.
Li H, Li J, Zhang X, Feng C, Fan C, Yang X, et al. DC591017, a phosphodiesterase-4 (PDE4) inhibitor with robust anti-inflammation through regulating PKA-CREB signaling. Biochem Pharmacol. 2020;177:113958.
Li H, Fan C, Feng C, Wu Y, Lu H, He P, et al. Inhibition of phosphodiesterase-4 attenuates murine ulcerative colitis through interference with mucosal immunity. Br J Pharmacol. 2019;176:2209–26.
Kelly MP. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal. 2018;42:281–91.
Taylor AE, Carey AN, Kudira R, Lages CS, Shi T, Lam S, et al. Interleukin 2 promotes hepatic regulatory T cell responses and protects from biliary fibrosis in murine sclerosing cholangitis. Hepatology. 2018;68:1905–21.
Meng M, Tan J, Chen W, Du Q, Xie B, Wang N, et al. The fibrosis and immunological features of hypochlorous acid induced mouse model of systemic sclerosis. Front Immunol. 2019;10:1861.
Cutolo M, Soldano S, Smith V. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol. 2019;15:753–64.
Henderson J, Distler J, O’Reilly S. The role of epigenetic modifications in systemic sclerosis: a druggable target. Trends Mol Med. 2019;25:395–411.
Arumugham VB, Baldari CT. cAMP: a multifaceted modulator of immune synapse assembly and T cell activation. J Leukoc Biol. 2017;101:1301–16.
Reed M, Crosbie D. Apremilast in the treatment of psoriatic arthritis: a perspective review. Ther Adv Musculoskelet Dis. 2017;9:45–53.
Onuora S. Systemic sclerosis: antifibrotic effects of PDE4 blockade? Nat Rev Rheumatol. 2017;13:198.
Elhai M, Avouac J, Kahan A, Allanore Y. Systemic sclerosis: recent insights. Jt Bone Spine. 2015;82:148–53.
Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, et al. The good and the bad collagens of fibrosis—their role in signaling and organ function. Adv Drug Deliv Rev. 2017;121:43–56.
Lichtenberger BM, Mastrogiannaki M, Watt FM. Epidermal beta-catenin activation remodels the dermis via paracrine signalling to distinct fibroblast lineages. Nat Commun. 2016;7:10537.
Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors. 2011;29:196–202.
Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–9.
Morikawa M, Derynck R, Miyazono K. TGF-beta and the TGF-beta family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8:1–25.
Do NN, Eming SA. Skin fibrosis: models and mechanisms. Curr Res Transl Med. 2016;64:185–93.
Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68-69:106–21.
Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS−) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084.
Zhang Z, Wu Y, Wu B, Qi Q, Li H, Lu H, et al. DZ2002 ameliorates fibrosis, inflammation, and vasculopathy in experimental systemic sclerosis models. Arthritis Res Ther. 2019;21:1–15.
Kalekar LA, Rosenblum MD. Regulatory T cells in inflammatory skin disease: from mice to humans. Int Immunol. 2019;31:457–63.
Kalekar LA, Cohen JN, Prevel N, Sandoval PM, Mathur AN, Moreau JM, et al. Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci Immunol. 2019;4:eaaw2910.
Guttman-Yassky E, Hanifin JM, Boguniewicz M, Wollenberg A, Bissonnette R, Purohit V, et al. The role of phosphodiesterase 4 in the pathophysiology of atopic dermatitis and the perspective for its inhibition. Exp Dermatol. 2019;28:3–10.
Zhang X, Dong G, Li H, Chen W, Li J, Feng C, et al. Structure-aided identification and optimization of tetrahydro-isoquinolines as novel PDE4 inhibitors leading to discovery of an effective antipsoriasis agent. J Med Chem. 2019;62:5579–93.
Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3’,5’-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discov. 2019;18:770–96.
Delyon J, Servy A, Laugier F, Andre J, Ortonne N, Battistella M, et al. PDE4D promotes FAK-mediated cell invasion in BRAF-mutated melanoma. Oncogene. 2017;36:3252–62.
Acknowledgements
This work was granted by the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program,” China (2018ZX09711002–006–011), CAS Key Laboratory of Receptor Research (SIMM1904YKF-01), Science & Technology Commission of Shanghai Municipality, China (No. 18431907100), and “Personalized Medicines-Molecular Signature-based Drug Discovery and Development,” Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12020231).
Author information
Authors and Affiliations
Contributions
QKL, HML, and WT designed research and contributed to the conception. QKL, CF, CGX, BW, HML, CLF, XQY, and HL performed research. QKL and HL analyzed data. QKL, HL, and WT wrote the paper. All the authors contributed to the collection and interpretation of data and approved the final draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Lu, Qk., Fan, C., Xiang, Cg. et al. Inhibition of PDE4 by apremilast attenuates skin fibrosis through directly suppressing activation of M1 and T cells. Acta Pharmacol Sin 43, 376–386 (2022). https://doi.org/10.1038/s41401-021-00656-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00656-x
