
Abstract
Recent evidence shows that when ischemic stroke (IS) occurs, the BBB would be destructed, thereby promoting the immune cells to migrate into the brain, suggesting that the immune responses can play a vital role in the pathology of IS. As an essential subpopulation of immunosuppressive T cells, regulatory T (Treg) cells are involved in maintaining immune homeostasis and suppressing immune responses in the pathophysiological conditions of IS. During the past decades, the regulatory role of Treg cells has attracted the interest of numerous researchers. However, whether they are beneficial or detrimental to the outcomes of IS remains controversial. Moreover, Treg cells exert distinctive effects in the different stages of IS. Therefore, it is urgent to elucidate how Treg cells modulate the immune responses induced by IS. In this review, we describe how Treg cells fluctuate and play a role in the regulation of immune responses after IS in both experimental animals and humans, and summarize their biological functions and mechanisms in both CNS and periphery. We also discuss how Treg cells participate in poststroke inflammation and immunodepression and the potential of Treg cells as a novel therapeutic approach.
文章介绍
免疫细胞浸润是调控卒中后神经损伤与修复的核心机制。调节性T (Treg) 细胞作为免疫抑制T细胞的重要亚群, 在缺血性脑卒中的病理进程中, 参与维持免疫稳态, 调控免疫反应等作用。既往研究表明, Treg细胞因缺血性脑卒中病理阶段不同而功能各异, 例如, Liesz等人证实, 脑卒中发生7天后, Treg细胞具有神经保护作用; 而Kleinschnitz等人发现, Treg 细胞缺失的DEREG小鼠在脑卒中1天后的梗死体积小于对照组, 证明Treg细胞在其中扮演有害角色。因此, 阐明Treg细胞如何调节缺血性脑卒中后免疫反应十分重要。在本篇综述中, 我们描述了Treg细胞在实验动物和人类发生缺血性脑卒中后的变化特征, 以及该细胞在调节免疫应答中的作用, 并讨论了Treg细胞如何参与缺血性脑卒中后炎症和免疫抑制的分子机制, 提出了Treg细胞作为一种新型细胞治疗方法的潜力及未来的研究方向。
Similar content being viewed by others


Introduction
Ischemic stroke (IS) is a cerebrovascular disease resulting in high mortality and disability [1], which causes over six million deaths per year worldwide [2]. As thrombolytic therapy (tissue-plasminogen activator) is the only current treatment for IS and is time-limited (4.5–6 h) [3], it is urgent to explore novel treatments that contribute to neurologic protection against IS. Researchers have recently highlighted the important roles of inflammation and the immune response in IS, as inflammation is a fast-reacting and rate-determining step that lasts for several days after IS [1, 4, 5]. For decades, due to blockade by the blood-brain barrier (BBB), it has been mainly accepted that the entry of immune cells into the brain is restricted [6]. However, it has been recently shown that when IS occurs, the BBB is destroyed, thereby allowing immune cells to migrate into the brain, indicating that immune responses can take place in both the central nervous system (CNS) and the periphery [7].
Regulatory T cells (Treg cells) are a subset of T cells that have been demonstrated to be vital for maintaining self-tolerance and limiting inflammatory collateral damage by suppressing active (self-reactive) immunity [8, 9]. Treg cells also consist of several subpopulations, among which natural Treg cells are the most studied. These cells are generally characterized as CD25+CD4+ and coexpress the transcription factor forkhead box P3 (Foxp3) [10]. The depletion of CD25+CD4+ Treg cells causes autoimmune diseases, while the reconstitution of Treg cells prevents this process [11]. However, there are several other types of Treg cells known as inducible Treg cells, such as Th3, Tr1, and natural killer Treg cells, that acquire their functions by stimulation with specific antigens [12]. Treg cells have been shown to exist mainly in lymphoid tissues [13]. However, it has been suggested that these cells also reside in various tissues [13]. According to different phenotypically and functionally distinct Treg cell subpopulations, Treg cells show site-specific functions in circulation and tissues [14].
In this article, we review how Treg cells fluctuate and play a role in the regulation of immune responses after IS in both experimental animals and humans and summarize the biological functions and mechanisms of Treg cells in both the CNS and the periphery. We also discuss how Treg cells are involved in poststroke inflammation and immunodepression and the potential of Treg cells as a novel therapeutic approach.
Treg cells in IS
Treg cells play a crucial immunomodulatory role in IS. However, the contribution of Treg cells can be either beneficial or detrimental and remains controversial. Many studies have investigated how Treg cells fluctuate in both human patients and experimental models.
Clinical observation of Treg cells in IS patients
Previous studies have shown that compared to those of controls, the levels of circulating Treg cells declined in stroke patients [15, 16], and the clinical observations of Urra et al. showed that circulating Treg cell levels decreased noticeably in the 2 days after stroke [17]. However, although the decrease in Treg cells was demonstrated to be related to apoptosis, there was no correlation with the outcomes of stroke [17, 18]. In contrast, other studies demonstrated that although the suppressive effects of Treg cells were attenuated, the proportion of Treg cells in the peripheral blood was higher in IS patients than in controls [19, 20]. Yan et al. examined the number of Treg cells, and the results showed that Treg cells were enhanced in the blood of IS patients compared to those in healthy controls on the 7th day after IS onset [20]. Moreover, Yan et al. discovered that the number of Treg cells increased beginning on the 7th day and lasted until the 3rd week after IS [19]. Interestingly, Pang et al. identified a relationship between infarct size and the trend in Treg cell fluctuations [21]. IS patients were divided into two groups according to infarct size (A group: <28.6 mL; B group: ≥28.6 mL). Treg cells were increased in the A group, while in the B group, cells were decreased in 3 days but increased after the 7th day [21]. Although there were some limitations in these studies, such as the small sample size (46 patients with acute stroke) [17] (67 subjects of 25 with acute IS) [20], this controversial finding led to the hypothesis that the fluctuation in Treg cell numbers could be a type of evolutionarily adaptive response, and these cells may leave the circulation and migrate to target tissues during the time after stroke occurs to protect against the inflammation triggered by IS [12, 16].
Treg cells in experimental models of IS
There is evidence confirming that Treg cells are recruited to the spleen, distal and proximal lymph nodes, and ischemic hemisphere in experimental models [22]. Treg cells located in different sites are heterogeneous, indicating that the function of Treg cells might be dependent on the environment [23].
Changes in Treg cells in the spleen
Treg cells in peripheral tissues have reparative functions in IS. As T cells patrol for cognate antigen in draining lymph nodes, and although T cells do not cross the brain, they significantly affect the severity of IS via immune signaling and the secretion of molecules such as interferon gamma (IFN-γ) [24, 25]. Kleinschnitz et al. discovered that Treg cells in the circulation were markedly decreased at the 24-h time point after IS and returned to a normal level after 3 days [26]. Interestingly, it was also shown that the fluctuation in Treg cells was accompanied by splenic atrophy [27]. The results of Offner et al. showed significant splenic atrophy in MCAO mice compared to sham-operated mice within 96 h, while Treg cells remained stable within 22 h but increased at the 96-h time point [27]. Moreover, Hilary et al. discovered a significant reduction in splenic volume at 24–48 h, accompanied by a rapid decrease in Treg cells in the blood and spleen after MCAO [28]. However, splenic atrophy and the number of splenocytes returned to normal 96 h after stroke [28]. In contrast, another study demonstrated that Treg cells doubled the day after IS onset and continued to increase 3 days after IS, while these cells could not be detected in the cerebral parenchyma until the 5th day [29].
In summary, these findings illustrate that Treg cells fluctuate in different locations in the brain and periphery and accumulate 3 days following the onset of IS [12]. Furthermore, these cells do not cross the BBB until the 5th day after IS [12].
Changes in Treg cells in the brain
As reviewed previously, only a few Treg cells infiltrate the brain during the acute phase; hence, whether Treg cells exert an effect at that time remains questionable [13, 14]. Gelderblom et al. discovered that in the first week after tMCAO, only a small number of CD25+Foxp3+ Treg cells could be observed [30]. In contrast, in the distal pMCAO model, Llovera et al. detected a large number of T cells and Treg cells in the ischemic hemisphere, and Foxp3+ Treg cells accounted for ~20% of all CD4+ T cells [31], which indicated that the inflammatory reaction induced by pMCAO is stronger than that induced by tMCAO [32]. Moreover, Stubbe et al. discovered that in the acute phase in the tMCAO model, there was only a low level of Foxp3+ Treg cells in the brain with no expression of tissue Treg cell-specific genes, while Ito et al. and Stubbe et al. demonstrated that 14 and 30 days after IS, which is the later stage, substantial numbers of Treg cells could be detected in the ischemic hemisphere and continued to increase thereafter [33], suggesting that there was a delayed response of Treg cells in the adaptive immune response [22, 33].
Paradoxical roles of Treg cells in IS
As reviewed previously, the dynamics of Treg cells in IS seem to be controversial. Moreover, the biological activities of Treg cells remain unclear, among which neuroprotective effects remain the mainstream view (Table 1).
Neuroprotective effects of Treg cells
Liesz et al. demonstrated that Treg cells served as neuroprotective modulators of inflammation in the brain after IS, and these cells were further confirmed to protect against secondary infarct growth. In their study, the researchers depleted Treg cells with a CD25-specific antibody. Compared to the controls, Treg cell-depleted mice showed significantly larger infarct sizes on the 7th day after pMCAO, accompanied by worse neurological performance. Moreover, to investigate whether these consequences resulted from the loss of Treg cells, the researchers administered CD4+CD25− cells, total CD4+ cells, or Treg cells to lymphocyte-deficient Rag2−/− mice. The mice that received CD4+CD25− cells showed larger infarct sizes than the mice that received other cell types, indicating that CD25+ Treg cells play a crucial role in neuroprotection after IS [34]. Zhang et al. illustrated the similar neuroprotective role of Treg cells. First, C57BL/6 mice were intraperitoneally administered interleukin (IL)-2/IL-2 Abs or isotype IgG 3 days before or 2 h after tMCAO. IL-2/IL-2 Abs enhanced the number of Treg cells, further leading to decreased infarct size and improved sensorimotor outcomes compared with those of the controls. Furthermore, the researchers adoptively transferred IL-2/IL-2 Abs to depletion of regulatory T cells (DEREG) mice. The results showed no additional protective effects of IL-2/IL-2 Abs. When Treg cells were administered to IL-2/IL-2 Abs-treated and isotype-treated mice, although the mice all showed improved neuroprotective outcomes, the protection was more robust in the former group, suggesting that Treg cells played a neuroprotective role after IS and that the effects of Treg cells were boosted in the presence of IL-2/IL-2 Abs [35].
Detrimental roles of Treg cells
Although numerous studies have shown beneficial effects of Treg cells after IS, some recent research findings have shown the opposite effects. Kleinschnitz et al. established the Treg cell-depleted DEREG mouse model via gene modification rather than using a CD25 antibody. After 30 or 60 min of tMCAO in DEREG mice, Rag1−/− mice, or C57BL/6 mice, the brain infarct volumes of DEREG mice induced with 60 min of tMCAO were markedly reduced 24 h poststroke compared to those of controls and were enlarged again when Treg cells were reconstituted over 3 weeks. Furthermore, when CD4+CD25+ Treg cells were transferred into C57BL/6 mice 1 day before tMCAO, the infarct size was enhanced significantly 1 day after IS onset with 30 min of tMCAO. More specifically, the transfer of FoxP3+ Treg cells into Rag1−/− mice resulted in similar infarctions sizes compared to those of the Rag1+/+ controls at 1 day after IS onset with 60 min of tMCAO [26]. Consistent with this finding, Schuhmann et al. administered the superagonist anti-CD28 antibody (CD28 SA) to C57BL/6, DEREG or Rag1−/− mice induced with 30 min of tMCAO to expand Treg cells. The results illustrated that in wild-type mice, the infarct volumes in the experimental group were larger than those in the controls 1 day after IS, while the other two groups of mice showed no significant differences, confirming that Treg cells enhanced infarct size and worsened neurological function after IS [36].
Neutral effects
Apart from the results discussed in the former two sections, several findings indicate that Treg cells do not affect poststroke outcomes. Ren et al. inserted the coding sequence of the diphtheria toxin receptor into the Foxp3 allele to specifically target Treg cells. By treating the resulting mice with diphtheria toxin, the researchers achieved almost complete Foxp3 depletion in the mice. The results showed that there was no significant differences in the infarct volumes and behavioral performance between vehicle- and diphtheria toxin-treated mice challenged by 60 min of tMCAO [37]. Interestingly, in the study of Stubbe et al., as previously described, although Treg cells fluctuated after the onset of IS, when CD4+CD45RBhigh cells were adoptively transferred into Rag1−/− mice 1 day before MCAO, the mice showed no de novo generation of Treg cells 2 weeks after occlusion. Moreover, to clarify the role of CD25+ Treg cells in the late phase after MCAO, the researchers injected anti-CD25 mAbs 3 and 14 days after tMCAO. Additionally, the researchers measured the infarct size 3 and 27 days after IS and evaluated neural function at 14 and 27 days after IS onset. However, the results showed no significant difference between the Treg depletion group and the controls [22].
Probable reasons for the paradoxical functions of Treg cells
The extent of brain damage
Emerging evidence has shown that infarct size could be a major determinant of changes in Treg cells. Liesz et al. demonstrated that only experimental models with extensive infarcts showed reduced lymphocyte counts; similarly in stroke patients, only severe IS patients went through poststroke peripheral immunosuppression [38, 39]. Moreover, splenic lymphocyte apoptosis and the production of blood cytokines were more significant in extensive infarcts than in small infarcts [38]. Consistent with that finding, Hug et al. indicated that infarct size was the major factor associated with the reduction in T helper cell counts on days 1 and 4 poststroke, which was further associated with infections [40]. The paradoxical findings reviewed here could be explained partly by the different types of IS models. With different infarct volumes, the lesions in the hemisphere could be diverse, thus resulting in the different effects of Treg cells.
The timing of Treg cell analysis
Generally, the neuroprotective effect of Treg cells was described in the later phase of infarct development, focusing on tissue reorganization and repair, while detrimental effects were observed during the acute phase [26, 34]. Moreover, Schumann et al. injected CD28 SA into tMCAO model animals and showed a positive correlation between Treg cell expansion and enhanced stroke size at 24 h after IS [36]. Na et al. also injected CD28 SA into tMCAO model animals and observed decreased infarct sizes and alleviated functional deficits 7 days after stroke onset [41]. However, Schumann et al. injected CD28 SA 3 days before or immediately after occlusion while the injection time in Na et al. was 3 or 6 h after IS onset; thus, the timing of Treg cell expansion is also a factor that cannot be ignored regarding this paradoxical phenomenon [36, 41, 42].
The inflammatory environment
It has been demonstrated that the role of Treg cells is associated with the specific inflammatory environment to maintain homeostasis [43]. Poststroke neuroinflammation is dissimilar among murine pMCAO and tMCAO models; that is, the role of Treg cells may depend on the experimental model [44]. It has been shown that neuroinflammation is more significant in pMCAO mice than in tMCAO mice, as more T-cell infiltration, microglial activation, and the expression of cytokines such as IL-1 and TNF-α could be observed 5 days after pMCAO than tMCAO [44]. Furthermore, compared to pMCAO models, tMCAO mice have been suggested to have secondary microthrombosis and potential endothelial damage within minutes after reperfusion [45, 46]. Secondary microthrombi were demonstrated to be responsible for ~70% of the final ischemic lesion size [47]. Moreover, microthrombosis was demonstrated to be partially independent of the inflammatory mechanism, which might contribute to pathophysiology and thus have a crucial impact on the outcomes of Treg cells [14]. In addition, the occurrence of secondary microthrombosis might affect local tissue-specific pathophysiological cascades associated with endothelial activation, thromboinflammation, and microvascular dysfunction, resulting in secondary ischemic infarctions and altered neuroinflammatory responses [14]. Ito et al. proved that tissue Treg cells exert tissue-specific effects, contributing to the maintenance of tissue homeostasis and repair by interacting with both inflammatory cells and tissue cells [13]. However, clinical studies have illustrated that secondary microthrombosis is modest or nonexistent after reperfusion [46, 48]. Therefore, the abovementioned differences between the tMCAO and pMCAO models might directly impact Treg cell function.
Mechanisms of Treg cells in IS progression
Cytokines-based secretion pathways
Through the secretion of anti-inflammatory cytokines, Treg cells are known to attenuate brain injury after IS [18]. Interleukin-10 (IL-10) is an essential cytokine that has been indicated to have pleiotropic immunosuppressive functions [49]. IL-10 plays a crucial role in cytokine-mediated modulation of neuroinflammation after IS by targeting antigen-presenting cells (APCs), such as targeting macrophages to inhibit the release of proinflammatory mediators [14, 49]. IL-10 is derived from both innate and adaptive immune cells, such as microglia and several subtypes of T cells, including Treg cells and Th2 cells, while T cells are the main sources of IL-10 [49, 50]. Furthermore, it has been verified that IL-10 plays a crucial role in the protective mechanism of Treg cells [50]. Several studies have described improved outcomes with the production of lymphocyte-derived IL-10 or the administration of IL-10 in mouse models [41, 51], indicating the protective effects of IL-10 on IS.
Moreover, interleukin-33 (IL-33), a novel cytokine of the IL-1 family, was reported to activate Treg cells associated with CNS repair and is known to facilitate the recovery of tissue after CNS injury and induce M2 macrophage-related genes [13, 52]. Several studies have shown that immediate intraperitoneal injection of IL-33 after tMCAO resulted in the enhancement of CD4+CD25+Foxp3+ Treg cells and a reduction in IFN-γ+ Treg cells in the spleen and brain compared to those in controls [52, 53]. Moreover, IL-33 upregulated Foxp3 mRNA levels, suggesting a protective role of IL-33 through promoting the Treg response [53]. Treg cell culture supernatant was treated with IL-33, the results showed increased production of IL-10, IL-35, and transforming growth factor-β (TGF-β), all of which are Treg cell cytokines [52], accompanied by decreased infarct sizes after IS [53]. However, the effects of IL-33 are suppressed when suppression of tumorigenicity 2 (ST2) is blocked [54]. The mice that were administered the anti-CD25 antibody showed a reduced protective effect of IL-33 [52]. Thus, IL-33 plays a neuroprotective role after IS onset and enhances Treg cells through the secretion of cytokines and a reduction in apoptosis-related proteins in an ST2-dependent manner [54].
In addition, there are several Treg cell-related mechanisms that impact the outcomes after IS through the secretion of other cytokines such as IL-35 and TGF-β [50].
Perforin- and granzyme-mediated cytolytic pathways
The suppressive activity of Treg cells includes inducing the death of effector cells by cytolysis [55]. Activated human Foxp3+ Treg cells were shown to express granzyme A but little granzyme B [56, 57]. Furthermore, Treg cell-mediated killing occurred in a perforin-dependent, granzyme A-dependent, and Fas-FasL-independent manner to activate certain subtypes of T cells, such as CD4+ and CD8+ T cells, and other kinds of cells [56]. In contrast, the expression of granzyme B was reported to be upregulated after the activation of Treg cells in mice [58]. Gondek et al. claimed that granzyme B-deficient mice showed suppressive Treg cell activity in vitro, while this response seemed to be perforin-independent [59]. Another study by Cao et al. confirmed that Treg cells could kill NK cells and CTLs in a perforin- and granzyme B-dependent manner, leading to the suppression of these cells in vivo [60]. Overall, Treg cell-mediated suppression works through perforin- and granzyme-mediated cytolytic mechanisms.
The PD-1/PD-L1 pathway
Programmed death-1 (PD-1) is an inducible immunomodulatory receptor expressed on B and T cells, as well as myeloid-derived cells, that regulates the adaptive immune response [61]. PD-L1 is a ligand of PD-1, and the interaction between PD-1 and PD-L1 can have an effect on the maintenance of peripheral tolerance [62]. Li et al. demonstrated the neuroprotective role of PD-L1 via the Treg cell-mediated inhibition of neutrophil-derived metalloproteinase-9 [63]. In contrast, Bodhankar et al. showed that PD-L1 enhanced CNS inflammation and infarct size, as evidenced by the decreased expression of CD80 by APCs and increased PD-1 and PD-L2 expression in PD-L1−/− mice, indicating the presence of an alternative suppressor T-cell signaling pathway [64]. When the researchers further blocked the PD-L1 checkpoint, the infarct volumes were reduced markedly, and CD8+ Treg cells were increased in the lesioned CNS hemisphere [65]. Hence, more work is required to determine how the PD-1/PD-L1 pathway precisely affects Treg cells.
The Htr7-based pathway
Serotonin (5-HT) is an important neurotransmitter that is involved in a wide range of physiological processes in the CNS, including pain, mood, anxiety, cardiovascular function, circadian rhythms, and feeding, and has been shown to participate in almost all neuropsychiatric disorders [66, 67]. 5-HT receptors have been proven to play a crucial role in enabling 5-HT to exert its effects [67]. Htr7 is a kind of G protein-coupled receptor for 5-HT that is generally expressed in the CNS and links to adenylate cyclase, which was further demonstrated to enhance the level of cyclic adenosine monophosphate (cAMP) [68, 69]. Intracellular cAMP was shown to promote the proliferation of Treg cells and enable Treg cells to maintain immune homeostasis by transmitting cAMP through membrane gap junctions to effector T cells, resulting in the suppression of these cells [58, 70]. There were improvements in neurological symptoms and an elevated level of Treg cells after the administration of 5-HT, which was not observed in Htr7-deficient mice [13]. In general, Treg cells can proliferate and be activated via an Htr7-dependent pathway [13].
Treg cell crosstalk with other cells
Treg cell crosstalk with microglia
Microglia are resident macrophage-like neuroimmune cells that are involved in the innate immune response in the CNS [71, 72]. After IS, microglia are activated, accumulate in the brain and differentiate into two subsets: the M1 proinflammatory phenotype and the M2 anti-inflammatory phenotype [73]. A lack of Treg cells induces microglia to transform into the M1 phenotype; in contrast, enhancing Treg cells promotes the transition toward the M2 phenotype [74]. Furthermore, M2 microglia were shown to promote Treg cell polarization by secreting IL-10 and TGF-β, resulting in reductions in infarct sizes after experimental IS [75]. Conversely, Treg cells play an important role in promoting the development of M2 microglia by elevating the IL-10-mediated expression of glycogen synthase kinase 3 beta [74, 76], which indicates that the crosstalk between M2 microglia and Treg cells via the secretion of IL-10 and TGF-β protects against brain damage after IS [73]. Additionally, Shu et al. found that microglia significantly induced hypoxia-inducible factor 1-alpha (HIF-1α) expression in Treg cells, and the inhibition of HIF-1α suppressed microglia-induced Sirt2 upregulation, which was proven to attenuate the anti-inflammatory effects of Treg cells [77].
Treg cell crosstalk with astrocytes
Astrocytes are crucial immune cells that maintain CNS homeostasis through neurotransmitters, synaptic plasticity, pH, cerebral blood flow, and water balance in pathological conditions such as IS [33, 78]. In recent decades, astrocytes have been shown to play pathogenic roles in disease processes due to their transformation of hyperplastic and hypertrophic cells; however, recent studies have demonstrated that astrocytes might exert neuroprotective effects [33, 78], which are influenced by the context and time of brain or spinal cord injury [33, 78]. Tobias et al. discovered an increase in astrocytes in DEREG mice, which was consistent with the results of Minako et al, indicating that both a reduction in T-cell infiltration into the brain and the depletion of Treg cells significantly elevated reactive astrocyte markers [33, 79]. In summary, Treg cells suppress astrogliosis, and the crosstalk between Treg cells and astrocytes affects neurological recovery.
Treg cell crosstalk with dendritic cells
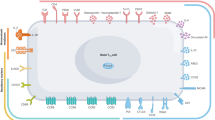
Dendritic cells (DCs) are a group of specialized APCs with crucial roles in the response to foreign and self-antigens by initiating and regulating the T-cell response [80, 81]. The crosstalk between DCs and Treg cells has been demonstrated to be crucial in immunomodulation. Cytotoxic T lymphocyte antigen 4 (CTLA-4) is a molecule in the CD28 family that is secreted by DCs and plays an important role in activating Treg cells [82, 83]. Several studies have shown that through the binding of CTLA-4 to CD80 and CD86 on DCs, Treg cells can modulate DC biology via the phosphoinositide 3-kinase/Akt/mechanistic target of rapamycin (mTOR) pathway, thus exerting tolerogenic effects [84]. Furthermore, indoleamine 2,3-dioxygenase (IDO), a rate-limiting enzyme in tryptophan catabolism, was proven to be induced by the CTLA-4-CD80/86 axis [85]. The metabolites derived by IDO were demonstrated to promote DCs to express ILT3 and ILT4, thus enhancing the induction of Treg cells [86]. In summary, DCs and Treg cells can affect each other to exert effects on the immune response and maintain tolerance [81] (Fig. 1).

Ischemic stroke leads to the damage of the BBB and neuron cells. The CNS antigen would be generated, pass the BBB and drain to the peripheral lymphoid nodes where they are taken up by APCs and further presented to naïve T cells. Treg cells as crucial immunomodulators, are transferred into the brain and exert a neuroprotective effect through the cytokine-dependent pathway via IL-10, IL-33, etc. Moreover, Treg cells modulate effector cells through the granzyme-dependent pathway, the crosstalk with several other cells, and the inhibition of neutrophil-derived MMP-9. However, Treg cells can be detrimental to cerebral injury with microthrombosis and microvascular dysfunction.
Treg cells participate in immunodepression and associated infections
There is homeostasis between the immune system and the nervous system under physiological conditions [87]. This balance can be interrupted, causing the activation and infiltration of immune cells into the brain [88]. However, although the immune response in the CNS contributes to neuronal injury in the early phase, the subsequent decrease in immune cells in the circulation causes stroke-induced immunodepression (SIID) as early as 12 h after IS and can last for months and years [89, 90], and SIID enhances host susceptibility to infection [91]. Recent studies have shown that ~30% of stroke patients experience infectious complications, with the most prevalent complications being pneumonia and urinary tract infections [91], which contribute to the ~20% mortality and high morbidity in stroke survivors [92]. Importantly, Treg cells have been proven to have long-term effects on SIID by promoting IL-10 secretion [89, 93]. Interestingly, Pang et al. discovered a relationship between infarct size and infection, and patients with larger infarct sizes showed a higher probability of infection [21].
Treg cells in human patients
Recent preclinical evidence attributed the risk of infection to stroke-induced immunosuppression. Generally, a significantly decreased spleen size has been observed in some patients in clinical studies after stroke [94, 95]. Moreover, Jiang et al. investigated that on days 1, 3, and 7 after stroke onset, the number of B cells, Th cells, NK cells, and cytotoxic T cells decreased, but the results showed an increase in the number of Treg cells, indicating a correlation between the splenic volumes and immune cells; B cells, Th cells, and cytotoxic T cells correlated positively with spleen size, while Treg cells correlated negatively with spleen volumes after stroke onset [95]. Furthermore, the balance between Th cells and Treg cells has attracted attention in the context of immunosuppression. Th cells are a crucial subtype of T cells that participate in the adaptive immune response by initiating and exacerbating the inflammatory response, whereas Treg cells play an immunosuppressive role in the immune response [96]. Compared to those of healthy controls, the levels of TNF-α and IL-10 were enhanced significantly, while IFN-γ was reduced in the serum of stroke patients [95]. IFN-γ secreted by Th cells plays an essential role in host survival, and IL-10 and TGF-β promote the differentiation of Treg cells [97, 98]; thus, changes in these cytokines in the circulation after IS may contribute to functional changes in the immune system and influence poststroke infection [95].
Treg cells in animal models
Splenic atrophy was also observed in an experimental study conducted by Offner et al. and was accompanied by a drastic reduction in splenocyte numbers and an increase in the number and activation of splenic CD4+FoxP3+ Treg cells secreting IL-10 [27]. As mentioned previously, the imbalance between Th cells and Treg cells can contribute to poststroke infection [96]. Dang et al. showed that the transcription factor HIF-1α exerted a regulatory effect on the balance between Treg cells and Th17 cells by directly binding to the loci encoding RORγt and histone modification enzymes such as p300 affecting the promoter regions of these target genes, thereby inducing their transcription and inhibiting FoxP3 [99, 100]. Furthermore, the sympathetic nervous system is one branch of the autonomic nervous system and has been demonstrated to modulate poststroke immune structures [91]. One of the factors mediating systemic immunosuppression after IS was that the hypothalamic-pituitary-adrenal axis was activated, leading to excessive secretion of glucocorticoids and thus inducing T cells to decrease the production of IFN-γ, further resulting in the enhanced secretion of IL-10 derived from Treg cells and apoptotic cell death [93].
IS therapy associated with Treg cells
Several clinical studies have used the expansion of peripheral Treg cells as a potential treatment for diseases such as Alzheimer’s disease (NCT01409915), Parkinson’s disease (NCT01882010), and amyotrophic lateral sclerosis (NCT04055623) [25]. Although it remains controversial whether Treg cells are beneficial or detrimental to the brain after IS, the therapeutic potential of Treg cells after IS has attracted the attention of numerous researchers due to their long therapeutic window after the onset of IS [101]. Although several studies have suggested an increased level of Treg cells in the late phase of IS, a reduction in circulating Treg cells soon after IS has been revealed [17, 20], providing a therapeutic strategy to increase the number of Treg cells in the early phase after IS. Li et al. enriched Treg cells in mice or rats at 2, 6, and 24 h after reperfusion [29]. The results illustrated that adoptively transferring Treg cells at all three time points significantly reduced brain infarcts, indicating that treatment with Treg cells might be effective for up to 24 h after IS [29]. In general, the application of Treg cells as a treatment for IS has arisen recently (Table 2).
Sources of Treg cells
As mentioned previously, the most direct approach for Treg cell therapy is adoptive transfer of purified Treg cells. However, as Treg cells are a small subset of T cells and constitute 5% of CD4+ T cells in circulation, it is necessary to expand Treg cells in vivo or in vitro [102]. The most accessible source of Treg cells is peripheral blood [101]. As the rate of the expansion of Treg cells in vitro is limited, Treg cells derived from umbilical cord blood (UCB) were proven to be another often used source [101]. Moreover, Dijke et al. showed that discarded pediatric thymuses could be a novel source of Treg cells with the potential to overcome the limitations of UCB and peripheral blood [103].
Stimulants to expand Treg cells
Direct stimulants
The most frequently used stimulants to expand Treg cells are anti-CD3 and anti-CD28 antibodies [104, 105]. In the presence of IL-2, Treg cells can maintain their suppressive function and phenotype [105]. Moreover, to ensure highly selective Treg cell expression, rapamycin, a mTOR inhibitor, is often added to prevent the proliferation of T effector cells [106, 107]. However, this suppression could affect Treg cells, which could result in requiring a long time for Treg cells to expand. Therefore, manufacturing practice-licensed artificial antigen-presenting cells are frequently used together with anti-CD3/CD28 antibodies and IL-2 to stimulate the pure expansion of Treg cells [102, 108].
Moreover, high concentrations of IL-2 have been used to expand Treg cells, and Ethan noticed that IL-2 coupled to anti-IL-2 mAbs could also inhibit the expansion of other T-cell subsets by blocking the binding site on IL-2, thereby selectively expanding Treg cells [109, 110]. Furthermore, the engagement of DR3, a member of the tumor necrosis factor receptor superfamily, was proven to facilitate the expansion of Treg cells [111, 112]. Therefore, an agonistic antibody against DR3 might be an excellent choice for expanding FoxP3+ Treg cells in vivo [111, 112].
Indirect stimulants
In addition to stimulants that directly act on Treg cells, FMS-like receptor tyrosine kinase ligand (Flt3L) can expand Treg cells in an indirect manner [113, 114]. This strategy has been revealed to work by expanding plasmacytoid dendritic cells [113]. As reviewed previously, DCs crosstalk with Treg cells, further leading to an increased number of Treg cells [101]. However, as Flt3L does not induce Treg cells, Flt3L is generally used in combination with rapamycin [113].
Pathological approaches
Previous studies suggested that Treg cells could be expanded through a pathological approach of “mucosal immunization” by the administration of cerebrovascular antigens [115,116,117]. E-selectin is a glycoprotein adhesion molecule that plays a role in maintaining immune tolerance, is specifically expressed on endothelial cells and recruits immunomodulatory factors to the vascular tree where thrombosis or hemorrhage occur [115, 116]. Researchers have proven that the repeated intranasal administration of recombinant E-selectin could enhance the number of Treg cells and thus induce mucosal tolerance and promote brain repair after stroke [115, 116]. Moreover, Gee et al. showed that intranasal administration of myelin basic protein could induce the response of Treg cells, leading to the prevention of deleterious autoimmunity after IS [117].
Conclusion
In summary, accumulating evidence from experimental and clinical studies has demonstrated that Treg cells play a complicated role in IS. As one of the potent immune cells that participate in the immune response after IS, Treg cells are capable of modulating several immunological pathways by secreting cytokines, cytolysis, essential receptor-based pathways, and interacting with other cells. Although there are still some controversial views on the functions of Treg cells, current studies of adoptive transfer or the expansion of Treg cells in vivo and in vitro have suggested that Treg cells might be an innovative therapeutic strategy for the treatment of IS. However, as Treg cells can exert different effects in different phases after IS onset, it is necessary to further evaluate the proper time, dose, and site of injection of the expanded Treg cells. Moreover, as the survival time of Treg cells is limited, it is necessary to investigate the safety of this strategy in humans. Overall, a comprehensive understanding of Treg cells is still urgently needed.
References
Nakamura K, Shichita T. Cellular and molecular mechanisms of sterile inflammation in ischaemic stroke. J Biochem. 2019;165:459–64.
Zhang R, Zhang Z, Chopp M. Function of neural stem cells in ischemic brain repair processes. J Cereb Blood Flow Metab. 2016;36:2034–43.
dela Pena IC, Yoo A, Tajiri N, Acosta SA, Ji X, Kaneko Y, et al. Granulocyte colony-stimulating factor attenuates delayed tPA-induced hemorrhagic transformation in ischemic stroke rats by enhancing angiogenesis and vasculogenesis. J Cereb Blood Flow Metab. 2015;35:338–46.
Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–12.
Chen C, Chu SF, Ai QD, Zhang Z, Chen NH. CKLF1/CCR5 axis is involved in neutrophils migration of rats with transient cerebral ischemia. Int Immunopharmacol. 2020;85:106577.
Guerrini MM, Okamoto K, Komatsu N, Sawa S, Danks L, Penninger JM, et al. Inhibition of the TNF family cytokine RANKL prevents autoimmune inflammation in the central nervous system. Immunity. 2015;43:1174–85.
Jian Z, Liu R, Zhu X, Smerin D, Zhong Y, Gu L, et al. The involvement and therapy target of immune cells after ischemic stroke. Front Immunol. 2019;10:2167.
Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101:455–8.
Duffy SS, Keating BA, Perera CJ, Moalem-Taylor G. The role of regulatory T cells in nervous system pathologies. J Neurosci Res. 2018;96:951–68.
Spitz C, Winkels H, Burger C, Weber C, Lutgens E, Hansson GK, et al. Regulatory T cells in atherosclerosis: critical immune regulatory function and therapeutic potential. Cell Mol Life Sci. 2016;73:901–22.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64.
Xu X, Li M, Jiang Y. The paradox role of regulatory T cells in ischemic stroke. Sci World J. 2013;2013:174373.
Ito M, Komai K, Nakamura T, Srirat T, Yoshimura A. Tissue regulatory T cells and neural repair. Int Immunol. 2019;31:361–9.
Liesz A, Kleinschnitz C. Regulatory T Cells in post-stroke immune homeostasis. Transl Stroke Res. 2016;7:313–21.
Hu Y, Zheng Y, Wu Y, Ni B, Shi S. Imbalance between IL-17A-producing cells and regulatory T cells during ischemic stroke. Mediators Inflamm. 2014;2014:813045.
Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. 2016;13:167–79.
Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009;158:1174–83.
Chen S, Wu H, Klebe D, Hong Y, Zhang J, Tang J. Regulatory T cell in stroke: a new paradigm for immune regulation. Clin Dev Immunol. 2013;2013:689827.
Yan J, Read SJ, Henderson RD, Hull R, O’Sullivan JD, McCombe PA, et al. Frequency and function of regulatory T cells after ischaemic stroke in humans. J Neuroimmunol. 2012;243:89–94.
Yan J, Greer JM, Etherington K, Cadigan GP, Cavanagh H, Henderson RD, et al. Immune activation in the peripheral blood of patients with acute ischemic stroke. J Neuroimmunol. 2009;206:112–7.
Pang X, Qian W. Changes in regulatory T-cell levels in acute cerebral ischemia. J Neurol Surg A Cent Eur Neurosurg. 2017;78:374–9.
Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2013;33:37–47.
Chaudhry A, Rudensky AY. Control of inflammation by integration of environmental cues by regulatory T cells. J Clin Invest. 2013;123:939–44.
Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature. 2016;535:425–9.
Machhi J, Kevadiya BD, Muhammad IK, Herskovitz J, Olson KE, Mosley RL, et al. Harnessing regulatory T cell neuroprotective activities for treatment of neurodegenerative disorders. Mol Neurodegener. 2020;15:32.
Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Gobel K, Schuhmann MK, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121:679–91.
Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176:6523–31.
Seifert HA, Hall AA, Chapman CB, Collier LA, Willing AE, Pennypacker KR. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J Neuroimmune Pharmacol. 2012;7:1017–24.
Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74:458–71.
Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–57.
Llovera G, Roth S, Plesnila N, Veltkamp R, Liesz A. Modeling stroke in mice: permanent coagulation of the distal middle cerebral artery. J Vis Exp. 2014:e51729. https://doi.org/10.3791/51729.
Llovera G, Hofmann K, Roth S, Salas-Perdomo A, Ferrer-Ferrer M, Perego C, et al. Results of a preclinical randomized controlled multicenter trial (pRCT): anti-CD49d treatment for acute brain ischemia. Sci Transl Med. 2015;7:299ra121.
Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 2019;565:246–50.
Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–9.
Zhang H, Xia Y, Ye Q, Yu F, Zhu W, Li P, et al. In vivo expansion of regulatory T cells with IL-2/IL-2 Antibody complex protects against transient ischemic stroke. J Neurosci. 2018;38:10168–79.
Schuhmann MK, Kraft P, Stoll G, Lorenz K, Meuth SG, Wiendl H, et al. CD28 superagonist-mediated boost of regulatory T cells increases thrombo-inflammation and ischemic neurodegeneration during the acute phase of experimental stroke. J Cereb Blood Flow Metab. 2015;35:6–10.
Ren X, Akiyoshi K, Vandenbark AA, Hurn PD, Offner H. CD4+FoxP3+ regulatory T-cells in cerebral ischemic stroke. Metab Brain Dis. 2011;26:87–90.
Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, et al. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009;40:2849–58.
Liesz A, Ruger H, Purrucker J, Zorn M, Dalpke A, Mohlenbruch M, et al. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS One. 2013;8:e74839.
Hug A, Dalpke A, Wieczorek N, Giese T, Lorenz A, Auffarth G, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke. 2009;40:3226–32.
Na SY, Mracsko E, Liesz A, Hunig T, Veltkamp R. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke. 2015;46:212–20.
Zhuang R, Feinberg MW. Regulatory T cells in ischemic cardiovascular injury and repair. J Mol Cell Cardiol. 2019;147:1–11.
Danese S, Rutella S. The Janus face of CD4+CD25+ regulatory T cells in cancer and autoimmunity. Curr Med Chem. 2007;14:649–66.
Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N, et al. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol. 2013;23:34–44.
Gauberti M, Martinez de Lizarrondo S, Orset C, Vivien D. Lack of secondary microthrombosis after thrombin-induced stroke in mice and non-human primates. J Thromb Haemost. 2014;12:409–14.
Bai J, Lyden PD. Revisiting cerebral postischemic reperfusion injury: new insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke. 2015;10:143–52.
Pham M, Kleinschnitz C, Helluy X, Bartsch AJ, Austinat M, Behr VC, et al. Enhanced cortical reperfusion protects coagulation factor XII-deficient mice from ischemic stroke as revealed by high-field MRI. Neuroimage. 2010;49:2907–14.
Lansberg MG, Straka M, Kemp S, Mlynash M, Wechsler LR, Jovin TG, et al. MRI profile and response to endovascular reperfusion after stroke (DEFUSE 2): a prospective cohort study. Lancet Neurol. 2012;11:860–7.
Neumann C, Scheffold A, Rutz S. Functions and regulation of T cell-derived interleukin-10. Semin Immunol. 2019;44:101344.
Liesz A, Hu X, Kleinschnitz C, Offner H. Functional role of regulatory lymphocytes in stroke: facts and controversies. Stroke. 2015;46:1422–30.
Liesz A, Zhou W, Na SY, Hammerling GJ, Garbi N, Karcher S, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci. 2013;33:17350–62.
Liu X, Hu R, Pei L, Si P, Wang C, Tian X, et al. Regulatory T cell is critical for interleukin-33-mediated neuroprotection against stroke. Exp Neurol. 2020;328:113233.
Xiao W, Guo S, Chen L, Luo Y. The role of Interleukin-33 in the modulation of splenic T-cell immune responses after experimental ischemic stroke. J Neuroimmunol. 2019;333:576970.
Zhang C, Li L, Feng K, Fan D, Xue W, Lu J. ‘Repair’ treg cells in tissue injury. Cell Physiol Biochem. 2017;43:2155–69.
Shalev I, Schmelzle M, Robson SC, Levy G. Making sense of regulatory T cell suppressive function. Semin Immunol. 2011;23:282–92.
Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–45.
Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601.
Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32.
Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–6.
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–46.
Yao S, Chen L. PD-1 as an immune modulatory receptor. Cancer J. 2014;20:262–4.
Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014;20:265–71.
Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, et al. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke. 2014;45:857–64.
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. PD-L1 enhances CNS inflammation and infarct volume following experimental stroke in mice in opposition to PD-1. J Neuroinflammation. 2013;10:111.
Bodhankar S, Chen Y, Lapato A, Dotson AL, Wang J, Vandenbark AA, et al. PD-L1 monoclonal antibody treats ischemic stroke by controlling central nervous system inflammation. Stroke. 2015;46:2926–34.
Leopoldo M, Lacivita E, Berardi F, Perrone R, Hedlund PB. Serotonin 5-HT7 receptor agents: structure-activity relationships and potential therapeutic applications in central nervous system disorders. Pharmacol Ther. 2011;129:120–48.
Wei YB, McCarthy M, Ren H, Carrillo-Roa T, Shekhtman T, DeModena A, et al. A functional variant in the serotonin receptor 7 gene (HTR7), rs7905446, is associated with good response to SSRIs in bipolar and unipolar depression. Mol Psychiatry. 2020;25:1312–22.
Vigli D, Rusconi L, Valenti D, La Montanara P, Cosentino L, Lacivita E, et al. Rescue of prepulse inhibition deficit and brain mitochondrial dysfunction by pharmacological stimulation of the central serotonin receptor 7 in a mouse model of CDKL5 deficiency disorder. Neuropharmacology. 2019;144:104–14.
Lenglet S, Louiset E, Delarue C, Vaudry H, Contesse V. Activation of 5-HT7 receptor in rat glomerulosa cells is associated with an increase in adenylyl cyclase activity and calcium influx through T-type calcium channels. Endocrinology. 2002;143:1748–60.
Klein M, Bopp T. Cyclic AMP represents a crucial component of Treg cell-mediated immune regulation. Front Immunol. 2016;7:315.
McDonough A, Weinstein JR. The role of microglia in ischemic preconditioning. Glia. 2020;68:455–71.
Chen C, Ai QD, Chu SF, Zhang Z, Chen NH. NK cells in cerebral ischemia. Biomed Pharmacother. 2019;109:547–54.
Wang S, Zhang H, Xu Y. Crosstalk between microglia and T cells contributes to brain damage and recovery after ischemic stroke. Neurol Res. 2016;38:495–503.
Zhou K, Zhong Q, Wang YC, Xiong XY, Meng ZY, Zhao T, et al. Regulatory T cells ameliorate intracerebral hemorrhage-induced inflammatory injury by modulating microglia/macrophage polarization through the IL-10/GSK3beta/PTEN axis. J Cereb Blood Flow Metab. 2017;37:967–79.
Brea D, Agulla J, Rodriguez-Yanez M, Barral D, Ramos-Cabrer P, Campos F, et al. Regulatory T cells modulate inflammation and reduce infarct volume in experimental brain ischaemia. J Cell Mol Med. 2014;18:1571–9.
Klebe D, McBride D, Flores JJ, Zhang JH, Tang J. Modulating the immune response towards a neuroregenerative peri-injury milieu after cerebral hemorrhage. J Neuroimmune Pharmacol. 2015;10:576–86.
Shu L, Xu CQ, Yan ZY, Yan Y, Jiang SZ, Wang YR. Post-stroke microglia induce Sirtuin2 expression to suppress the anti-inflammatory function of infiltrating regulatory T cells. Inflammation. 2019;42:1968–79.
Nutma E, van Gent D, Amor S, Peferoen LAN. Astrocyte and oligodendrocyte cross-talk in the central nervous system. Cells. 2020;9:600.
Kramer TJ, Hack N, Bruhl TJ, Menzel L, Hummel R, Griemert EV, et al. Depletion of regulatory T cells increases T cell brain infiltration, reactive astrogliosis, and interferon-gamma gene expression in acute experimental traumatic brain injury. J Neuroinflammation. 2019;16:163.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
Bourque J, Hawiger D. Immunomodulatory bonds of the partnership between dendritic cells and T cells. Crit Rev Immunol. 2018;38:379–401.
Halpert MM, Konduri V, Liang D, Chen Y, Wing JB, Paust S, et al. Dendritic cell-secreted cytotoxic T-lymphocyte-associated protein-4 regulates the T-cell response by downmodulating bystander surface B7. Stem Cells Dev. 2016;25:774–87.
Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302.
Alissafi T, Banos A, Boon L, Sparwasser T, Ghigo A, Wing K, et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J Clin Invest. 2017;127:2789–804.
Harden JL, Egilmez NK. Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol Invest. 2012;41:738–64.
Brenk M, Scheler M, Koch S, Neumann J, Takikawa O, Hacker G, et al. Tryptophan deprivation induces inhibitory receptors ILT3 and ILT4 on dendritic cells favoring the induction of human CD4+ CD25+ Foxp3+ T regulatory cells. J Immunol. 2009;183:145–54.
Chavan SS, Pavlov VA, Tracey KJ. Mechanisms and therapeutic relevance of neuro-immune communication. Immunity. 2017;46:927–42.
Gan Y, Liu Q, Wu W, Yin JX, Bai XF, Shen R, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci USA. 2014;111:2704–9.
Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32:598–611.
Zhang S, Jin Y, Liu X, Yang L, Ge Z, Wang H, et al. Methamphetamine modulates glutamatergic synaptic transmission in rat primary cultured hippocampal neurons. Brain Res. 2014;1582:1–11.
Malone K, Amu S, Moore AC, Waeber C. Immunomodulatory therapeutic strategies in stroke. Front Pharmacol. 2019;10:630.
Shi K, Wood K, Shi FD, Wang X, Liu Q. Stroke-induced immunosuppression and poststroke infection. Stroke Vasc Neurol. 2018;3:34–41.
Santos Samary C, Pelosi P, Leme Silva P, Rieken Macedo Rocco P. Immunomodulation after ischemic stroke: potential mechanisms and implications for therapy. Crit Care. 2016;20:391.
Vahidy FS, Parsha KN, Rahbar MH, Lee M, Bui TT, Nguyen C, et al. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J Cereb Blood Flow Metab. 2016;36:1012–21.
Jiang C, Kong W, Wang Y, Ziai W, Yang Q, Zuo F, et al. Changes in the cellular immune system and circulating inflammatory markers of stroke patients. Oncotarget. 2017;8:3553–67.
Kleinewietfeld M, Hafler DA. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol. 2013;25:305–12.
Green AM, Difazio R, Flynn JL. IFN-gamma from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol. 2013;190:270–7.
Gao L, Lu Q, Huang LJ, Ruan LH, Yang JJ, Huang WL, et al. Transplanted neural stem cells modulate regulatory T, gammadelta T cells and corresponding cytokines after intracerebral hemorrhage in rats. Int J Mol Sci. 2014;15:4431–41.
Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–84.
Tsun A, Chen Z, Li B. Romance of the three kingdoms: RORgammat allies with HIF1alpha against FoxP3 in regulating T cell metabolism and differentiation. Protein Cell. 2011;2:778–81.
Xia Y, Cai W, Thomson AW, Hu X. Regulatory T cell therapy for ischemic stroke: how far from clinical translation? Transl Stroke Res. 2016;7:415–9.
Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discov. 2019;18:749–69.
Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transpl. 2016;16:58–71.
Mathew JM, Voss JH, LeFever A, Konieczna I, Stratton C, He J, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. 2018;8:7428.
Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI. In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood. 2007;109:827–35.
Rossetti M, Spreafico R, Saidin S, Chua C, Moshref M, Leong JY, et al. Ex vivo-expanded but not in vitro-induced human regulatory T cells are candidates for cell therapy in autoimmune diseases thanks to stable demethylation of the FOXP3 regulatory T cell-specific demethylated region. J Immunol. 2015;194:113–24.
Xie L, Sun F, Wang J, Mao X, Xie L, Yang SH, et al. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol. 2014;192:6009–19.
Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. 2011;3:83ra41.
Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transpl. 2013;13:3010–20.
Shevach EM. Application of IL-2 therapy to target T regulatory cell function. Trends Immunol. 2012;33:626–32.
Kim BS, Nishikii H, Baker J, Pierini A, Schneidawind D, Pan Y, et al. Treatment with agonistic DR3 antibody results in expansion of donor Tregs and reduced graft-versus-host disease. Blood. 2015;126:546–57.
Rodriguez-Barbosa JI, Schneider P, Graca L, Buhler L, Perez-Simon JA, Del Rio ML. The role of TNFR2 and DR3 in the in vivo expansion of tregs in T cell depleting transplantation regimens. Int J Mol Sci. 2020;21:3347.
Biswas M, Sarkar D, Kumar SR, Nayak S, Rogers GL, Markusic DM, et al. Synergy between rapamycin and FLT3 ligand enhances plasmacytoid dendritic cell-dependent induction of CD4+CD25+FoxP3+ Treg. Blood. 2015;125:2937–47.
Klein O, Ebert LM, Zanker D, Woods K, Tan BS, Fucikova J, et al. Flt3 ligand expands CD4+FoxP3+ regulatory T cells in human subjects. Eur J Immunol. 2013;43:533–9.
Ishibashi S, Maric D, Mou Y, Ohtani R, Ruetzler C, Hallenbeck JM. Mucosal tolerance to E-selectin promotes the survival of newly generated neuroblasts via regulatory T-cell induction after stroke in spontaneously hypertensive rats. J Cereb Blood Flow Metab. 2009;29:606–20.
Ishibashi S. Immunomodulation by inducing tolerance to E-selectin and adult neurogenesis after stroke. Rinsho Shinkeigaku Clin Neurol. 2010;50:882–5.
Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke. 2008;39:1575–82.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81730096, 82074044, 81873026, 81973499, and 81773924), the Natural Science Foundation of Beijing (7192135), the Drug Innovation Major Project (2018ZX09711001-002-007, 2018ZX09711001-003-005, and 2018ZX09711001-009-013), and the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2 M-1-004).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Wang, Hy., Ye, Jr., Cui, Ly. et al. Regulatory T cells in ischemic stroke. Acta Pharmacol Sin 43, 1–9 (2022). https://doi.org/10.1038/s41401-021-00641-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00641-4
Keywords
This article is cited by
-
Chronic vascular pathogenesis results in the reduced serum Metrnl levels in ischemic stroke patients
Acta Pharmacologica Sinica (2024)
-
Tregs dysfunction aggravates postoperative cognitive impairment in aged mice
Journal of Neuroinflammation (2023)
-
Temporal changes in regulatory T cell subsets defined by the transcription factor Helios in stroke and their potential role in stroke-associated infection: a prospective case–control study
Journal of Neuroinflammation (2023)
-
Humanized cerebral organoids-based ischemic stroke model for discovering of potential anti-stroke agents
Acta Pharmacologica Sinica (2023)
-
Emerging Targets for Modulation of Immune Response and Inflammation in Stroke
Neurochemical Research (2023)
