
Abstract
Bromodomain and extra-terminal proteins (BETs) are potential targets for the therapeutic treatment of prostate cancer (PC). Herein, we report the design, the synthesis, and a structure−activity relationship study of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one derivative as novel selective BET inhibitors. One representative compound, 19 (Y06014), bound to BRD4(1) in the low micromolar range and demonstrated high selectivity for BRD4(1) over other non-BET bromodomain-containing proteins. This molecule also potently inhibited cell growth, colony formation, and mRNA expression of AR-regulated genes in PC cell lines. Y06014 also shows stronger activity than the second-generation antiandrogen enzalutamide. Y06014 may serve as a new small molecule probe for further validation of BET as a molecular target for PC drug development.
Similar content being viewed by others
Introduction
Prostate cancer (PC) is one of the most common malignancies diagnosed among men [1, 2]. Since Huggins and Hodges’s discovery in the 1940s that androgens promote PC growth [3], the central therapy for patients with locally advanced or metastatic disease is targeting the androgen receptor (AR). The standard first-line treatment for metastatic PC is androgen deprivation therapy (ADT), which can induce marked tumor regression and normalize serum prostate-specific antigen (PSA). Initially, patients are sensitive to ADT. However, most patients become insensitive to these therapies after long-term treatment and eventually progress to castration-resistant prostate cancer (CRPC), which is predominantly driven by deregulated AR signaling [4,5,6,7]. Unfortunately, most patients who develop CRPC succumb to the disease [8]. Several drugs targeting AR signaling against CRPC such as abiraterone acetate, enzalutamide, and darolutamide have been approved by the FDA. However, secondary resistance has been observed in patients [9,10,11,12]. Taken together, these data strongly argue for the development of new strategies to block AR function in CRPC.
The bromodomain and extra-terminal (BET) family is an important class of epigenetic readers [13]. It consists of bromodomain-containing protein 2 (BRD2), BRD3, BRD4, and BRDT and recognizes acetylated histones by tandem domains (BD1 and BD2) to regulate gene transcription [14]. Moreover, BRD2/3/4 can bind to the N-terminal domain of AR directly and this interaction can be disrupted by BET inhibitors, resulting in abrogation of AR-mediated transcription and inhibition of tumor growth in CRPC xenograft mouse models [8, 15,16,17,18]. These studies revealed that inhibition of BET proteins represents a promising approach to block AR signaling and overcome antiandrogen resistance. Thus, targeting BET proteins is an alternative strategy for the treatment of CRPC.
A number of small molecules BET inhibitors have been reported to date [19,20,21,22,23,24,25,26,27] (Fig. 1). JQ1 (1) was the first reported BET bromodomain inhibitor. This discovery motivated drug development efforts in this area. Since then, several BET inhibitors have been discovered, such as I-BET-762 (2) and I-BET-151 (3) [23, 24]. Recently, our group reported several selective BET inhibitors, such as Y02224, Y02234, Y06137, and Y08060 [16,17,18, 28]. However, compounds that have entered clinical trials for CRPC are still limited. Several BET inhibitors, including I-BET-762, OTX-015, ZEN003694, and GS-5829, have recently advanced into human clinical trials for CRPC as a single agent or in combination with AR antagonists (enzalutamide or ARN-509) [29,30,31,32]. Despite the discovery of these BET inhibitors, finding new, potent, and specific BET inhibitors with different chemotypes is still highly desirable to exploit the therapeutic potential of BET inhibition for CRPC.

Structures of representative BET bromodomain inhibitors.
In this study, we report that Y06014, a benzo[cd]indol-2(1H)-one derivative, binds to the BRD4 bromodomain. We found that Y06014 could inhibit cell growth, proliferation, and colony formation of PC cell lines. Y06014 also reduces the expression of the AR-regulated genes PSA, KLK2, and TMPRSS2. Overall, Y06014 represents a new class of compounds to develop BET inhibitors against PC.
Materials and methods
Computational methods
In the molecular docking study, the crystal structure of BRD4(1) in complex with the inhibitor Y02234 (PDB ID: 5YQX) [17] was used as the reference structure. Protein structure preparation for docking studies includes water deletion, hydrogen atom addition, and protonation state adjustment. All of the ligand and protein preparation were performed in Maestro (version 9.4, Schrödinger, LLC, New York, NY, 2013) implemented in the Schrödinger program (http://www.schrödinger.com). In this study, a molecular docking study was performed with the Glide program (version 6.1, Schrödinger, LLC, New York, NY, 2013) using the SP score mode.
General chemistry
All reagents and solvents were obtained from commercial sources and used without further purification. Flash chromatography was performed using silica gel (200–300 mesh). All reactions were monitored by TLC, using silica gel plates with fluorescence F254 and UV light visualization. 1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 MHz and the Bruker AV-500 spectrometer at 125 MHz. Coupling constants (J) are expressed in hertz (Hz). Chemical shifts (δ) of NMR are reported in parts per million (ppm) units relative to internal control (TMS). Low-resolution ESI-MS were recorded on an Agilent 1200 HPLC-MSD mass spectrometer and high-resolution ESI-MS on an Applied Biosystems Q-STAR Elite ESI-LC-MS/MS mass spectrometer. The purity of compounds was determined by reverse-phase high-performance liquid chromatography (HPLC) analysis to be >95%. HPLC instrument, Dionex Summit HPLC (column: Diamonsil C18, 5.0 μm, 4.6 × 250 mm (Dikma Technologies); detector, PDA-100 photodiode array; injector, ASI-100 autoinjector; pump, p-680A). A flow rate of 1.0 mL/min was used with a mobile phase of MeOH in H2O (v/v, 85:15).
1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl 4-methylbenzenesulfonate (8)
Benzo[de]isochromene-1,3-dione (7) (6 g, 30 mmol) and hydroxylamine hydrochloride (2 g, 30 mmol) were combined as a solution in pyridine (40 mL). The reaction was conducted under reflux for 1 h followed by cooling to 80 °C. To the reaction, the system mixture was added powdered p-toluenesulfonyl chloride (11.5 g, 60 mmol). After the addition, the reaction was performed under reflux for 1 h. After cooling to room temperature, the reaction mixture was poured into ice water (300 mL) and stirred to precipitate crystals. The precipitate was filtered and rinsed with additional cool water (100 mL) and saturated NaHCO3 (100 mL) to give the title compound (8.6 g, 78%) as a yellow solid.
Benzo[cd]indol-2(1H)-one (9)
To a solution of compound 8 (8.6 g, 23.5 mmol) in ethanol (50 mL) and water (40 mL) was added an aqueous solution of sodium hydroxide (2.7 mol/L, 30 mL) at room temperature. The mixture was heated to reflux temperature for 3 h while distilling the ethanol. After the reaction was completed, the reaction mixture was cooled to 75 °C, concentrated hydrochloric acid was added dropwise, and a yellow precipitate was formed. Then, the mixture was further cooled. The precipitate was collected by filtration and washed with water (100 mL × 2). The resulting crude product was purified by silica gel chromatography with dichloromethane (DCM) to give the title compound (3.3 g, 83%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 6.8 Hz, 1H), 7.78 (t, J = 7.6 Hz, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.48 (t, J = 7.2 Hz, 1H), 6.98 (d, J = 7.2 Hz, 1H).
6-Bromobenzo[cd]indol-2(1H)-one (10)
Benz[c,d]indol-2(1H)-one (3.38 g, 20 mmol) was dissolved in DCM (50 mL). Bromine (3.2 g, 20 mmol) was added to the solution and stirred at room temperature for 24 h. A saturated sodium thiosulfate aqueous solution (30 mL) was poured into the reaction mixture. The resulting precipitate was filtered off and washed with DCM to give the title compound a yellow solid (4.0 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.11 (d, J = 6.8 Hz, 1H), 8.09 (d, J = 5.2 Hz, 1H), 7.93 (t, J = 6.0 Hz, 1H), 7.50 (d, J = 6.4 Hz, 1H), 7.48 (t, J = 7.2 Hz, 1H), 6.90 (d, J = 6.0 Hz, 1H).
6-(3,5-Dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (11)
A mixture of 6-bromobenzo[cd]indol-2(1H)-one (10) (500 mg, 2.01 mmol), 3,5-dimethylisoxazole-4-boronic acid (426 mg, 3.02 mmol), Pd (pph3)4 (350 mg, 0.3 mmol), and K2CO3 (834 mg, 6.03 mmol) in DMF/H2O = 30:1 was stirred under argon at 100 °C overnight. After the reaction was complete, the reaction mixture was cooled to room temperature. The reaction mixture was extracted with EtOAc (50 mL × 3). The organic layer was washed with brine and dried over Na2SO4. The solid was filtered off, and the filtrate was concentrated under reduced pressure. The resulting crude product was purified by silica gel chromatography with petroleum ether/EtOAc (3/1, v/v) to yield the title compound (170 mg, 32%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.73 (s, 1H), 8.15 (dd, J = 5.5, 2.0 Hz, 1H), 7.81 (m, 2H), 7.30 (d, J = 7.2 Hz, 1H), 7.08 (d, J = 7.2 Hz, 1H), 2.32 (s, 3H), 2.16 (s, 3H). MS (ESI) m/z for C16H12N2O2 [M + H]+calcd. 264.28; found 265.8. HPLC, tR = 22.38 min, 99.00% purity.
1-(Cyclopentylsulfonyl)-6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (12)
General procedure for syntheses of 12, 13, 16. To a stirred solution of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (11) (150 mg, 0.57 mmol) in DMF (5 mL) was added NaH (68 mg, 1.71 mmol) at 0 °C. One hour later, cyclopentanesulfonyl chloride (144 mg, 0.85 mmol) was added to the solution and the reaction mixture was stirred at 0 °C for 3 h. Water was added, the aqueous layer was extracted with ethyl acetate (30 mL × 3), and the combined organic layer was washed with water and brine, dried with Na2SO4, and evaporated. The residue was purified by silica gel chromatography with petroleum ether/EtOAc (3/1, v/v) to afford the title compound (101 mg, 45%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.22 (d, J = 6.8 Hz, 1H), 7.84 (ddd, J = 15.2, 12.3, 7.5 Hz, 3H), 7.40 (d, J = 7.5 Hz, 1H), 4.42 (m, 1H), 2.32 (s, 3H), 2.26 (td, J = 14.5, 7.2 Hz, 2H), 2.16 (s, 3H), 2.09 (dt, J = 13.1, 7.5 Hz, 2H), 1.96 (m, 2H), 1.76 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 166.69, 165.81, 159.50, 135.97, 131.00, 130.87, 129.39, 129.02, 126.31, 126.02, 124.33, 123.63, 113.13, 111.02, 63.41, 27.47, 25.93, 11.60, 10.64. MS (ESI) m/z for C21H20N2O4S [M + H]+, calcd: 396.11; found 396.9. HPLC, tR = 37.62 min, 96.11% purity.
1-(Cyclohexylsulfonyl)-6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (13)
Yellow solid, 86% yield. 1H NMR (500 MHz, CDCl3) δ 8.22 (d, J = 6.9 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.82 (dd, J = 9.0, 7.4 Hz, 2H), 7.39 (d, J = 7.5 Hz, 1H), 3.77 (tt, J = 12.1, 3.4 Hz, 1H), 2.32 (s, 3H), 2.22 (d, J = 11.1 Hz, 2H), 2.15 (s, 3H), 1.91 (d, J = 13.6 Hz, 2H), 1.76 (ddd, J = 24.6, 12.3, 3.1 Hz, 2H), 1.39 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 166.68, 165.77, 159.48, 136.10, 130.98, 130.88, 129.38, 129.03, 126.26, 126.04, 124.30, 123.62, 113.13, 111.14, 62.96, 25.72, 24.95, 24.88, 11.60, 10.64. MS (ESI) m/z for C22H22N2O4S [M + H]+, calcd: 410.13; found 411.2. HPLC, tR = 41.17 min, 99.62% purity.
1-((2,4-Difluorophenyl)sulfonyl)-6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (16)
Yellow solid, 45% yield. 1H NMR (400 MHz, CDCl3) δ 8.42 (m, 1H), 8.12 (d, J = 7.0 Hz, 1H), 7.99 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.78 (dd, J = 8.2, 7.1 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.13 (ddd, J = 9.0, 7.9, 1.4 Hz, 1H), 6.89 (ddd, J = 10.5, 8.4, 2.4 Hz, 1H), 2.33 (s, 3H), 2.16 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 167.96, 166.73, 165.89, 164.61, 161.57, 159.49, 134.87, 134.20, 134.12, 131.03, 129.32, 129.09, 126.50, 126.02, 123.88, 123.82, 122.87, 113.11, 112.32, 112.18, 111.54, 105.99, 105.78, 105.59, 11.61, 10.65. MS (ESI) m/z for C22H14F2N2O4S [M + H]+, calcd: 440.06; found 441.6. HPLC, tR = 26.73 min, 95.40% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-(phenylsulfonyl)benzo[cd]indol-2(1H)-one (14)
To a stirred solution of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (11) (100 mg, 0.38 mmol) in DMF (5 mL) was added NaH (45 mg, 1.14 mmol) at 0 °C. One hour later, Benzenesulfonyl chloride (100 mg, 0.57 mmol) was added to the solution and the reaction mixture was stirred at 75 °C for 2 h. Water was added, the aqueous layer was extracted with ethyl acetate (30 mL × 3), and the combined organic layer was washed with water and brine, dried with Na2SO4, and evaporated. The residue was purified by silica gel chromatography with petroleum ether/EtOAc (3/1, v/v) to afford the title compound (60 mg, 40%) as a yellow solid.1H NMR (500 MHz, CDCl3) δ 8.22 (d, J = 7.9 Hz, 2H), 8.12 (d, J = 7.0 Hz, 1H), 8.01 (d, J = 7.5 Hz, 1H), 7.83 (d, J = 8.2 Hz, 1H), 7.79 (m, 1H), 7.65 (t, J = 7.4 Hz, 1H), 7.56 (t, J = 7.8 Hz, 2H), 7.44 (d, J = 7.5 Hz, 1H), 2.31 (s, 3H), 2.14 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.69, 164.79, 159.48, 138.47, 135.12, 134.37, 130.96, 130.74, 129.37, 129.25, 129.06, 128.06, 126.50, 125.90, 124.29, 123.78, 113.12, 111.01, 11.59, 10.64. MS (ESI) m/z for C22H16N2O4S [M + H]+, calcd: 404.08; found 405.1. HPLC, tR = 28.16 min, 98.03% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-((4-fluorophenyl)sulfonyl)benzo[cd]indol-2(1H)-one (15)
General procedure for syntheses of 15, 17, and 18. To a stirred solution of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (11) (90 mg, 0.34 mmol) in THF (10 mL) was added NaH (45 mg, 1.42 mmol) at 0 °C. One hour later, 4-fluorobenzenesulfonyl chloride (162 mg, 0.57 mmol) was added to the solution and the reaction mixture was stirred at rt for 3 h. Water was added, the aqueous layer was extracted with ethyl acetate (30 mL × 3), and the combined organic layer was washed with water and brine, dried with Na2SO4, and evaporated. The residue was purified by silica gel chromatography with petroleum ether/EtOAc (2/1, v/v) to afford the title compound (60 mg, 42%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.28 (m, 2H), 8.12 (d, J = 7.0 Hz, 1H), 7.99 (d, J = 7.5 Hz, 1H), 7.85 (d, J = 8.2 Hz, 1H), 7.81 (m, 1H), 7.44 (d, J = 7.5 Hz, 1H), 7.22 (t, J = 8.5 Hz, 2H), 2.31 (s, 3H), 2.14 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 167.23, 166.72, 165.18, 164.82, 159.46, 134.98, 134.45, 131.18, 131.10, 130.96, 130.86, 129.43, 129.10, 126.52, 126.00, 124.19, 123.95, 116.68, 116.50, 113.08, 111.02, 11.58, 10.63. MS (ESI) m/z for C22H15FN2O4S [M + H]+, calcd: 422.07; found 423.4. HPLC, tR = 33.17 min, 96.16% purity.
1-((5-Chloro-2-methoxyphenyl)sulfonyl)-6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (17)
Yellow solid, 56% yield. 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 2.6 Hz, 1H), 8.12 (d, J = 6.9 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.78 (dd, J = 8.2, 7.1 Hz, 1H), 7.53 (dd, J = 8.8, 2.6 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 6.86 (d, J = 8.9 Hz, 1H), 3.54 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.62, 164.67, 159.45, 156.01, 135.95, 135.82, 131.56, 130.86, 130.73, 129.24, 128.95, 127.53, 126.4, 125.93, 125.83, 124.16, 123.32, 113.60, 113.21, 111.48, 56.27, 11.63, 10.64. MS (ESI) m/z for C23H17ClN2O5S [M + H]+, calcd: 468.05; found 469.2. HPLC, tR = 22.69 min, 98.69% purity.
1-((5-Bromo-2-methoxyphenyl)sulfonyl)-6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (18)
Yellow solid, 41% yield. 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 2.5 Hz, 1H), 8.12 (d, J = 7.0 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.78 (dd, J = 8.2, 7.1 Hz, 1H), 7.67 (dd, J = 8.8, 2.5 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 6.80 (d, J = 8.8 Hz, 1H), 3.54 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.61, 164.66, 159.45, 156.50, 138.74, 135.96, 134.31, 130.86, 130.72, 129.24, 128.95, 127.89, 126.42, 125.83, 124.17, 123.32, 114.00, 113.21, 112.66, 111.48, 56.22, 11.63, 10.64. MS (ESI) m/z for C23H17BrN2O5S [M + H]+, calcd: 512.00; found 513.1. HPLC, tR = 30.81 min, 98.75% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-ethylbenzo[cd]indol-2(1H)-one (19)
General procedure for the synthesis of 19, 20, and 21. To a stirred solution of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (11) (80 mg, 0.3 mmol) in DMF (5 mL) was added NaH (35 mg, 0.9 mmol) at rt. 30 min later, chloroethane (71 mg, 0.45 mmol) was added to the solution and the reaction mixture was stirred at rt for 3 h. Water was added, the aqueous layer was extracted with ethyl acetate (20 mL × 3), and the combined organic layer was washed with water and brine, dried with Na2SO4, and evaporated. The residue was purified by silica gel chromatography with petroleum ether/EtOAc (2/1, v/v) to yield the desired product (80 mg, 90%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.13 (m, 1H), 7.72 (d, J = 3.3 Hz, 2H), 7.30 (d, J = 7.2 Hz, 1H), 6.98 (d, J = 7.2 Hz, 1H), 4.01 (q, J = 7.2 Hz, 2H), 2.31 (s, 3H), 2.15 (s, 3H), 1.41 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 167.58, 166.41, 159.71, 139.51, 130.45, 129.14, 128.98, 128.58, 127.31, 125.59, 124.56, 121.24, 113.68, 104.76, 35.04, 14.04, 11.56, 10.64. MS (ESI) m/z for C18H16N2O2 [M + H]+, calcd: 292.12; found 293.0. HPLC, tR = 21.95 min, 98.52% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-propylbenzo[cd]indol-2(1H)-one (20)
Yellow solid, 75% yield. 1H NMR (400 MHz, CDCl3) δ 8.13 (m, 1H), 7.75 (m, 2H), 7.29 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 3.92 (t, J = 7.2 Hz, 2H), 2.31 (s, 3H), 2.16 (s, 3H), 1.91 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 167.93, 166.41, 159.72, 139.97, 130.44, 129.13, 128.98, 128.53, 127.21, 125.51, 124.58, 121.19, 113.68, 104.94, 41.98, 22.13, 11.58, 11.51, 10.66. MS (ESI) m/z for C19H18N2O2 [M + H]+, calcd: 306.14; found 307.1. HPLC, tR = 25.79 min, 99.35% purity.
1-Butyl-6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (21)
Yellow solid, 52% yield. 1H NMR (400 MHz, CDCl3) δ 8.13 (m, 1H), 7.76 (m, 2H), 7.29 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 3.95 (t, J = 7.2 Hz, 2H), 2.31 (s, 3H), 2.16 (s, 3H), 1.85 (m, 2H), 1.52 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 167.88, 166.41, 159.72, 139.94, 130.44, 129.13, 128.97, 128.53, 127.23, 125.53, 124.56, 121.19, 113.69, 104.91, 40.13, 30.91, 20.24, 13.78, 11.57, 10.65. MS (ESI) m/z for C20H20N2O2 [M + H]+, calcd: 320.15; found 321.1. HPLC, tR = 29.68 min, 97.74% purity.
Methyl 2-(6-(3,5-dimethylisoxazol-4-yl)-2-oxobenzo[cd]indol-1(2H)-yl)acetate (22)
To a stirred solution of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (11) (200 mg, 0.76 mmol) in DMF (10 mL) was added K2CO3 (314 mg, 2.27 mmol) and iodoethane (174 mg, 1.14 mmol) at rt. The reaction mixture was stirred at 80 °C for 4 h. Water was added, the aqueous layer was extracted with ethyl acetate (20 mL × 3), and the combined organic layer was washed with water and brine, dried with Na2SO4, and evaporated. The residue was purified by silica gel chromatography with petroleum ether/EtOAc (3/1, v/v) to yield the desired product (200 mg, 78%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.15 (dd, J = 4.6, 2.9 Hz, 1H), 7.78 (m, 2H), 7.30 (d, J = 7.3 Hz, 1H), 6.91 (d, J = 7.3 Hz, 1H), 4.73 (s, 2H), 3.81 (s, 3H), 2.31 (s, 3H), 2.15 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 168.60, 167.61, 166.49, 159.67, 138.99, 130.41, 129.46, 129.26, 128.68, 126.41, 125.79, 125.08, 121.87, 113.56, 104.99, 52.62, 41.43, 11.56, 10.64.MS (ESI) m/z for C19H16N2O4 [M + H]+, calcd: 336.11; found 337.1. HPLC, tR = 24.06 min, 97.27% purity.
2-(6-(3,5-Dimethylisoxazol-4-yl)-2-oxobenzo[cd]indol-1(2H)-yl)acetic acid (23)
Methyl 2-(6-(3,5-dimethylisoxazol-4-yl)-2-oxobenzo[cd]indol-1(2H)-yl)acetate (150 mg, 0.45 mmol) was dissolved in MeOH (5 mL) and 2 mol/L NaOH aqueous solution (5 mL). The mixture was stirred at room temperature for 2 h. The solvent was removed and diluted hydrochloric acid was added dropwise, and a yellow precipitate was formed. The precipitate was collected by filtration and washed with water (10 mL × 2). The resulting crude product was purified by recrystallization with petroleum and ether/ethyl acetate to afford the final product (100 mg, 70%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 13.12 (s, 1H), 8.15 (d, J = 6.2 Hz, 1H), 7.86 (q, J = 8.0 Hz, 2H), 7.49 (d, J = 7.3 Hz, 1H), 7.29 (d, J = 7.3 Hz, 1H), 4.72 (s, 2H), 2.29 (s, 3H), 2.10 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 171.60, 167.95, 166.60, 159.70, 138.81, 130.45, 129.60, 129.34, 128.63, 126.23, 125.69, 125.32, 121.94, 113.61, 105.29, 41.36, 11.56, 10.61. MS (ESI) m/z for C18H14N2O4 [M + H]+, calcd: 322.10; found 323.1. HPLC, tR = 14.59 min, 98.98% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-(2-(pyrrolidin-1-yl)ethyl)benzo[cd]indol-2(1H)-one (24)
General procedure for the synthesis of 24–28. To a stirred solution of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one (11) (100 mg, 0.38 mmol) in DMF (5 mL) was added NaH (46 mg, 1.14 mmol) at 0 °C. 30 min later, 1-(2-chloroethyl)pyrrolidine (97 mg, 0.57 mmol) was added to the solution and the reaction mixture was stirred at 100 °C overnight. Water was added, the aqueous layer was extracted with ethyl acetate (20 mL × 3), and the combined organic layer was washed with water and brine, dried with Na2SO4, and evaporated. The residue was purified by silica gel chromatography with petroleum ether/EtOAc (1/1, v/v) to yield the desired product (59 mg, 43%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.13 (m, 1H), 7.72 (d, J = 3.6 Hz, 2H), 7.29 (d, J = 7.2 Hz, 1H), 7.03 (d, J = 7.2 Hz, 1H), 4.11 (t, J = 7.3 Hz, 2H), 2.89 (t, J = 7.4 Hz, 2H), 2.67 (s, 4H), 2.31 (s, 3H), 2.15 (s, 3H), 1.82 (s, 4H). 13C NMR (125 MHz, CDCl3) δ 167.80, 166.42, 159.71, 139.77, 130.48, 129.13, 129.05, 128.57, 127.11, 125.60, 124.64, 121.30, 113.66, 105.02, 54.40, 54.26, 39.75, 23.59, 11.56, 10.64.MS (ESI) m/z for C22H23N3O2 [M + H]+, calcd: 361.18; found 362.1. HPLC, tR = 30.69 min, 99.89% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-(2-morpholinoethyl)benzo[cd]indol-2(1H)-one (25)
Yellow solid, 52% yield. 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 3.4 Hz, 1H), 7.73 (d, J = 3.3 Hz, 2H), 7.29 (d, J = 7.1 Hz, 1H), 7.00 (d, J = 7.2 Hz, 1H), 4.09 (t, J = 6.8 Hz, 2H), 3.70 (s, 4H), 2.76 (t, J = 6.8 Hz, 2H), 2.59 (s, 4H), 2.31 (s, 3H), 2.15 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 167.83, 166.41, 159.67, 139.74, 130.39, 129.18, 129.10, 128.57, 127.05, 125.60, 124.67, 121.37, 113.63, 104.92, 66.96, 56.77, 53.84, 37.81, 11.58, 10.65.MS (ESI) m/z for C22H23N3O3 [M + H]+, calcd: 377.17; found 378.1. HPLC, tR = 29.17 min, 99.48% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-(3-morpholinopropyl)benzo[cd]indol-2(1H)-one (26)
Yellow solid, 43% yield. 1H NMR (400 MHz, CDCl3) δ 8.13 (m, 1H), 7.73 (d, J = 3.6 Hz, 2H), 7.28 (d, J = 7.2 Hz, 1H), 7.02 (d, J = 7.3 Hz, 1H), 4.03 (t, J = 6.8 Hz, 2H), 3.69 (m, 4H), 2.47 (t, J = 7.0 Hz, 2H), 2.42 (s, 4H), 2.31 (s, 3H), 2.15 (s, 3H), 2.04 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 168.01, 166.41, 159.68, 139.95, 130.40, 129.18, 129.07, 128.54, 127.18, 125.54, 124.60, 121.29, 113.64, 104.97, 66.91, 55.98, 53.66, 38.48, 25.53, 11.57, 10.65.MS (ESI) m/z for C23H25N3O3 [M + H]+, calcd: 391.19; found 392.1. HPLC, tR = 21.93 min, 95.72% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-(3-phenylpropyl)benzo[cd]indol-2(1H)-one (27)
Yellow solid, 69% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.09 (dd, J = 5.1, 2.4 Hz, 1H), 7.86 (m, 2H), 7.47 (d, J = 7.3 Hz, 1H), 7.24 (q, J = 6.9 Hz, 5H), 7.15 (td, J = 6.2, 2.6 Hz, 1H), 3.96 (t, J = 7.1 Hz, 2H), 2.73 (m, 2H), 2.29 (s, 3H), 2.09 (s, 3H), 2.04 (dd, J = 14.9, 7.4 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 167.92, 166.42, 159.70, 141.10, 139.72, 130.42, 129.16, 129.04, 128.54, 128.42, 128.32, 127.14, 126.04, 125.54, 124.63, 121.30, 113.66, 104.92, 39.96, 33.19, 30.15, 11.57, 10.65. MS (ESI) m/z for C25H22N2O2 [M + H]+, calcd: 382.17; found 383.2. HPLC, tR = 36.10 min, 99.88% purity.
6-(3,5-Dimethylisoxazol-4-yl)-1-(1-phenylethyl)benzo[cd]indol-2(1H)-one (28)
Yellow solid, 64% yield. 1H NMR (400 MHz, CDCl3) δ 8.15 (dd, J = 6.0, 1.5 Hz, 1H), 7.78 (m, 2H), 7.48 (d, J = 7.7 Hz, 2H), 7.36 (t, J = 7.5 Hz, 2H), 7.29 (t, J = 7.2 Hz, 1H), 7.08 (d, J = 7.3 Hz, 1H), 6.61 (d, J = 7.3 Hz, 1H), 6.04 (q, J = 7.1 Hz, 1H), 2.27 (d, J = 4.4 Hz, 3H), 2.12 (d, J = 3.4 Hz, 3H), 1.95 (d, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 167.77, 166.41, 159.69, 140.11, 138.02, 130.33, 129.20, 129.00, 128.64, 128.61, 127.52, 126.96, 126.69, 125.84, 124.70, 121.12, 113.57, 107.40, 49.26, 17.50, 11.57, 10.66.MS (ESI) m/z for C24H20N2O2 [M + H]+, calcd: 368.15; found 369.3. HPLC, tR = 42.40 min, 99.35% purity.
Plasmid construction, protein expression, and purification
The bromodomains were expressed as a His6-fusion protein with a TEV cleavage site between His6 tag and bromodomain using the pET24a expression vector (Novagen). cDNA encoding bromodomain of human BRD4(1) (residues N44-E168), EP300 (residues A1040-G1161), CREBBP (residues R1081-G1197), BAZ2B (residues S1858-S1972), TIF(1) (residues G896-E1014), PCAF (residues G715-D831), BRD9 (residues L14-Q134), and TAF1(1) (residues R1377-D1503) were synthesized by Genscript. BL21 (DE3) cells transformed with these expression plasmids were grown in LB broth at 25 °C to an OD600 nm of approximately 1.0 and then induced with 0.1 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) at 16 °C for 16 h. Cells were harvested by centrifugation (6000 × g for 15 min at 4 °C, JLA 81000 rotor, on a Beckman Coulter Avanti J-20 XP centrifuge) and were frozen at −80 °C as pellets for storage. Cells were resuspended in extract buffer (50 mM HEPES, pH 7.5 at 25 °C, 500 mM NaCl, 5 mM imidazole, 5% glycerol, and 0.5 mM TCEP (Tris(2-carboxyethyl)phosphine hydrochloride)) and high-pressure homogenized using a JN3000 PLUS high-pressure homogenizer (JNBIO, Guangzhou, China) at 4 °C. The lysate was collected on ice and centrifuged at 12000 × g for 40 min. The supernatant was loaded onto a 5 mL NiSO4-loaded HisTrap HP column (Ni-NTA, GE Healthcare, NJ). The column was washed with 20 mL of extract buffer (50 mM HEPES, pH 7.5 at 25 °C, 500 mM NaCl, 50 mM imidazole). The protein was eluted with a 50–500 mM imidazole gradient in elute buffer with 50 mM HEPES, pH 7.5 at 25 °C, 500 mM NaCl, 500 mM imidazole. The protein was concentrated and further purified by a gel filtration column (HiLoad, Superdex 75, 16/60, GE Healthcare). The sample purity of each fraction was examined by SDS-PAGE, and the sample concentration was determined by Bradford assay. Purified proteins were concentrated and stored in the gel filtration buffer (10 mM Hepes pH 7.5 at 25 °C, 150 mM NaCl, 0.5 mM TCEP) and were used for crystallization or stored at −80 °C for AlphaScreen or TSA assays.
AlphaScreen assay
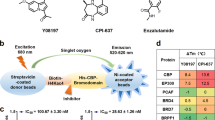
Interactions between bromodomain-containing proteins (BCP) and ligands were assessed by luminescence-based AlphaScreen technology (Perkin Elmer) as previously described in refs. [17, 18] using a histidine detection kit from PerkinElmer (Norwalk, CT). All of the reactions contained bromodomain-containing protein bound to nickel acceptor beads (5 μg/mL) and biotinylated acetylated histone H4 peptide bound to streptavidin donor beads (5 μg/mL) in the presence or absence of the indicated amounts of control compound (+)-JQ1 (1), or candidate compounds. The C-terminal biotinylated tetra-acetylated histone peptide H4 (bH4KAc4) sequence was H-SGRGK(Ac)GGK(Ac)GLGK(Ac)GGAK(Ac)RHRK-Biotin-OH (synthesized by Genscript). The experiment was conducted with protein/peptide ratio as follows for sensitive signal: BRD4(1): bH4KAc4 = 50 nM: 50 nM. The reagents were diluted in the buffer (50 mM MOPS, pH 7.4, 50 mM NaF, 50 μM CHAPS, and 0.1 mg/mL bovine serum albumin) and allowed to equilibrate at room temperature prior to addition to low-volume 384-well plates (ProxiPlate-384 Plus, PerkinElmer, USA). Plates were foil-sealed to protect from light, incubated at room temperature for 2.5 h, and read on an EnSpire plate reader (PerkinElmer, USA). When excited by a laser beam of 680 nm, the donor beam emits singlet oxygen that activates thioxene derivatives in the acceptor beads, which releases photons of 520–620 nm as the binding signal. All experiments were carried out in triplicate on the same plate. The results were based on an average of three experiments with standard errors typically less than 10% of the measurements.
Thermal shift assay (TSA)
TSAs were carried out using the Bio-Rad CFX96 Real-Time PCR system. All reactions were buffered in 10 mM HEPES, pH 7.5, 150 mM NaCl at a final concentration of 10 μM proteins and 200 μM compounds. The 20 μL reaction mix was added to the wells of the 96-well PCR plate. SYPRO Orange (ABI, Sigma) was added as a fluorescence probe at a dilution of 1:1000 and incubated with compounds on ice for 30 min. Total DMSO concentration was restricted to 1% or less. Excitation and emission filters for the SYPRO orange dye were set to 465 and 590 nm, respectively. The temperature was raised with a step of 0.3 °C per minute from 30 to 75 °C, and fluorescence readings were taken at each (0.3 °C) interval. All experiments were performed in triplicates. Melting temperatures (Tm) were calculated by fitting the sigmoidal melt curve to the Boltzmann equation using GraphPad Prism. ΔTm is the difference in Tm values calculated for reactions with and without compounds.
Cell culture, cell viability, and cell colony formation assays
Prostate cell lines C4–2B, LNCaP, 22Rv1, Du145, and leukemia cell line MV4;11 were cultured in RPMI-1640, plus 10% FBS. Cells were grown at 37 °C in 5% CO2 incubators. For cell viability, cells were seeded in 384-well plates at 500–1000 cells per well (optimum density for growth) in a total volume of 20 μL of media. After 12 h, 10 μL chemical compounds with two fold serial dilution was added in a final volume of 30 μL of media each well with a final concentration from 5 nM to 100 μM. The measurement was conducted 96 h after seeded for LNCaP, C4–2B, and 22Rv1, 72 h after seeded for Du145, and 120 h after seeded for MV4;11. Then, 25 μL of Cell-Titer GLO reagents (Promega) was added, and luminescence was measured on GLOMAX microplate luminometer (Promega), according to the manufacturer’s instructions. The estimated in vitro half-maximal inhibitory concentration (IC50) values were calculated using GraphPad Prism 6 software.
For colony formation assay, 1500 cells per well were seeded in a 6-well plate for C4–2B, respectively. Cells were cultured with vehicle or indicated concentrations of compounds for 14 days with a 3 mL medium. When the cell colony grew visible, the medium was removed and the plates were washed with 2 mL PBS for one time. The cell colonies were stained with 2.5% crystal violet (in MeOH) for 2 h. The plates were scanned with an HP scanner.
RNA isolation and quantitative real-time PCR
22Rv1 cells were plated at 200,000 cells per well in 12-well plates. Twenty-four hours later, cells were treated with vehicle or 19 (5 μM) for 48 h. Total RNA was then isolated with the Eastep® Super Total RNA Extraction Kit and cDNA was synthesized from 1000 ng total RNA using the All-in-oneTM First-Strand cDNA Synthesis Kit. qPCRs were performed in triplicate using standard SYBR green reagents on a Bio-Rad CFX96 real-time PCR system. The analysis was performed on triplicate PCR data for each biological duplicate (normalized to β-actin). The primer sequences for qPCR used are as follows: AR-FL_qPCR_fwd, 5′-ACA TCA AGG AAC TCG ATC GTA TCA TTG C-3′; AR-FL_qPCR_rev, 5′-TTG GGC ACT TGC ACA GAG AT-3′; AR-V7_qPCR_fwd, 5′-CCA TCT TGT CGT CTT CGG AAA TGT TAT GAA GC-3′; AR-V7_qPCR_rev, 5′-TTT GAA TGA GGC AAG TCA GCC TTT CT-3′. PSA_fwd, 5′-CAC AGG CCA GGT ATT TCA GGT-3′; PSA_rev, 5′-GAG GCT CAT ATC GTA GAG CGG-3′; KLK2_fwd, 5′-CAA CAT CTG GAG GGG AAA GGG-3′; KLK2_rev, 5′-AGG CCA AGT GAT GCC AGA AC-3′; TMPRSS2_fwd, 5′-CAA GTG CTC CAA CTC TGG GAT-3′; TMPRSS2_rev, 5′-AAC ACA CCG ATT CTC GTC CTC-3′; C-MYC_fwd, 5′-GGC TCC TGG CAA AAG GTC A-3′; C-MYC_rev, 5′-CTG CGT AGT TGT GCT GAT GT-3′; β-Actin_fwd, 5′-GAG AAA ATC TGG CAC CAC ACC-3′; β-Actin_rev, 5′-ATA CCC CTC GTA GAT GGG CAC-3′.
Results and discussion
In-house library screening
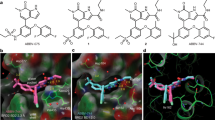
To screen potent BET inhibitors, a TSA was performed on an in-house chemical collection. Compound 6 with a 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one scaffold was shown to have a temperature shift of 2.1 °C. The predicted binding mode of 6 bound to BRD4(1) showed that the 3,5-dimethylisoxazole group interacted with the conserved residues Asn140 and Tyr97 through direct and water-mediated hydrogen bonds (Fig. 2a). The methyl group of the 3,5-dimethylisoxazole group mimicked the terminal methyl group of acetyl-lysine (KAc) and occupied the small pocket surrounded by the residues Pro82, Phe83, Val87, and Ile146. Moreover, the tricyclic aromatic core of compound 6 could also fill the KAc binding pocket. The butylsulfonyl group of 6 was able to interact with the WPF shelf (composed of Trp81, Pro82, and Phe83) by means of hydrophobic interactions (Fig. 2a). The carbonyl O could form hydrogen bonds with water, contributing to its activity. These important interactions indicated that 6 may serve as a good starting point for new BET inhibitors. To develop compounds with improved affinity for BRD4, an extensive structure−activity relationship investigation was conducted.

The crystal structure 5YQX from the PDB was used as a template for molecular docking for compounds (a) 6, (b) 14, and (c) 19. All compounds are shown as sticks, and all residues are shown as lines. Water molecules are shown as spheres. In addition, an electrostatic potential surface is shown. Hydrogen bonds are shown as yellow dashed lines.
Chemistry
The 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one derivatives were designed and synthesized as shown in Scheme 1. Briefly, benzo[de]isochromene-1,3-dione (7) reacted with hydroxylamine hydrochloride and p-toluene sulfonic acid (PTSA) in the presence of pyridine to produce compound 8, which was further reacted with NaOH followed by HCl treatment to produce intermediate 9. Direct bromination of 9 with bromine provided fragment 10, which was followed by coupling with 3,5-dimethylisoxazole-4-boronic acid under palladium catalysis to deliver 11. The final sulfonamides (12–18) were synthesized from compound 11 using the corresponding sulfonyl chlorides. For compounds 19–22 and 24–28, the R group was introduced by reaction with chloro-, bromo- or iodo-substituents. Compound 23 was synthesized by a simple hydrolysis reaction.

Synthesis of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one derivatives. Reagents and conditions: (a) NH4OH·HCl, pyridine, PTSA, 95 °C, 1 h, 78%; (b) (i) 2.7 mol/L NaOH, EtOH, H2O, reflux, 3 h, (ii) HCl, rt, 30 min, 83%; (c) Br2, DCM, rt, 24 h, 80%; (d) 3,5-dimethylisoxazole-4-boronic acid, Pd(PPh3)4, K2CO3, DMF/H2O = 30:1, 100 °C, overnight, 32%; (e) sulfonyl chloride, NaH, DMF 0 °C, 3 h, 40%–86%; (f) RI, NaH, DMF, rt, 3 h, 52%–90% or RX (X = Br, Cl), NaH, DMF, 100 °C, overnight, 43%–78%; (g) 2 mol/L NaOH aqueous solution, MeOH, rt, 2 h, 70%.
Structure-activity relationship (SAR) studies
To optimize the interactions with the WPF shelf, various substituents at the R position of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one were designed to explore the chemical space for affinity improvement (Table 1). All synthesized compounds were evaluated for their BRD4(1) inhibitory activities via a TSA and an AlphaScreen assay.
To enhance the hydrophobic interactions with residues Trp81, Pro82, and Phe83 of the WPF shelf, cycloalkyl substituents were introduced at the R positions, and compounds 12 and 13 were synthesized (Table 1). Compounds 12 and 13 displayed BRD4(1) inhibitory activity similar to that of 6 with temperature shifts of 1.8 and 1.2 °C in the TSA, respectively. Aromatic substituents were also introduced to extend the SAR study. Replacement of the cyclohexyl group in 13 with a phenyl group led to 14, which exhibited a similar inhibitory effect to that of 13 (TSA: 1.2 °C vs 1.2 °C; inhibition rate: 28.6% vs 28.4% for 13 and 14, respectively). The results suggested that cyclopentyl, cyclohexyl, and phenyl groups have similar affinities for BRD4(1).
The predicted binding mode of compound 14 in complex with BRD4(1) obviously showed that the phenyl group is directly above the WPF hydrophobic area formed by the hydrophobic residues Trp81, Pro82, Met149, and Ile146 (Fig. 2b). Thus, various phenyl-substituted derivatives were designed and synthesized for biological evaluation. The results revealed that substitutions to different positions of the phenyl ring in 14 could achieve diverse impacts on BRD4(1) bromodomain inhibitory activity. As shown in Table 1, para-F-substituted derivative 15 had no activity. However, when disubstituted derivatives were designed and synthesized, improved potency was obtained. 2,4-Difluorophenyl compound 16 exhibited improved activity with a maximum inhibition of 18.9% at 20 μM. Compound 17 binds to BRD4(1) with a temperature shift of 1.2 °C in the TSA and was equipotent to 14. Further study showed that the meta-Cl group in 17 could be replaced by a bromo moiety (18) to display almost identical potency against BRD4(1), with an inhibition ratio value of 11.7% at 20 μM. The TSA also demonstrated a stabilizing effect from 17 on the BRD4(1) protein with a temperature shift of 1.2 °C. All of the above results suggested that these sulfonamide derivatives demonstrated weak binding affinity to the BRD4(1) protein. It seems that the sulfonamide linker might provide an unfavorable angle and distance range for substitution and thus cannot form favorable interactions with the WPF shelf.
Next, to find potent 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one derivative, the sulfonamide linker was replaced by an amide moiety and its derivatives to give compounds 11 and 19–28. These compounds displayed significant improvements in the inhibition of BRD4(1) (Table 1). Interestingly, the sulfonamide moiety in compound 6 was removed to yield compound 11, which displayed an obvious potency improvement against BRD4(1) with a thermal shift of 6.3 °C and an IC50 value of 4.87 μM. Further investigation revealed that the hydrogen atom in 11 could be replaced by an ethyl moiety (19) to display almost identical potency against BRD4(1), with a thermal shift of 5.9 °C and an IC50 value of 3.79 μM. All of the results showed that compound 19 is a good starting point for bromodomain inhibitor development.
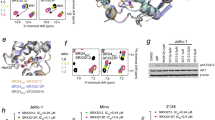
To better design potent compounds, we predicted the complex structure of 19 with the BRD4(1) protein to gain a structural understanding of their interactions (Fig. 2c). The modeled structure of 19 and BRD4(1) shows that the binding mode of 19 is similar but not identical to that of 6 (Fig. 2a). Analysis of the binding model for compound 19 in complex with the BRD4(1) domain suggested that replacement of the ethyl group in 19 with a larger group such as a propyl or phenyl group might further enhance the hydrophobic interactions with BRD4(1). This discovery encouraged us to design larger substituted compounds. However, when the ethyl moiety in 19 was replaced with the relatively large hydrophobic groups propyl (20), butyl (21), or methyl acetate (22), the resulting molecules displayed a 1- to 2-fold potency loss. For instance, propyl group-substituted analog 20 exhibited an IC50 value of 9.17 μM against BRD4(1), which was approximately twofold less potent than 19. The compound with a hydrophilic substituent (23) showed a dramatic loss of inhibition, and its maximum inhibition was less than 20% at 20 μM. This result indicated that the hydrophobic interaction between the ligand and the protein is potentially a critical interaction. To illustrate this point, flexible linkers with 2–3 methylenes were used to attach polar substituents (24, 25, and 26). Not surprisingly, these compounds exhibited moderate or weak activity. Replacement of the polar substituent in 26 with a phenyl group resulted in 27 showing a sharp decrease in BRD4(1) inhibitory potency (ΔTm = 1.1 °C). Compound 28 was also synthesized. It was found that 28 bound to BRD4(1) with a temperature shift of 2.7 °C and a maximum inhibition ratio of less than 30% at 20 μM. Luckily, the crystal structure of the 28-BRD4(1) complex was obtained (Fig. 3a, b). We can see that 28 showed an orientation of the 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one scaffold that was almost identical to that of the other compounds (6, 14, or 19, Fig. 2a–c). From the above SAR, we can see that among these various groups explored, the ethyl moiety remained the best for binding to the BRD4(1) WPF shelf. Hence, our modifications at the WPF shelf position yielded compounds 11 and 19 with much improved binding affinities over initial compound 6.

a Fo-fc omits map overlapped with the ligand. The density contour is shown, and the contour level is set to 1.0 sigma. b Compound 28 is shown as sticks, and all residues are shown as lines. Water molecules are shown as spheres. In addition, an electrostatic potential surface is shown. Hydrogen bonds are shown as yellow dashed lines.
Evaluation of the binding specificity over other non-BET bromodomain-containing proteins
Bromodomains have been reported to be a part of numerous large protein architectures, many of which share a conserved module and have the common 3D structure pattern of three long helices (helices A–C) and one short helix (helix Z). The selectivity of diverse bromodomain inhibitors is critical for the success of drug discovery. To study the selectivity profile, representative compounds were tested against BRD4(1) domain proteins and seven representative non-BET bromodomain-containing proteins from six other subfamilies by thermal stability shift assay. Compound 1 was included in these experiments as a control compound, and the results are shown in Fig. 4. The results showed that compound 19 displayed excellent selectivity for the BET subfamily over other non-BET families.

The heat map shows the relative ΔTm, where red indicates a large ΔTm and yellow and green indicate small ΔTm values. Compound concentration, 200 µM; protein concentration, 10 µM.
Evaluation of the inhibitory effects on cell growth and gene expression in PC cell lines
The BET family has been identified as a novel therapeutic target for PC. To evaluate the antiproliferative activities of these 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one derivative, we tested our representative compounds for their cellular activity in AR-positive and AR-negative PC cell lines (C4–2B, LNCaP, 22Rv1, and Du-145) (Table 2, Fig. 5a, b).

BET inhibitors suppress the growth of (a) LNCaP and (b) 22Rv1 prostate cancer cells
All compounds exhibited reasonable potency against these PC cell lines. For example, 19 showed cell viability inhibition IC50 values of 1.93, 2.07, and 2.28 μM in the C4–2B, LNCaP, and 22Rv1 cell lines, respectively (Fig. 6a). These activities were stronger than those of the second-generation antiandrogen enzalutamide (36.66 μM in 22Rv1 cells) [18, 33]. It is interesting to note that compounds 20 and 25 displayed less potent inhibitory activity than 19 at the protein level but showed similar potencies to 19 in the same cell lines. All of the tested compounds were also active against MV4;11 leukemia cells (Table 2). It seems that the present compounds are more sensitive to MV4;11 cells than prostate cells. The results showed that our BET inhibitors effectively inhibited PC cell proliferation.

a Inhibition curves for prostate cell growth. b Compound 19 inhibits C4–2B cell colony formation. c qRT−PCR analysis of full-length AR, AR-V7, PSA, KLK2, TMPRSS2, and c-MYC expression in 22Rv1 cells treated with vehicle or 19 (5 μM) for 48 h. Data represent the mean ± SD (n = 3) from one of three independent experiments. NS not significant. *P ≤ 0.05, **P ≤ 0.005 by two-tailed Student’s t test.
To further confirm the long-term cell growth inhibitory effects, a colony formation assay was performed with representative compound 19. As shown in Fig. 6b, compound 19 inhibited C4–2B cell colony formation in a dose-dependent manner. Colony formation was markedly decreased by compound 19 at 2 μM. The results showed that 19 significantly inhibited the growth of the C4–2B cell line.
To investigate whether the antiproliferative effects were caused by the inhibition of BRD4(1), we further performed real-time quantitative fluorescent PCR (qRT-PCR) in 22Rv1 cells to detect the expression of the AR, AR-regulated genes, and other oncogenes. As shown in Fig. 6c, the AR-regulated genes PSA (also known as KLK3), KLK2, and TMPRSS2 were suppressed at the mRNA level upon treatment with 19 in 22Rv1 cells. The oncogenic driver MYC was also downregulated by the BET bromodomain inhibitor. The results showed that c-Myc mRNA levels were suppressed upon treatment with 19. The results demonstrated that BET inhibitor 19 could effectively suppress cell growth and the related gene expression in PC cells. Based on these data, compound 19 and its derivatives may serve as a promising therapeutic strategy for PC by targeting BRD4(1).
Conclusions
In summary, we reported our in-house library screening and the synthesis and evaluation of 6-(3,5-dimethylisoxazol-4-yl)benzo[cd]indol-2(1H)-one analog as new and selective BET bromodomain inhibitors. The cocrystal structure of this kind of scaffold with BRD4(1) provides a solid foundation for further medicinal chemistry optimization. One of the most promising compounds, 19, inhibited BRD4(1) activity with the promising IC50 value of 3.79 μM and selectively inhibited non-BET bromodomain-containing proteins. Furthermore, compound 19 demonstrated inhibitory activities against the growth of AR-positive PC cell lines and a dose-dependent reduction in the colony formation of PC cells. Compound 19 also suppressed gene expression in 22Rv1 cells. The results further proved that compound 19 might serve as a new lead compound for the discovery of anti-PC compounds and other tumor drugs. Further structural optimization of 19 is in progress, and the results will be reported in due course.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Huggins C, Stevens RE, Hodges CV. Studies on prostate cancer II the effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209–23.
Balk SP. Androgen receptor as a target in androgen-independent prostate cancer. Urology. 2002;60:132–8.
Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9.
Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao YH, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22.
Asangani IA, Dommeti VL, Wang XJ, Malik R, Cieslik M, Yang RD, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82.
De Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005.
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97.
Fizazi K, Albiges L, Loriot Y, Massard C. ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev Anticanc. 2015;15:1007–17.
Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408–18.
Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta. 2014;1839:676–85.
Sahai V, Redig AJ, Collier KA, Eckerdt FD, Munshi HG. Targeting BET bromodomain proteins in solid tumors. Oncotarget. 2016;7:53997–4009.
Welti J, Sharp A, Yuan W, Dolling D, Rodrigues DN, Figueiredo I, et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin Cancer Res. 2018;24:3149–62.
Xiang QP, Zhang Y, Li JG, Xue XQ, Wang C, Song M, et al. Y08060: a selective BET inhibitor for treatment of prostate cancer. ACS Med Chem Lett. 2018;9:262–7.
Xue XQ, Zhang Y, Wang C, Zhang MF, Xiang QP, Wang JJ, et al. Benzoxazinone-containing 3,5-dimethylisoxazole derivatives as BET bromodomain inhibitors for treatment of castration-resistant prostate cancer. Eur J Med Chem. 2018;152:542–59.
Zhang M, Zhang Y, Song M, Xue X, Wang J, Wang C, et al. Structure-based discovery and optimization of benzo[d]isoxazole derivatives as potent and selective BET inhibitors for potential treatment of castration-resistant prostate cancer (CRPC). J Med Chem. 2018;61:3037–58.
Zhang GT, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, et al. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem. 2013;56:9251–64.
Zhao LL, Cao DY, Chen TT, Wang YQ, Miao ZH, Xu YC, et al. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. J Med Chem. 2013;56:3833–51.
Li Z, Xiao S, Yang Y, Chen C, Lu T, Chen Z, et al. Discovery of 8-methyl-pyrrolo[1,2-a]pyrazin-1(2H)-one derivatives as highly potent and selective bromodomain and extra-terminal (BET) bromodomain inhibitors. J Med Chem. 2020;63:3956–75.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73.
Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F, et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A). Bioorg Med Chem Lett. 2012;22:2968–72.
Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem. 2013;56:7501–15.
Bui MH, Lin XY, Albert DH, Li LM, Lam LT, Faivre EJ, et al. Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. 2017;77:2976–89.
McDaniel KF, Wang L, Soltwedel T, Fidanze SD, Hasvold LA, Liu DC, et al. Discovery of N-(4-(2,4-difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/mivebresib), a potent and orally available bromodomain and extraterminal domain (BET) family bromodomain inhibitor. J Med Chem. 2017;60:8369–84.
Hewings DS, Fedorov O, Filippakopoulos P, Martin S, Picaud S, Tumber A, et al. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. J Med Chem. 2013;56:3217–27.
Xue X, Zhang Y, Liu Z, Song M, Xing Y, Xiang Q, et al. Discovery of benzo[cd]indol-2(1H)-ones as potent and specific BET bromodomain inhibitors: structure-based virtual screening, optimization, and biological evaluation. J Med Chem. 2016;59:1565–79.
A study to investigate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of GSK525762 in subjects with NUT midline carcinoma (NMC) and other cancers. Clinical Trials Apr 30 2012: https://clinicaltrials.gov/ct2/show/NCT01587703 (2012). Accessed 11 Mar 2018.
A study of ZEN003694 in patients with metastatic castration-resistant prostate cancer. Clinical Trials Mar 10, 2016: https://clinicaltrials.gov/ct2/show/NCT02705469 (2016). Accessed 11 Mar 2018.
Safety, tolerability, pharmacokinetics, and pharmacodynamics of GS-5829 as a single agent and in combination with enzalutamide in participants with metastatic castrate-resistant prostate cancer. Clinical Trials Nov 17, 2015: https://clinicaltrials.gov/ct2/show/NCT02607228 (2015). Accessed 11 Mar 2018.
A dose-finding study of OTX105/MK-8628, a small molecule inhibitor of the bromodomain and extra-terminal (BET) proteins, in adults with selected advanced solid tumors (MK-8628-003). Clinical Trials Oct 8, 2014: https://clinicaltrials.gov/ct2/show/NCT02259114 (2014). Accessed 11 Mar 2018.
Xiang QP, Wang C, Zhang Y, Xue XQ, Song M, Zhang C, et al. Discovery and optimization of 1-(1H-indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain inhibitors for the treatment of castration-resistant prostate cancer. Eur J Med Chem. 2018;147:238–52.
Acknowledgements
We gratefully acknowledge financial support from the Chinese National Programs for Key Research and Development (grant 2019YFE0123700, 2016YFB0201701), the Key International Cooperation Projects of the Chinese Academy of Sciences (grant 154144KYSB20180044 and 154144KYSB20180063), the Chinese Academy of Sciences STS Program (grant KFJ-STS-QYZX-090), the National Natural Science Foundation of China (grant 81673357), the Natural Science Foundation of Guangdong province (grant 2015A030312014, 2019A1515110592), the State Key Laboratory of Respiratory Disease (grant SKLRD-Z-202018), Guangdong Provincial Key Laboratory of Biocomputing (grant 2016B030301007), Frontier Research Program of Bioland Laboratory (Guangzhou Regenerative Medicine and Health Guangdong Laboratory, grant 2018GZR110105016), the Natural Science Foundation of Jiangsu Province (grant BK20190246) and the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (grant 19KJB350011). The authors also gratefully acknowledge support from the Guangzhou Branch of the Supercomputing Center of the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
YX and LJX designed the research. TBW, QPX, CW, CW, CZ, MFZ, and ZXL conduct the research; TBW, QPX, YZ, LJX, and YX analyzed the date and wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Wu, Tb., Xiang, Qp., Wang, C. et al. Y06014 is a selective BET inhibitor for the treatment of prostate cancer. Acta Pharmacol Sin 42, 2120–2131 (2021). https://doi.org/10.1038/s41401-021-00614-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00614-7
