
Abstract
Mantle cell lymphoma (MCL) is a lymphoproliferative disorder lacking reliable therapies. PI3K pathway contributes to the pathogenesis of MCL, serving as a potential target. However, idelalisib, an FDA-approved drug targeting PI3Kδ, has shown intrinsic resistance in MCL treatment. Here we report that a p300/CBP inhibitor, A-485, could overcome resistance to idelalisib in MCL cells in vitro and in vivo. A-485 was discovered in a combinational drug screening from an epigenetic compound library containing 45 small molecule modulators. We found that A-485, the highly selective catalytic inhibitor of p300 and CBP, was the most potent compound that enhanced the sensitivity of MCL cell line Z-138 to idelalisib. Combination of A-485 and idelalisib remarkably decreased the viability of three MCL cell lines tested. Co-treatment with A-485 and idelalisib in Maver-1 and Z-138 MCL cell xenograft mice for 3 weeks dramatically suppressed the tumor growth by reversing the unsustained inhibition in PI3K downstream signaling. We further demonstrated that p300/CBP inhibition decreased histone acetylation at RTKs gene promoters and reduced transcriptional upregulation of RTKs, thereby inhibiting the downstream persistent activation of MAPK/ERK signaling, which also contributed to the pathogenesis of MCL. Therefore, additional inhibition of p300/CBP blocked MAPK/ERK signaling, which rendered maintaining activation to PI3K-mTOR downstream signals p-S6 and p-4E-BP1, thus leading to suppression of cell growth and tumor progression and eliminating the intrinsic resistance to idelalisib ultimately. Our results provide a promising combination therapy for MCL and highlight the potential use of epigenetic inhibitors targeting p300/CBP to reverse drug resistance in tumor.
Similar content being viewed by others


Introduction
Mantle cell lymphoma (MCL) is a non-Hodgkin lymphoma (NHL) of the pregerminal center of mature B cells characterized by aggressive clinical courses and poor survival. The 10-year survival rate is only 5%–10%, and the median overall survival time is only ~3 years, which is the shortest among all subtypes of NHL [1]. MCL remains incurable due to the paucity of reliable curative therapies [2]. Surgery is rarely indicated, and unlike most NHLs, MCL typically progresses after chemotherapy; the median time to treatment failure is less than 18 months [3]. Despite the marked success of targeted therapy, the treatment outcomes of MCL patients remain poor due to the emergence of drug resistance and the unresolved mechanisms underlying resistance [4]. Identifying the molecular pathways resulting in resistance is pivotal for the treatment of not only MCL but also some other malignancies.
Numerous studies have identified that activation of the phosphoinositide-3 kinase (PI3K) signaling axis, one of the most frequently hyperactivated pathways in human malignancies [5], contributes to the pathogenesis of MCL, and this pathway has become a potential therapeutic target [6,7,8]. RAC-alpha serine/threonine-protein kinase (AKT) phosphorylation often serves as a surrogate readout for the PI3K signaling pathway [9]. PI3K and AKT are involved in the activation of mammalian target of rapamycin (mTOR) and mediate the phosphorylation cascade of multiple downstream effectors. P70 ribosomal protein S6 kinase (S6K) and eIF4E-binding proteins (4E-BPs) are direct substrates that control cell proliferation and survival by regulating protein and nucleotide synthesis [10]. The PI3K family is divided into four different classes. In mammals, class I PI3Ks have 4 catalytic subunit isoforms, p110α, p110β, p110δ, and p110γ, that dimerize with the p85 regulatory subunit. The p110δ isoform is a key messenger and is highly enriched in cells of hematologic origin, predominantly leukocytes [11]. Dysfunction of the PI3K p110δ isoform is implicated in B cell malignancies. Therefore, PI3Kδ is considered an attractive drug target. However, in some lymphomas, the clinical outcomes have not been satisfactory. Idelalisib (Zydelig), an approved PI3Kδ inhibitor, has been reported to produce limited responses in some NHLs, including MCL [12]. In particular, preclinical studies reported limited activity toward MCL cells with a half-maximal inhibitory concentration (IC50) higher than 10 μM in proliferation assays [13], suggesting intrinsic resistance to idelalisib treatment and PI3Kδ inhibition. In addition, a large phase I trial of idelalisib in individuals with hematologic malignancies reported inferior responses in MCL patients compared with other patient subsets [12]. In addition to the disappointing clinical outcomes, the reason underlying the poor response to idelalisib in some hematologic malignancies remains elusive. Given the limited efficacy of idelalisib as a monotherapy, combinatorial studies are ongoing to identify alternative treatment options and characterize the underlying mechanisms of resistance.
Epigenetic modifications are generally observed in all of the recognized hallmarks of cancer and underlie multiple necessary biological events during tumorigenesis and tumor progression. In particular, epigenetic dysregulation makes an important contribution to the formation and progression of MCL. For example, some tumor suppressor genes are silenced by epigenetic modifications and are involved in the pathogenesis of MCL. Whole-genome expression profiling of MCL cell lines identified the hypermethylation in promoter regions of suppressor genes [14]. In addition, epigenetic modifications are involved in the deregulation of cyclin D1 in MCL. For example, abnormal histone acetylation and DNA methylation were found in the cyclin D1 promoter in MCL [15, 16]. In clinical trials, the histone deacetylase inhibitor vorinostat and hypomethylating agent cladribine have been used as part of combination regimens in MCL treatment [17,18,19].
In recent years, it has become clear that epigenetic dysfunction not only is a well-established driver of cancers but also mediates intrinsic and acquired resistance to targeted therapies [20, 21]. However, the epigenetic mechanisms underlying drug resistance have not been fully elucidated, although some patterns of how epigenetic modifications ultimately drive bypass resistance mechanisms have been reported [21,22,23]. In particular, no studies have determined the role of epigenetic modification in mediating resistance to PI3Kδ inhibitors. It is important to investigate whether epigenetic regulation engages in crosstalk with the PI3K pathway and to discover new strategies for overcoming resistance to PI3Kδ inhibitors. In this study, we explored the epigenetic mechanism of resistance to an approved PI3Kδ inhibitor, idelalisib, in MCL and identified a new combination therapy containing an epigenetic agent that sensitizes MCL to idelalisib.
Materials and methods
Cell lines and cell culture
Human MCL cell lines, including Z-138, Maver-1, and Jeko-1, were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Human normal cell lines, including HUVECs and RPTECs, were purchased from ATCC. The human normal cell line L02 was a gift from Xin Hua Hospital Affiliated with Shanghai Jiao Tong University School of Medicine. Cells were cultured in the appropriate medium following the instructions of ATCC and maintained at 37 °C in a 5% CO2 incubator. Cell lines were authenticated by short tandem repeat (STR) profiling at regular intervals. Mycoplasma detection was routinely performed throughout the study.
Cell viability assays and combinatorial drug screening
Experiments were carried out in 96-well plates in triplicate. The viable cell number in each well was determined using CellTiter-Glo reagent (Promega, Madison, WI, USA), and the plates were read in an automated Envision plate reader (PerkinElmer, Waltham, Mass) after 96 h of incubation. The percent inhibition values were calculated using the formula (sample result/mean value of the entire plate) × 100%. For the combinatorial drug screen, resistant cell lines were treated with increasing doses of each of 45 epigenetic agents with or without 2 μM idelalisib. The area under the curve was calculated for each agent. The combination index (CI) was calculated using CalcuSyn software (Biosoft, Cambridge, MA, USA), which defines CI < 1 as synergistic, CI = 1 as additive, and CI > 1 as antagonistic.
Compounds
Forty-five epigenetic compounds were purchased from MedChemExpress (Monmouth Junction, NJ, USA) and are listed in Supplementary Table 1 with detailed information. For in vitro experiments, idelalisib (BD224801, CSNpharm, Chicago, USA), BI-D1870 (Monmouth Junction), LJH-685 (Monmouth Junction) and epigenetic compounds were dissolved in DMSO and stored until use, and the final concentration of DMSO in the culture medium was <0.1%.
Cell cycle analysis
Cells were plated in 12-well plates at a concentration of 1 × 106 cells/well and treated with the indicated concentrations of drugs for 72 h. Cells were then washed with PBS, permeabilized with 70% ethanol, treated with RNase A, and stained with propidium iodide (Vazyme Biotech Co., Ltd., Nanjing, China) for 30 min at room temperature. The cell cycle distribution was analyzed with a FACSCalibur flow cytometer (BD Pharmingen, San Diego, CA, USA).
Western blotting
Total protein was obtained from cultured cells. Samples containing equal amounts of protein were subjected to 4%–16% SDS-PAGE, and proteins were transferred onto nitrocellulose membranes (Millipore, Temecula, CA, USA). After blocking with 5% nonfat milk, membranes were incubated at 4 °C overnight with primary antibodies and then with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h. The antibodies used are listed in Supplementary Table 2. Target protein bands were detected by the enhanced chemiluminescence (ECL) method in a ChemiScope 3400 imaging system using ECL substrate (Clinx, Shanghai, China).
Mouse xenograft studies
Four- to six-week-old female CB17/Icr-Prkdcscid/IcrlcoCrl (SCID) mice provided by Shanghai Laboratory Animal Center, Chinese Academy of Sciences, were used for establishment of Maver-1 and Z-138 xenografts. A total of 5 × 106 cells suspended in 100 μL of 1 × PBS and Matrigel matrix (Corning, Shanghai, China) were inoculated subcutaneously in the right flank. Mice bearing xenografts were randomly assigned to four groups when the tumor volume reached ~100 mm3. Each group of mice (n = 6) was treated daily with vehicle, 30 mg/kg idelalisib (by oral gavage), 50 mg/kg A-485 (intraperitoneally), or a combination of both agents. The animals’ body weights and tumor sizes were measured every 3 days, and the tumor volume was calculated as length × width2/2. All mice were humanely sacrificed by euthanasia after almost 3 weeks of drug treatment, and the final tumor volumes and tumor weights were measured. Tumors were processed for H&E staining and immunohistochemical staining. All animal experiments were conducted in compliance with the Institutional Animal Care and Use Committee guidelines of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
H&E staining and immunohistochemical analysis
Xenograft tumors were fixed in 4% paraformaldehyde overnight before being embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry, anti-p-S6 (#4858, Cell Signaling Technology, MA, USA) and anti-p-4E-BP1 (#2855, Cell Signaling Technology) antibodies were used. Images were acquired under a fluorescence microscope, and immunoreactivity was measured and quantified with Image-Pro Plus 6.0.
RNA sequencing and bioinformatic analysis
Maver-1 cells were treated with two treatments—control and a combination of idelalisib and A-485—for 8 h. Three biological replicates were performed. Total RNA was extracted to prepare cDNA libraries, which were sequenced on the Illumina HiSeq 2000 platform using paired-end approaches. ViaSTAR 2.5, the sequencing reads were mapped to hg19, and gene expression was quantified by featureCounts software [24]. Differential gene expression analysis was performed by the DESeq2 R package [25], and significantly changed genes were identified as those with a P value < 0.05 and a log2 (fold change) >1.2. Differentially expressed genes (DEGs) were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. All gene readouts were subjected to gene set enrichment analysis (GSEA) using GSEA 4.0.3 software available online (www.gsea-msigdb.org).
RNA extraction and quantitative RT-PCR
Total RNA from cells was extracted using TRIzol reagent (Vazyme Biotech Co., Ltd., Nanjing, China) and was then subjected to reverse transcription using HiScript II qRTSuperMix (Vazyme Biotech Co., Ltd.) according to the manufacturer’s protocol. Real-time PCR was performed using AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Ltd.), and gene expression was detected using quantitative RT-PCR in a Quant Studio 6 Flex Real-Time PCR system (ABI, Waltham, USA). The expression levels of target genes were normalized to those of GAPDH, ACTB, and B2M and calculated using the ∆∆Ct method. The detailed primer sequences are listed in Supplementary Table 3.
ChIP-qPCR assay
The chromatin immunoprecipitation (ChIP) assay was performed using a Simple ChIP Plus Enzymatic Chromatin IP Kit (#9005, Cell Signaling Technology) according to the procedures provided by the manufacturer. Cells were fixed with formaldehyde and lysed. Harvested chromatin was fragmented using sonication and was then incubated overnight with antibodies against H3K27ac (#8173, Cell Signaling Technology) or with normal rabbit IgG. Immunoprecipitated DNA was analyzed by qPCR to detect the enrichment of H3K27ac at the promoter regions of EPHA2 and ERBB2. The detailed sequences of the primers used for ChIP-qPCR are listed in Supplementary Table 4.
Cell transfection
Transfection of Z-138 cells was carried out using an Amaxa 4D-Nucleofector system (Lonza, Cologne, Germany). In brief, 1 million cells were resuspended in SF solution (Lonza) and mixed with 2 μM EphA2 and ErbB2 plasmids (Youbio Biological Technology Co., Ltd., Changsha, China). In each reaction, a GFP-expressing plasmid was used as a control.
Statistical analyses
Mean, SD and SEM values were calculated using GraphPad Prism software. Comparisons between the indicated groups were made using two-tailed Student’s t test, one-way analysis of variance (ANOVA) and two-way ANOVA with Dunnett’s multiple comparisons test. P values < 0.05 were considered statistically significant for all experiments.
Results
Combinatorial drug screen identifies P300/CBP inhibitors as sensitizers to idelalisib in MCL cell lines
Through recent research, a combinatorial drug screening method has been designed and a new combination of CDK4/6i and PI3Ki has been identified to overcome PI3Ki resistance in breast cancer [26]. Inspired by this work and aiming to identify potential epigenetic sensitizers to idelalisib in MCL cells, we performed a combinatorial drug screen using an epigenetic compound library containing 45 small molecule modulators. The collection consists of agents targeting epigenetic writers, including DNA methyltransferases (DNMTs), histone acetyltransferases (HATs), lysine methyltransferases (KMTs), and protein arginine methyltransferases (PRMTs); epigenetic erasers, encompassing histone deacetylases (HDACs), sirtuins, and histone demethylases (KDM); and epigenetic readers (Fig. 1a). The bioactivity and safety of these agents have been confirmed in preclinical studies and/or clinical trials. Resistant MCL Z-138 cells were seeded in 96-well plates and treated with increasing doses of each candidate drug in the presence or absence of 2 μM idelalisib [13]. Epigenetic compounds were applied at doses across a broad spectrum to ensure that the single-agent activities were well reflected. Cell proliferation was assessed after 96 h, and drugs were ranked based on the difference between the combination treatment and the single-agent treatment (Fig. 1b). Compounds with the greatest difference were considered potential sensitizers to idelalisib.

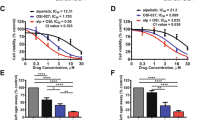
a Epigenetic compound libraries used for the screen. The numbers in parentheses indicate the numbers of compounds in each category used for the screen. b A schematic showing the approach used for the combinatorial drug screen. Z-138 cells were seeded in triplicate in 96-well plates and treated with increasing doses of each of the 45 candidates in the presence or absence of 2 μM idelalisib (idela). c Histogram showing the results of the idelalisib screen. Agents were ranked according to the largest difference in AUC between the two dose–response curves in Z-138 cells. d Chemical structure of the hit compound A-485. e Validation of the hit compound in MCL cells. A growth assay in Maver-1, Z-138, and Jeko-1 cells was performed following 96 h of exposure to the indicated agents at progressively increasing doses. f Combination index (CI) analysis. The survival fraction affected (Fa) and CI value are shown for Maver-1, Z-138, and Jeko-1 cells. The CI quantitatively determines synergy (CI < 1), an additive effect (CI = 1), or antagonism (CI > 1). The data shown are the mean ± SD of experiments completed in triplicate. At least three biological replicates were performed for all assays.
Notably, for the first time, A-485, the highly selective catalytic inhibitor of p300 and CBP [27], was identified as the most potent sensitizer to the PI3Kδ inhibitor in Z-138 MCL cells (Fig. 1c). Additionally, most of the inhibitors targeting HDACs also showed certain cooperative effects on Z-138 cells. In particular, the first-in-class alkylating HDAC inhibitor EDO-S101 ranked second in our assay, combined treatment of HDACi and PI3Ki has been previously reported to be effective in particular types of cancer, such as MYC-driven medulloblastoma [28]. To confirm the initial screening results, three MCL cell lines in which the IC50 value was higher than 10 μM were exposed to increasing doses of A-485 and idelalisib as single agents or in combination. Similarly, the combination of A-485 and idelalisib markedly decreased the viability of MCL cells (Fig. 1d) and exhibited a synergistic effect with a CI of 0.2–0.8 as assessed by the CalcuSyn model [29] (Fig. 1e). To further evaluate the safety of the drug combinations, we evaluated the effects of the combinations on HUVECs, RPTECs, and L02 cells, which are normal human cells. When treated with the same concentration of the drug combination, normal human cells exhibited significantly higher viability than MCL cells (Fig. S1). These results suggest that these two drugs have combinatorial inhibitory effects on MCL cells but have greatly reduced effects on normal cells.
Combined A-485 and idelalisib-induced cell cycle arrest in MCL cells is mediated by coinhibition of PI3K downstream signaling
Having confirmed that cotreatment with A-485 overcomes resistance to idelalisib in MCL cell lines, we examined the effect on the cell cycle to determine how this combination results in growth inhibition. Z-138 and Maver-1 cells, the most resistant cell lines, were treated with either 10 μM idelalisib or/and 0.5 μM A-485 for 72 h. Cells were analyzed by flow cytometry using PI-DAPI staining. Of note, an increase in the population of cells in G0/G1 phase was observed upon combination treatment with Z-138 (Fig. 2a, c), which offered more insight into the intense combination effect in MCL cell lines. Similarly, an increasing proportion of Maver-1 cells accumulated in G0/G1 phase (Fig. 2b, d) upon cotreatment.

a, b Cells were treated with DMSO (control), idelalisib (10 μM), A-485 (0.5 μM), or a combination of two compounds for 72 h. c, d The percentages of cells in G1, S, and G2 phases were quantified. The data shown here are the mean ± SD of three independent experiments. ****P < 0.0001. e, f Immunoblotting of phosphorylation changes in PI3K pathway molecules. Z-138 and Maver-1 cells treated with the indicated doses of idelalisib were exposed to A-485 for 24 h. α-Tubulin and β-actin were used as controls.
To gain more insight into the underlying mechanism responsible for the combination effect, we focused on signal transduction along the PI3K-AKT-mTOR axis. Z-138 and Maver-1 cells were treated with increasing doses of idelalisib, A-485, or the combination for 24 h and were then harvested. Treatment with idelalisib alone led to prominent suppression of AKT phosphorylation, but the phosphorylation levels of rpS6 and 4E-BP-1 were not decreased in resistant Maver-1 and Z-138 cells in response to idelalisib. Only modest inhibition was seen in the higher-dose setting of >3.3 μM. Remarkably, the addition of A-485 blocked the maintenance of p-S6 and p-4E-BP-1 (Fig. 2e, f), both of which are critical mediators of mTOR-dependent G1-phase progression [30] and control cell proliferation and survival. Collectively, these results suggested that the combination effect resulted from suppression of cell cycle arrest through coinhibition of PI3K-AKT-mTOR downstream signaling. Furthermore, failure of mTOR downstream inhibition may be responsible for the resistance to single-agent idelalisib treatment, and a cotreatment strategy with p300/CBP inhibition could reverse this escape.
Combined therapy synergistically suppresses mantle cell lymphoma in xenograft models
To determine whether the combination of idelalisib and A-485 can synergize to suppress MCL tumor growth in xenograft models, we induced tumor formation in immunodeficient mice via transplantation of Maver-1 cells. When the tumors became measurable, the mice were randomly assigned to four groups and were then treated with vehicle, A-485 at 50 mg·kg−1·d−1, idelalisib at 30 mg·kg−1·d−1 or the combination at the same doses. Notably, treatment with idelalisib or A-485 alone moderately slowed tumor growth, while treatment with the combination of idelalisib and A-485 significantly inhibited tumor growth, with a reduction of ~60% relative to the control group in the Maver-1 xenograft model (Fig. 3a). After treatment for ~24 days, when the vehicle tumors had reached the allowed maximum tumor size, the animals were sacrificed, and tumor samples were harvested and weighed. Compared with the control treatment, idelalisib and A-485 alone had partial therapeutic effects, as displayed by the decreased tumor weight. However, tumors in the combination group had significantly lower tumor weights than tumors in the other groups (Fig. 3b). We also performed a similar tumor xenograft experiment using the Z-138 cell line. Consistent with the above results, treatment of mice bearing Z-138 cell-derived tumors with either idelalisib or A-485 alone only partially inhibited tumor growth, while the combination treatment led to a significant reduction in Z-138 tumor growth (Fig. 3c). Furthermore, after treatment for 21 days, the harvested tumor samples from the combination group demonstrated markedly lower tumor weights (Fig. 3d). Importantly, we found that the combination therapy was well tolerated in the animals, as there were no substantial changes in the body weights of the mice in the four groups throughout the experiment (Fig. S2a, b).

a Tumor growth curve in the Maver-1 model. Mice were treated with A-485 via i.p. administration of 50 mg/kg, idelalisib via p.o. administration of 30 mg/kg, or the indicated combination at the same doses daily for 24 days. b Representative Maver-1 xenografts harvested from mice. Tumor samples were weighed after the mice were sacrificed. c Tumor growth curve in the Z-138 model. Mice were treated with A-485 via i.p. administration of 50 mg/kg, idelalisib via p.o. administration of 30 mg/kg, or the indicated combination at the same doses daily for 21 days. d Representative Z-138 xenografts harvested from mice. Tumor samples were weighed after the mice were sacrificed. e Representative IHC image of molecular changes in resected tumor tissues. Tumor samples as described were collected 8 h after the last dosing, and intratumoral molecular changes were detected using immunohistochemical analysis. Scale bar, 50 μm. f Logarithmic summed IOD value of protein expression in the IHC image. The error bars indicate the SEMs (n = 6 mice per group). **P < 0.01; ****P < 0.0001.
At the end of the observation period, the tumors were collected and used for immunohistochemical analysis. Treatment with idelalisib alone resulted in modest suppression of PI3K-mTOR downstream signaling, as evidenced by the observation that sustained p-S6 and p-4E-BP1 signaling was observed in both allografted cell lines. Consistent with the observations at the cellular level, the maintenance of mTOR downstream signaling activation was blocked by p300/CBP inhibition in vivo, and sustained suppression of PI3K-mTOR signaling was observed in the collected tumor samples in the combination group (Fig. 3e, f). Taken together, these findings showed that the combination of idelalisib and A-485 synergistically inhibited tumor growth and progression in MCL preclinical models without increasing toxicity. The data confirmed the synergistic suppression of sustained PI3K-mTOR downstream signaling in vivo, which could be the underlying mechanism of resistance to idelalisib in MCL.
Gene expression profiling identifies cancer-related pathways and predicts a role for suppressing MAPK/ERK signaling in MCL by combined treatment
Aiming to investigate the molecular mechanisms leading to the lack of mTOR downstream signaling inhibition upon single-agent treatment with idelalisib, we analyzed the transcriptome of Maver-1 cells treated with combination therapy using RNA sequencing (RNA-seq) and compared this transcriptome to that of control cells. Maver-1 cells were treated with control or with the combination of 1 μM idelalisib and 3 μM A-485 for 8 h. As expected, combination treatment altered the global transcription profile, including upregulation of 833 genes and downregulation of 1096 genes (Fig. 4a).

a Volcano plot showingthe differentially expressed genes. The horizontal line corresponds to a P value of 0.05, and the vertical lines indicate upregulation and downregulation by 1.2-fold. The blue and red points indicate the differentially expressed genes with a >1.2-fold change between the two groups. b KEGG pathway analysis of differentially expressed genes (DEGs) between the combination group and control group. The most significantly enriched pathways are presented. c Gene ontology enrichment analysis of DEGs. The most significantly enriched biological events are presented. d–f GSEA of gene signatures regulated by the combination treatment. Normalized enrichment score (NES) and false discovery rate (FDR) values are shown. Expression datasets were generated from the two collections of the combination group and control group. n = 3 biological replicates per condition.
KEGG pathway analysis to identify more specific clustering of these DEGs revealed several pathways significantly affected by combination therapy (Fig. 4b). Among the enriched pathways, we focused on the top-ranked downregulated Rap1 signaling pathway. Rap1 was found to be activated by stimulation of multiple transmembrane receptors, including RTKs, GPCRs, cytokine receptors and cell adhesion molecules [31]. Previous studies have identified that the main function of Rap1 signaling is regulating the activation of the mitogen-activated protein kinase (MAPK) pathway, which is involved in cell proliferation and survival [32]. GO analysis indicated that these DEGs were enriched in cell proliferation, activation, and adhesion within lymphocytes, which could constitute the normal response of Maver-1 cells to drug stimulation, since MCL is characterized as a lymphocyte disorder; in addition, this result was consistent with the growth suppression effects found in the above proliferation assays. Importantly, regulation of MAPK was also identified in GO analysis and were highly ranked (Fig. 4c).
Using the complete mRNA expression profiles generated from the two collections instead of only the DEGs, we characterized the pathway features via GSEA. Consistent with the above results, the GSEA results demonstrated that the combination treatment markedly regulated genes involved in cell proliferation [normalized enrichment score (NES) [13] = −2.44] and the cell cycle (NES = −1.66), which was highly consistent with our findings from the in vitro and in vivo experiments described above (Fig. 4d, e). Notably, the similar significant downregulation of the ERK1/2 cascade (NES = −1.51), enriched in the GSEA results, attracted our attention (Fig. 4f).
The inhibitory effects of the combination therapy on the MAPK/ERK pathway are mediated by a reduction in upstream RTKs
MAPK signaling plays a crucial role in extracellular signal transduction and triggers cellular responses [33]. Abnormally activated MAPK/ERK signaling can impair the efficacy of PI3K signaling inhibitors [34,35,36,37,38,39]. A study identified that activated RSK3/4, which are directly regulated by ERK, can mediate the resistance of breast cancer to PI3K pathway blockade by regulating transcriptional regulators, including rpS6 and eIF4B [39].
To confirm the possible critical role that MAPK/ERK signaling plays in reversing the resistance of MCL to PI3Kδ inhibitors, Maver-1 and Z-138 cells were treated with 3 μM A-485, 1/0.1 μM idelalisib, a combination of both or DMSO for 24 h, and the phosphorylation levels of the crucial nodes of MAPK/ERK signaling, namely, MEK, ERK and RSK, were measured in harvested cells. Consistent with the above results, the results showed that combined treatment with A-485 completely inhibited MAPK/ERK pathway signaling. Moreover, the sustained activation of the PI3K-mTOR downstream signaling molecules p-S6 and p-4E-BP1, which was identified in vitro and in vivo as indicated above, was adequately suppressed (Fig. 5a, b). In addition, to further confirm that reactivation of MEK-ERK-RSK signaling mediates the unsustained sensitivity to idelalisib, we conducted a combinatorial study on Z-138 cells using idelalisib and RSK inhibitors. As expected, the combined treatment exhibited significant synergistic effects on reducing cell viability (Fig. S3a, b).

a, b Pathway phosphorylation changes. Z-138 and Maver-1 cells treated with the indicated doses of idelalisib were exposed to 3 μM A-485 for 24 h. Phosphorylation changes in ERK-RSK-mTOR signaling molecules were detected by immunoblotting. α-Tubulin and GAPDH were used as controls. c Heat map showing the 25 most significantly downregulated genes in the MAPK pathway. Hub genes are highlighted in red. d Table of RTK-related genes among the top 25 genes. Seven of the 25 genes are listed in the table and ranked according to the log2 (fold change) value. e RT-qPCR analysis was conducted to validate the repression of hub genes. Z-138, Maver-1, and Jeko-1 cells were treated with 3 μM A-485 and 1 μM idelalisib for 8 h. The data are shown as the mean ± SD of three independent experiments. **P < 0.01; ***P < 0.001.
To gain more insight into why MAPK/ERK signaling was regulated by the combination treatment, a more specific gene expression analysis was performed. Interestingly, the top 25 most downregulated genes in the MAPK/ERK pathway were RTK family members or their homologous ligands (Fig. 5c). Specifically, ~30% of the top 25 genes were RTK-related genes, including FGF8, EPHA2, FGFR3, MET, ERBB2, PDGFA and EGF. All of these were highly ranked, as shown (Fig. 5d). For further confirmation, Z-138, Maver-1, and Jeko-1 cells were treated with the combination of 3 μM A-485 and 1 μM idelalisib for 8 h and were then harvested to detect the variations in mRNA expression between the two groups. Similarly, the mRNA expression of RTKs was significantly downregulated by the combination treatment, among which FGF8, ERBB2, and EPHA2 were the most affected genes in all three MCL cell lines (Fig. 5e). Taken together, these results demonstrated that combination treatment with a PI3K inhibitor and p300/CBP inhibition led to effective anticancer effects, at least in part, via reprogramming of the MAPK/ERK pathway and transcriptional modulation of RTK-related genes.
RTK upregulation is required for feedback activation of MAPK/ERK signaling and is transcriptionally blocked by P300/CBP inhibition
To demonstrate whether downregulation of MAPK activity contributes to the reversal of idelalisib resistance, we treated Z-138 cells with 1 μM idelalisib at multiple time points for up to 24 h. The phosphorylation levels of MAPK signaling pathway components were measured in harvested cells. Notably, upregulation of the MAPK/ERK pathway over time was observed. Maximal suppression of this signaling was only transient, as phosphorylation began to rebound at 4–12 h, indicating that MAPK activation was triggered. In addition, a rebound in the phosphorylation of S6 and 4E-BP-1 was observed, demonstrating the crosstalk between activation of the MAPK pathway and downstream mTOR signaling. More importantly, additional treatment with A-485 interfered with the rebound of MAPK signaling and consequently attenuated reactivation of downstream signaling (Fig. 6a). These findings indicated that idelalisib-induced activation of MAPK signaling was responsible for the resistance of MCL cells and that p300/CBP inhibition blocked this escape. RTKs are a group of transmembrane receptors constitution one of the most important families of molecular sensors for perceiving extracellular stimuli and evoking cellular responses [40]. The MAPK pathway is triggered to regulate cell proliferation and survival in response to the activation of RTKs [41]. Homeostatic feedback upregulation of RTKs induced by PI3K/AKT inhibition to maintain the equilibrium of the PI3K pathway has been reported, as has compensatory activation of the downstream MAPK pathway in some tumors [20, 42]. To test whether idelalisib leads to feedback upregulation of RTKs, Maver-1, and Z-138 cells treated with 1 μM idelalisib were harvested at multiple time points up to 24 h. As expected, upregulation of EPHA2 and ERBB2 over time was observed in both cell lines (Fig. 6b, c), which confirmed that the MCL cells acquired resistance to PI3K inhibition due to upregulation of RTKs and downstream compensatory activation of MAPK signaling.

a Pathway phosphorylation changes. Z-138 cells were treated with 1 μM of idela with or without 3 μM of A-485 for the indicated times. Phosphorylation changes in ERK-RSK-mTOR signaling molecules were detected by immunoblotting. Hsp90 was used as the control. b, c RT-qPCR was conducted to measure the mRNA levels of EPHA2 and ERBB2. Z-138 and Maver-1 cells treated with 1 μM of idelalisib at multiple time points for up to 24 h were compared with DMSO-treated control cells. The error bars indicate the SD of three independent experiments. **P < 0.01. d, e Histone acetylation at the EPHA2 and ERBB2 gene promoters. Z-138 cells were treated for 24 h with 1 μM or 3 μM A-485, harvested, and subjected to a ChIP assay using the anti-ac-H3K27 antibody and qPCR analysis using primers targeting the promoter regions of the indicated genes. f A schematic representation of signaling responses to combined inhibition of PI3K and p300/CBP.
The next step was to understand how p300/CBP inhibitors cause transcriptional downregulation of EPHA2 and ERBB2 in cancer cells. p300/CBP are highly enriched in the enhancers and promoters of active genes, regulating gene transcription by relaxing the chromatin structure through their intrinsic histone acetyltransferase (HAT) activity, which is required for acetylation of H3K18 and H3K27 [43, 44]. To understand how p300/CBP inhibition causes transcriptional downregulation of RTKs in MCL cells, we conducted ChIP using anti-acetyl-histone (H3K27) antibodies in Z-138 cells followed by qPCR. Treatment with 1 or 3 μM A-485 for 24 h reduced the abundance of the RTK gene promoter region, as verified by three independent pairs of primers specifically targeting the EPHA2 or ERBB2 gene promoter, in the fraction precipitated with the anti-Ac-H3K27 antibody (Fig. 6d, e). These findings indicated that A-485 treatment reduced histone acetylation in the EPHA2 and ERBB2 promoters. To further confirm that these two genes can affect MAPK/ERK activation, we overexpressed EphA2 and ErbB2 in Z-138 cells, which displayed strong activation of MAPK/ERK signaling. Moreover, the addition of A-485 reduced the activation of MAPK/ERK signaling in RTK-overexpressing cells (Fig. S4a, b).
Collectively, these results showed that feedback upregulation of upstream RTKs induced by PI3K inhibition was responsible for the compensatory activation of MAPK/ERK signaling. Additional p300/CBP inhibition reversed idelalisib-induced upregulation of RTKs as well as the subsequent abnormal activation of downstream PI3K-mTOR signaling and eventually eliminated resistance to idelalisib in MCL cells.
Discussion
Targeted therapies for human cancers have achieved remarkable success; however, the development of drug resistance and the undefined underlying mechanisms remain significant challenges. In this study, we described a new combination approach that exhibited a synergistic effect in MCL, which lacks effective treatment. We screened an epigenetic compound library and first identified the p300/CBP inhibitor A-485 as a sensitizer to idelalisib in vitro and in vivo. Moreover, we were the first to elucidate the molecular mechanism by which p300/CBP inhibition sensitizes MCL to PI3Kδ inhibitors.
Epigenetic dysfunction has long been recognized as a critical driving factor in cancer cells, and the transcriptional and epigenetic changes underlying resistant phenotypes have recently been highlighted. Several studies have identified that despite their potential as monotherapies, epigenetic drugs can show potent synergistic effects with other targeted therapies or in reversing tumor resistance. For example, FTO upregulation-mediated N6-methyladenosine (m6A) hypomethylation improves mRNA stability and increases the translation of proliferation and survival proteins, resulting inintrinsic and acquired resistance in leukemia during tyrosine kinase inhibitor therapy [45]. Similarly, targeting histone deacetylase (HDAC) can block the expression of melanocyte lineage transcription factor (MITF), which was identified as a driver of MAPK inhibitor resistance in melanoma [46]. Based on the prominent preclinical successes, a set of combinations of epigenetic drugs and various targeted agents are currently entering early phase clinical trials and are showing promising efficacy [47,48,49].
However, few studies have investigated the synergistic effects of p300/CBP inhibitors combined with other targeted therapies, and the role of p300/CBP in drug resistance remains poorly understood. Here, an epigenetic mechanism-based rationale for combination therapy was provided. For the first time, we identified that targeting p300/CBP with A-485 can modulate intrinsic drug resistance in MCL. These preclinical observations are particularly encouraging since idelalisib is FDA-approved, and agents targeting p300/CBP are available for clinical use (NCT04068597, NCT03568656), allowing rapid evaluation of the potential efficacy of this combination in human clinical trials. Therapeutic studies using idelalisib and p300/CBP inhibitors in MCL could be promising.
Mechanistically, we showed that idelalisib treatment results in upregulation of RTKs and compensatory activation of MAPK/ERK signaling. As the model illustrated (Fig. 6f), idelalisib treatment particularly upregulated the expression of the RTK receptors EPHA2 and ERBB2 and activated downstream MEK/ERK/RSK signaling. Feedback activation of MAPK/ERK signaling can sustain PI3K-mTOR downstream signaling activity, leading to the activation of p-S6 and p-4E-BP-1 and resulting in uncontrolled cell growth and tumor formation. The results indicated that the feedback activation of upstream RTKs is responsible for idelalisib-induced resistance. In addition, we discovered that A-485 decreased histone acetylation at the EPHA2 and ERBB2 gene promoters and reduced transcriptional upregulation of RTKs, which could eventually overcome idelalisib-induced resistance.
Cotreatment with PI3K inhibitors and various agents targeting each RTK seems impractical due to the potential triple toxicity and the absence of potent inhibitors of some RTKs. PI3Ki-induced feedback upregulation of RTKs was observed in a broad range of malignant cells [20, 42, 50], indicating that the mechanism elucidated in this work may not be limited to MCL. However, RTK combination therapy is impossible due to the extreme difficulty of predicting receptor feedback in different tumor types. In particular, transcriptional upregulation of RTKs is increasingly reported as a major mechanism of resistance to monotherapies targeting the MAPK/ERK pathway in multiple cancers [51, 52]. Consequently, when seeking potential combination strategies to reverse kinase resistance, it is essential to target epigenetic alterations that drive the initial escape mechanism.
Based on our results, targeting the histone acetyltransferases p300/CBP is promising. p300/CBP inhibition reduces the levels of H3K27ac histone modification specifically at gene promoters, which reduces the PI3Ki-induced transcriptional upregulation of resistant RTKs, blocks the consequent activation of the MEK-ERK-RSK bypass, and ultimately leads to sensitization to idelalisib treatment. Notably, to accomplish sensitization to PI3Kis in various tumor types, targeting p300/CBP could be more efficacious than an RTK-specific inhibitorto block multiple resistance-related RTKs, since H3K27ac loss could be unbiased and mediated by p300/CBP inhibition. As histone acetyltransferases (HAT) and transcriptional coactivators, p300/CBP are typically recruited to gene promoters or enhancers and modulate transcriptional expression by acetylating chromatin, by which multiple cellular processes are influenced.
In summary, although some epigenetic modulators have been reported to exhibit synergetic effects with targeted kinase inhibitor therapies in resistant tumors, no studies have focused on the function of p300/CBP in reversing drug resistance. Here, using the potent inhibitor A-485, we demonstrated for the first time the requirement for p300/CBP histone acetyltransferase activity in the response to PI3Kδi resistance. This intriguing finding may have wider significance, since p300/CBP are frequently overexpressed and dysregulated in a broad spectrum of cancer types. Further studies investigating the potential function of p300/CBP in reversing resistance in other tumors may be promising.
References
Ghielmini M, Zucca E. How I treat mantle cell lymphoma. Blood. 2009;114:1469–76.
Weigert O, Unterhalt M, Hiddemann W, Dreyling M. Mantle cell lymphoma: state-of-the-art management and future perspective. Leuk Lymphoma. 2009;50:1937–50.
Hoster E, Dreyling M, Klapper W, Gisselbrecht C, van Hoof A, Kluin-Nelemans HC, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111:558–65.
Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122:3416–23.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554.
Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108:1668–76.
Dal Col J, Zancai P, Terrin L, Guidoboni M, Ponzoni M, Pavan A, et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood. 2008;111:5142–51.
Rizzatti EG, Falcao RP, Panepucci RA, Proto-Siqueira R, Anselmo-Lima WT, Okamoto OK, et al. Gene expression profiling of mantle cell lymphoma cells reveals aberrant expression of genes from the PI3K-AKT, WNT and TGFbeta signalling pathways. Br J Haematol. 2005;130:516–26.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405.
Malka-Mahieu H, Newman M, Desaubry L, Robert C, Vagner S. Molecular pathways: the eIF4F translation initiation complex-new opportunities for cancer treatment. Clin Cancer Res. 2017;23:21–5.
Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. 1997;94:4330–5.
Kahl B, Byrd JC, Flinn IW, Wagner-Johnston N, Spurgeon S, Benson D, et al. Clinical safety and activity in a phase 1 study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110d, in patients with relapsed or refractory non-Hodgkin lymphoma [abstract]. Blood. 2010;116:1777.
Jacobson C, Kopp N, Layer JV, Redd RA, Tschuri S, Haebe S, et al. HSP90 inhibition overcomes ibrutinib resistance in mantle cell lymphoma. Blood. 2016;128:2517–26.
Ripperger T, von Neuhoff N, Kamphues K, Emura M, Lehmann U, Tauscher M, et al. Promoter methylation of PARG1, a novel candidate tumor suppressor gene in mantle-cell lymphomas. Haematologica. 2007;92:460–8.
Hrgovic I, Doll M, Kleemann J, Wang XF, Zoeller N, Pinter A, et al. The histone deacetylase inhibitor trichostatin a decreases lymphangiogenesis by inducing apoptosis and cell cycle arrest via p21-dependent pathways. BMC Cancer. 2016;16:763.
Kitazawa S, Kitazawa R, Maeda S. Transcriptional regulation of rat cyclin D1 gene by CpG methylation status in promoter region. J Biol Chem. 1999;274:28787–93.
Spurgeon SE, Sharma K, Claxton DF, Ehmann C, Gallagher C, Shimko S, et al. Final results of a phase 1-2 STUDY OF VORINOSTAT (SAHA), cladribine, and rituximab (SCR) relapsed B-cell non-Hodgkin’s lymphoma and previously untreated mantle cell lymphoma. Blood. 2014;124:1714. https://ashpublications.org/blood/article/128/22/1792/91238/The-Results-of-a-Phase-I-Study-using-Velcade?searchresult=1.
Pu JJ, Ehmann WC, Liao J, Capper C, Levy M, Claxton DF, et al. The results of a phase I study using Velcade, Cladribine and Rituximab (VCR) in treating mantle cell lymphoma. Blood. 2016;128:1792. https://ashpublications.org/blood/article/124/21/1714/88341/Final-Results-of-a-Phase-1-2-Study-of-Vorinostat?searchresult=1.
Puvvada SD, Guillen-Rodriguez J, Kumar A, Inclan L, Heard K, Rivera XI, et al. Phase 2 open-label study of bortezomib, cladribine, and rituximab in advanced, newly diagnosed, and relapsed/refractory mantle-cell and indolent lymphomas. Clin Lymphoma Myeloma Leuk. 2018;18:58–64.
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74.
Leonard B, Brand TM, O’Keefe RA, Lee ED, Zeng Y, Kemmer JD, et al. BET inhibition overcomes receptor tyrosine kinase-mediated cetuximab resistance in HNSCC. Cancer Res. 2018;78:4331–43.
Paoluzzi L, Hanniford D, Sokolova E, Osman I, Darvishian F, Wang J, et al. BET and BRAF inhibitors act synergistically against BRAF-mutant melanoma. Cancer Med. 2016;5:1183–93.
Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33:401–16.e8.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–49.
Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128–32.
Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell. 2016;29:311–23.
Chou TC. The median effect principle and the combination index for quantitation of synergism and antagonism. San Diego, Calif: Academic Press; 1991.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–77.
Minato N, Kometani K, Hattori M. Regulation of immune responses and hematopoiesis by the Rap1 signal. Adv Immunol. 2007;93:229–64.
Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–80.
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93.
Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt cross-talk. J Biol Chem. 2002;277:31099–106.
Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30:777–82.
Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858–66.
Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011;10:558–65.
Serra V, Eichhorn PJ, Garcia-Garcia C, Ibrahim YH, Prudkin L, Sanchez G, et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J Clin Invest. 2013;123:2551–63.
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34.
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40.
Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–57.
Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–62.
Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31.
Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A dynamic N(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018;28:1062–76.
Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–42.
Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104:1828–35.
Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82.
Pawar A, Gollavilli PN, Wang S, Asangani IA. Resistance to BET inhibitor leads to alternative therapeutic vulnerabilities in castration-resistant prostate cancer. Cell Rep. 2018;22:2236–45.
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71.
Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007;26:3909–19.
Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169–79.
Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (81625022, 91853205, 81821005 to CL, 81803554 to YZ, 81703415 to SC, 81973166 to BZ, 21820102008 to HJ); the Chinese Academy of Science grant (XDA12020353 to CL), KC Wong Education Foundation to BZ and CL, the National Science and Technology Major Project (2018ZX09711002-008-005 to BZ, 2018ZX09711002 to CL), Science and Technology Commission of Shanghai Municipality (18431907100 to HJ and 19XD1404700 to CL), the Youth Innovation Promotion Association (2017333 to YY).
Author information
Authors and Affiliations
Contributions
XRZ, XL, LPL, JH, JH, and ZFC conducted experiments. XRZ, YYZ analyzed data and wrote the paper. XL, JCL, HRT, SJF, QL, SJC, HD edited the paper. YXY, BZ provided reagents. HLJ, KXC, YYZ, CXH, and CL supervised the project and revised the papers. All authors have read and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhou, Xr., Li, X., Liao, Lp. et al. P300/CBP inhibition sensitizes mantle cell lymphoma to PI3Kδ inhibitor idelalisib. Acta Pharmacol Sin 43, 457–469 (2022). https://doi.org/10.1038/s41401-021-00643-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00643-2
Keywords
This article is cited by
-
Protein acylation: mechanisms, biological functions and therapeutic targets
Signal Transduction and Targeted Therapy (2022)
