
Abstract
Cardiac fibrosis is a typical pathological change in various cardiovascular diseases. Although it has been recognized as a crucial risk factor responsible for heart failure, there is still a lack of effective treatment. Recent evidence shows that microRNAs (miRNAs) play an important role in the development of cardiac fibrosis and represent novel therapeutic targets. In this study we tried to identify the cardiac fibrosis-associated miRNA and elucidate its regulatory mechanisms in mice. Cardiac fibrosis was induced by infusion of angiotensin II (Ang II, 2 mg·kg−1·d−1) for 2 weeks via osmotic pumps. We showed that Ang II infusion induced cardiac disfunction and fibrosis accompanied by markedly increased expression level of miR-99b-3p in heart tissues. Upregulation of miR-99b-3p and fibrotic responses were also observed in cultured rat cardiac fibroblasts (CFs) treated with Ang II (100 nM) in vitro. Transfection with miR-99b-3p mimic resulted in the overproduction of fibronectin, collagen I, vimentin and α-SMA, and facilitated the proliferation and migration of CFs. On the contrary, transfection with specific miR-99b-3p inhibitor attenuated Ang II-induced fibrotic responses. Similarly, intravenous injection of specific miR-99b-3p antagomir could prevent Ang II-infused mice from cardiac dysfunction and fibrosis. We identified glycogen synthase kinase-3 beta (GSK-3β) as a direct target of miR-99b-3p. In CFs, miR-99b-3p mimic significantly reduced the expression of GSK-3β, leading to activation of its downstream profibrotic effector Smad3, whereas miR-99b-3p inhibitor caused anti-fibrotic effects. GSK-3β knockdown ameliorated the anti-fibrotic role of miR-99b-3p inhibitor. These results suggest that miR-99b-3p contributes to Ang II-induced cardiac fibrosis at least partially through GSK-3β. The modulation of miR-99b-3p may provide a new approach for tackling fibrosis-related cardiomyopathy.
Similar content being viewed by others

Introduction
Cardiac fibrosis, characterized by the activation of cardiac fibroblasts (CFs) and excessive accumulation of extracellular matrix (ECM), is a common pathological phenomenon in nearly all forms of heart diseases. It triggers myocardial stiffness and impairs left ventricular systolic and diastolic function, ultimately resulting in progression towards heart failure, a leading cause of sudden death [1, 2]. The pathogenesis of cardiac fibrosis is complicated, and a variety of pathophysiological stimuli, such as hemodynamic overload, oxidative stress, inflammation and aging, have been demonstrated to orchestrate the initiation of fibrotic responses and subsequent cardiac remodeling [3]. However, our understanding of the molecular mechanisms underlying cardiac fibrosis is still limited. Currently, there are no specific therapeutics that can satisfactorily reduce fibrosis-related morbidity and mortality [4, 5]. Thus, it is of the utmost importance to further identify novel strategies and agents for combating cardiac fibrosis.
MicroRNAs (miRNAs), a group of small endogenous noncoding RNA molecules, have recently attracted increasing attention due to their critical roles in controlling gene expression at the posttranscriptional level [6]. Most miRNAs are highly conserved across species and negatively regulate downstream target genes by inducing mRNA degradation or translational repression, thereby participating in various cellular processes, including proliferation, differentiation, survival and apoptosis [7]. Accumulating studies have revealed the essential impact of miRNA dysregulation on the pathogenesis of numerous diseases, especially cancer and cardiovascular disorders [8,9,10]. Patients and experimental animals with cardiac fibrosis exhibit altered miRNA expression profiles, suggesting that miRNAs may have potential implications as biomarkers or therapeutic targets [11]. Moreover, several miRNAs have been linked to the development and progression of cardiac fibrosis by manipulating certain fibrogenic pathways. For example, the upregulation of miR-21 can promote cardiac fibrosis by stimulating the mitogen-activated protein kinase cascade [12]. MiR-125b also contributes to fibrogenesis by functionally modulating apelin and p53 [13]. In contrast, miR-29b suppresses transforming growth factor beta (TGF-β) signaling, thereby playing a protective role against cardiac fibrosis [14]. These facts highlight miRNA-based therapy as a promising strategy to alleviate fibrotic damage in the diseased heart.
Recently, we performed miRNA sequencing on left ventricular tissues from angiotensin II (Ang II)-infused mice and observed that the expression of miR-99b-3p was significantly elevated along with extensive fibrotic changes, indicating its involvement in cardiac fibrosis. The dysregulation of miR-99b-3p has been previously reported to be associated with tumor development and progression. It promotes the metastasis and proliferation of hepatocellular carcinoma but functions as a suppressor of oral squamous cell carcinoma and gastric cancer [15, 16]. Nevertheless, little is known about the potential influence of miR-99b-3p on the cardiovascular system. The present study reveals that miR-99b-3p participates in Ang II-induced cardiac fibrosis by targeting glycogen synthase kinase-3 beta (GSK-3β), which may provide new insight into the pathophysiological roles of miR-99b-3p.
Materials and methods
Animal model and echocardiography
C57BL/6 mice (male, weighing 18–22 g, SPF grade, Certification No. 44008500019764) were provided by the Experimental Animal Center of Sun Yat-sen University. All animal experiments were approved by the Research Ethics Committee of Sun Yat-Sen University and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by National Institutes of Health. The mice were treated with Ang II (2 mg·kg−1·d−1) for two weeks via implanted Alzet osmotic pumps (Durect, Cupertino, CA, USA). The animals in the control group received normal saline (NS). To inhibit miRNA in vivo, a specific miR-99b-3p antagomir (ant-miR-99b-3p) and a negative control antagomir (ant-NC) were purchased from GenePharma (Shanghai, China). The mice were administered ant-miR-99b-3p (12 mg·kg−1·d−1) or an equal dose of ant-NC by tail vein injection on the day before Ang II infusion and on days 2, 4, 7, 9, 12, and 14 after Ang II infusion. Subsequently, cardiac function was evaluated by a Technos MPX ultrasound system (Esaote, Genoa, Italy) according to our previous report [17]. The echocardiographic parameters left ventricular posterior wall thickness in diastole and systole (LVPW;d and LVPW;s), left ventricular anterior wall thickness (LVAW;d and LVAW;s), left ventricular internal dimension (LVID;d and LVID;s), ejection fraction (EF), and fractional shortening (FS) were recorded.
Histological and immunohistochemical examinations
After the echocardiographic measurement, the mice were sacrificed by exposure to CO2. The hearts were quickly excised and weighed. The ratios of heart weight to tibia length (HW/TL), heart weight to body weight (HW/BW), and left ventricle weight to body weight (LVW/BW) were calculated. Furthermore, cardiac tissues were fixed with 4% paraformaldehyde, embedded in paraffin blocks and cut into 5-μm thick sections. Hematoxylin-eosin (H&E) staining and Sirius red staining were used to visualize the histological changes and collagen deposition, respectively. The expression and distribution of fibronectin, α-smooth muscle actin (α-SMA) and GSK-3β were detected by immunohistochemistry.
Cell culture and transfection
Sprague-Dawley rats (male, weighing 150–200 g, SPF grade, Certification No. 44008500019168) were obtained from the Experimental Animal Center of Sun Yat-sen University. CFs were isolated as described previously [18]. Cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum in an incubator at 37 °C with 5% CO2. MiR-99b-3p mimic, inhibitor, small interference RNA for GSK-3β (si-GSK-3β) and relevant negative controls were purchased from RiboBio (Guangzhou, China). Transfections of CFs were performed by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. For the miR-99b-3p mimic and inhibitor, the final concentrations were 50 nM and 100 nM, respectively. Cells were harvested at 6 h after transfection for subsequent treatments.
Cell proliferation and migration assays
CFs were seeded into 96-well plates at a density of 5 × 103 cells per well and then incubated with 10 μM 5-ethynyl-2-deoxyuridine (EdU, RiboBio) for 24 h. After that, cells were fixed and stained with Hoechst 33342. Images were captured by a fluorescence microscope (Thermo Fisher Scientific, Rockford, IL, USA). EdU-positive cells were counted to determine the proliferation of CFs. For the measurement of cell migration, CFs were seeded into 6-well plates, and then a scratch wound was generated using a pipette tip. Cells were further cultured in a humidified incubator at 37 °C and imaged under an inverted microscope. The wound closure rate was calculated.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cultured CFs or fresh cardiac tissues with TRIzol reagent (Invitrogen) and converted into cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Polymerase chain reaction was performed to determine the mRNA levels of fibronectin, collagen I, collagen III and α-SMA with SYBR Green qPCR Master Mix (Toyobo Life Science, Osaka, Japan) by a PikoReal qPCR system (Thermo Fisher Scientific). The results were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. To quantify miR-99b-3p, the RT product was obtained by using the Bulge-Loop miRNA RT-PCR Starter Kit (RiboBio). All PCR primers were synthesized by Sangon Biotech (Shanghai, China) as shown in Supplementary Table S1.
Western blotting
Proteins were extracted from CFs or cardiac tissues by using a mixture of phenylmethylsulfonyl fluoride (Sigma, Saint Louis, MO, USA) and RIPA lysis buffer (Beyotime, Nantong, China) supplemented with protease and phosphatase inhibitors. A total of 25 μg of protein was loaded onto a 10% sodium dodecyl sulfate-polyacrylamide gel. After being separated by electrophoresis, sample proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat dry milk solution and further probed with primary antibodies at 4 °C overnight followed by incubation with horseradish peroxidase-labeled secondary antibodies for 90 min at room temperature. Bands were visualized with High-sig ECL Western blotting substrate (Tanon, Shanghai, China). The band intensities were measured using ImageJ software (NIH, Bethesda, MD, USA). The information concerning the antibodies is listed in Supplementary Table S2.
Dual-luciferase reporter assay
A sequence of the GSK-3β untranslated region (3′UTR) containing the predicted miR-99b-3p binding sites or a sequence of the GSK-3β 3′UTR with point mutations in the putative miR-99b-3p binding sites was inserted into the pEZX-MT06 firefly/Renilla dual-luciferase vector (GeneCopoeia, Rockville, MD, USA). HEK293 cells seeded in 96-well plates were cotransfected with miR-99b-3p mimics or negative control together with reporter vectors (0.2 μg per well) by using Lipofectamine 2000. Forty-eight hours later, the cells were lysed and further tested with a dual-luciferase assay kit (GeneCopoeia). Luciferase activity was measured by a microplate reader (Molecular Devices, San Jose, CA, USA).
Statistical analysis
The data are presented as the mean ± the standard error of the mean (SEM). Unpaired Student’s t-test was performed to compare the statistical significance between two groups. The differences among multiple groups were measured by one-way analysis of variance (ANOVA) with Tukey’s post hoc test. In all cases, a P value less than 0.05 was considered statistically significant.
Results
Ang II induces cardiac fibrosis and upregulates miR-99b-3p expression
C57BL/6 mice received Ang II infusion (2 mg·kg−1·d−1) via osmotic pumps for 14 days. The hearts of Ang II-treated mice were enlarged compared with those from control animals (Fig. 1a). Ang II led to obvious histological changes, including inflammatory cell infiltration and extracellular matrix collagen deposition in the myocardium, as detected by H&E and Sirius red staining (Fig. 1b–d). The HW/BW, LVW/BW and HW/TL ratios were also elevated following Ang II treatment (Fig. 1f). Echocardiography revealed that the LVPW, EF and FS were increased by Ang II, while the LVID was decreased, indicating abnormalities of cardiac structure and function (Fig. 1e, g). In addition, the results of immunohistochemistry and qRT-PCR analyses showed that the levels of fibrotic biomarkers, such as fibronectin, collagen I, collagen III and α-SMA, were upregulated after Ang II infusion (Fig. 1h, i). This evidence demonstrated that the mouse model of cardiac fibrosis was successfully established. Moreover, miR-99b-3p expression was strongly increased in fibrotic cardiac tissues from Ang II-infused mice (Fig. 1j).

C57BL/6 mice were subjected to Ang II infusion (2 mg·kg−1·d−1) via osmotic minipumps for 14 days. a Gross morphology of the heart. b–d Pathologic changes as indicated by H&E and Sirius red staining. e Representative echocardiographic graphs. f Ratios of HW/BW, LVW/BW, and HW/TL. g Echocardiographic parameters were analyzed. h Immunohistochemical expression of fibronectin and α-SMA. i, j The expression of fibrotic markers and miR-99b-3p was determined by qRT-PCR. All data are presented as the mean ± SEM. *P < 0.05 vs. the normal saline (NS) group, n = 5. Cultured rat CFs were incubated with 100 nM Ang II for the indicated time points. k, l The levels of fibrotic biomarkers were measured by qRT-PCR and Western blotting. m MiR-99b-3p expression following Ang II treatment. *P < 0.05 vs. the control group, n = 3.
To further confirm the influence of Ang II on miR-99b-3p expression, cultured rat CFs were incubated with 100 nM Ang II for the indicated time points. The mRNA and protein contents of fibrotic biomarkers were determined by Western blotting and qRT-PCR. Compared to the control group, Ang II-stimulated cells showed a time-dependent increase in the expression of fibronectin, collagen I, vimentin and α-SMA (Fig. 1k, l). Similar to its upregulation in vivo, the level of miR-99b-3p was significantly elevated in CFs following Ang II treatment (Fig. 1m), suggesting the involvement of miR-99b-3p in the development of cardiac fibrosis.
MiR-99b-3p promotes fibrotic responses in CFs
Both gain-of-function and loss-of-function approaches were used to elucidate the functional role of miR-99b-3p in cardiac fibrosis. The results showed that forced expression of miR-99b-3p by miRNA mimic elevated the protein levels of fibronectin, collagen I, vimentin and α-SMA in cultured rat CFs (Fig. 2a–d). The expression of these fibrotic biomarkers was further enhanced when the cells were concurrently treated with Ang II. Moreover, an EdU incorporation assay was performed to examine the effect of miR-99b-3p on cell proliferation. The results showed that the miR-99b-3p mimic increased the proliferative ability of CFs (Fig. 2e). It also remarkably facilitated CF migration, as determined by the scratch test (Fig. 2f). Conversely, suppressing miR-99b-3p by transfection with its specific inhibitor, an antisense nucleotide sequence, could attenuate Ang II-induced expression of fibrotic biomarkers (Fig. 3a–d) and inhibit the proliferation and migration of CFs (Fig. 3e, f). These findings suggested that miR-99b-3p was sufficient to promote fibrotic responses, and the upregulation of miR-99b-3p expression contributed to Ang II-stimulated cardiac fibrosis.

Cultured CFs were transfected with miR-99b-3p mimic or its negative control (NC mimic) and subjected to Ang II treatment (100 nM, 48 h). a–d The protein levels of fibronectin, collagen I, vimentin, and α-SMA were detected by Western blotting. e An EdU incorporation assay was performed to examine CF proliferation. f Cell migration was determined by the scratch-wound assay. All data are presented as the mean ± SEM. *P < 0.05 vs. the NC group, #P < 0.05 vs. the NC plus Ang II treatment group, n = 3.

Cultured CFs were treated with miR-99b-3p inhibitor or its negative control (NC inhibitor). Cells were further stimulated with or without 100 nM Ang II for 48 h. a–d Western blotting was conducted to detect the protein expression of fibronectin, collagen I, vimentin, and α-SMA. e The proliferation of CFs was measured by EdU incorporation assay. f A scratch-wound assay was used to test the cell migration ability. All data are presented as the mean ± SEM. *P < 0.05 vs. the NC group, #P < 0.05 vs. the NC plus Ang II treatment group, n = 3.
The 3′UTR of GSK-3β mRNA is a direct target of miR-99b-3p
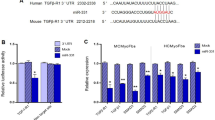
The sequence of miR-99b-3p is identical among rats, mice, and humans (Supplementary Table S3). Computational analysis of miRNA targets was performed by using the target prediction programs TargetScan, miRDB, and miRanda, which consistently identified a putative complementary seed region between miR-99b-3p and GSK-3β mRNA (Fig. 4a). Then, HEK293 cells were transfected with pEZX-MT06 dual-luciferase vectors containing the predicted miR-99b-3p binding sites or mutated binding sites in the 3′UTR of GSK-3β mRNA. Dual-luciferase reporter assay showed that the miR-99b-3p mimic efficiently suppressed the luciferase activity in cells expressing the wild-type GSK-3β 3′UTR but not in those expressing the mutated GSK-3β 3′UTR (Fig. 4b). Furthermore, the expression of GSK-3β was determined by qRT-PCR and Western blotting in rat CFs. The results revealed that both the miR-99b-3p mimic and its inhibitor had no detectable effects on GSK-3β mRNA levels (Fig. 4c, d). However, the protein level of GSK-3β was decreased by the miR-99b-3p mimic, while it was enhanced in the presence of the miR-99b-3p inhibitor (Fig. 4e, f), indicating that miR-99b-3p affected GSK-3β expression by blocking translation. In addition, Ang II also led to downregulation of GSK-3β protein expression, which was reversed by the miR-99b-3p inhibitor.

a Predicted binding sites for miR-99b-3p and the mutated binding sites in the GSK-3β 3′UTR. b HEK293 cells were transfected with pEZX-MT06 dual-luciferase reporter vectors containing wild-type or mutant GSK-3β 3′UTR. Cells were further treated with miR-99b-3p mimic or its negative control (NC), and luciferase activity was detected. c, d Cultured CFs were treated with miR-99b-3p mimic or inhibitor in the presence or absence of Ang II (100 nM) for 48 h. The mRNA expression of GSK-3β was measured by qRT-PCR. e, f Western blotting was performed to detect the protein level of GSK-3β. All data are presented as the mean ± SEM. n.s. no significant difference. *P < 0.05 vs. the NC group, #P < 0.05 vs. the NC plus Ang II treatment group, n = 3.
GSK-3β is involved in miR-99b-3p-mediated cardiac fibrosis
Since GSK-3β was identified as a candidate target of miR-99b-3p, we next investigated its involvement in miR-99b-3p-mediated fibrosis. In rat CFs, depletion of GSK-3β by siRNA resulted in obvious increases in the protein levels of fibronectin, collagen I, vimentin and α-SMA (Fig. 5a). This was in agreement with previous reports concerning the antifibrotic role of GSK-3β [19]. Treatment with the miR-99b-3p inhibitor attenuated the Ang II-promoted expression of these fibrotic markers, which was compromised by GSK-3β siRNA (Fig. 5b). Moreover, the inhibitory effects of miR-99b-3p inhibitor on Ang II-stimulated proliferation and migration of CFs were ameliorated when GSK-3β was concurrently silenced (Fig. 5c, d), indicating the involvement of GSK-3β in miR-99b-3p-mediated cardiac fibrosis.

a Cultured CFs were transfected with GSK-3β siRNA (si-GSK-3β) or negative control (NC) for 48 h. The expression of GSK-3β and fibrotic markers were determined by Western blotting. b CFs with or without GSK-3β knockdown were treated with miR-99b-3p inhibitor and further stimulated with 100 nM Ang II for 48 h. Western blotting was performed to detect the protein levels of GSK-3β and fibrotic markers. c An EdU incorporation assay was used to examine the proliferation of CFs. d The migration ability of cells was tested by the scratch-wound assay. All data are presented as the mean ± SEM. *P < 0.05 vs. the NC or no treatment group, #P < 0.05 vs. the Ang II treatment group, $P < 0.05 vs. the Ang II plus miR-99b-3p inhibitor treatment group, n = 3.
Smad3 is a well-characterized transcription factor contributing to the development of cardiac fibrosis [20, 21]. Emerging evidence has shown that GSK-3β exerts antifibrotic effects by directly interacting with Smad3 and negatively modulating its activation [22, 23]. Here, we also observed that the phosphorylation of Smad3 was notably elevated in CFs following GSK-3β knockdown (Fig. 6a). Administration of the miR-99b-3p mimic enhanced the level of phosphorylated Smad3, while the miR-99b-3p inhibitor markedly decreased Smad3 phosphorylation induced by Ang II (Fig. 6b, c). In addition, CFs were subjected to miR-99b-3p mimic treatment in the presence of SIS3, an inhibitor of Smad3 phosphorylation [24]. The results showed that forced expression of miR-99b-3p augmented the levels of fibronectin and collagen I, which was reversed by SIS3 (Fig. 6d). Thus, miR-99b-3p might promote cardiac fibrosis by suppressing GSK-3β and hence leading to activation of its downstream profibrotic effector Smad3.

a GSK-3β was knocked down in cultured CFs. GSK-3β expression and Smad3 phosphorylation were detected by Western blotting. b, c CFs were treated with miR-99b-3p mimic or inhibitor in the presence or absence of Ang II (100 nM) for 48 h. The level of phosphorylated Smad3 was determined. d CFs were treated with miR-99b-3p mimic and the Smad3 inhibitor SIS3 (1 μM) for 48 h. The phosphorylation of Smad3 and the protein expression of fibronectin and collagen I were analyzed. All data are presented as the mean ± SEM. *P < 0.05 vs. the negative control (NC) or no treatment (con) group, #P < 0.05 vs. the NC plus Ang II treatment group, $P < 0.05 vs. the group treated with miR-99b-3p mimic alone, n = 3.
Inhibition of miR-99b-3p rescues Ang II-induced cardiac fibrosis in mice
To explore the possibility that targeting miR-99b-3p could be utilized to counteract cardiac fibrosis in vivo, C57BL/6 mice were injected by the tail vein with miR-99b-3p antagomir, and subjected to Ang II infusion as described in Materials and methods. Different from the miR-99b-3p inhibitor used for in vitro tests, the antagomir is a single-strand antisense oligonucleotide with chemical modifications to enhance the delivery, stability and inhibiting effect in vivo. The expression of miR-99b-3p was elevated in heart tissues from Ang II-treated mice, which was significantly suppressed by antagomir administration (Supplementary Fig. S1). Morphological and histological examinations showed that the miR-99b-3p antagomir attenuated the enlargement of heart size and collagen deposition stimulated by Ang II (Fig. 7a–e). It also suppressed the increase in the HW/BW ratio following Ang II treatment (Fig. 7f). In addition, treatment with miR-99b-3p antagomir ameliorated Ang II-induced abnormalities in cardiac structure and function, which included elevation of EF, LVAW and LVPW accompanied by reduced LVID (Fig. 7g–k). In cardiac tissues from Ang II-infused mice, the increased mRNA and protein levels of fibrotic factors, such as fibronectin, collagen I, vimentin and α-SMA, were all alleviated by the miR-99b-3p antagomir (Fig. 8a–c). The above findings demonstrated that inhibition of miR-99b-3p by its antagomir could protect mice against Ang II-triggered cardiac fibrosis. In addition, the Ang II-induced reduction in GSK-3β protein expression was effectively restored by miR-99b-3p antagomir treatment, and suppression of miR-99b-3p effectively blunted the increase in Smad3 phosphorylation (Fig. 8d). These results were in line with our observations in vitro, indicating the involvement of the GSK-3β/Smad3 cascade in miR-99b-3p-mediated cardiac fibrosis.

C57BL/6 mice were injected with miR-99b-3p antagomir (ant-miR-99b-3p) or negative control (ant-NC) via the tail vein and subjected to Ang II infusion (2 mg·kg−1·d−1) for 14 days. a–e Pathologic changes in the hearts were observed by H&E staining, Sirius red staining and echocardiography. f, g The HW/BW ratio and ejection fraction were measured. h–k Echocardiographic parameters were recorded. All data are presented as the mean ± SEM. *P < 0.05 vs. the normal saline (NS) group, #P < 0.05 vs. the Ang II treatment group, n = 5. n.s. no significant difference.

C57BL/6 mice received Ang II infusion (2 mg·kg−1·d−1) with or without miR-99b-3p antagomir (ant-miR-99b-3p) for 14 days. a The expression of fibrotic markers was measured by Western blotting. b Immunohistochemical staining for fibronectin, α-SMA and GSK-3β. c qRT-PCR analysis of the mRNA levels of fibrotic markers. d The phosphorylation of Smad3 and the protein expression of GSK-3β were determined. All data are presented as the mean ± SEM. *P < 0.05 vs. the normal saline (NS) group, #P < 0.05 vs. the Ang II treatment group, n = 5.
Discussion
Emerging studies have demonstrated that miRNAs are essential regulators in cardiovascular biology [9, 25]. Dysregulation of miRNA expression causes aberrant gene expression in the heart and ultimately results in pathological conditions [26]. Recently, it has been recognized that miRNAs may serve as biomarkers or therapeutic targets for cardiac fibrosis [27,28,29]. Thus, identifying and characterizing fibrosis-associated miRNAs will not only help to better understand the pathogenesis of cardiac fibrosis but also contribute to the development of novel treatment methods. In the present work, we provide the first evidence that miR-99b-3p participates in Ang II-induced cardiac fibrosis, suggesting a potential therapeutic strategy by targeting miR-99b-3p.
CFs are the predominant cardiac cell types responsible for maintaining myocardial structure and ECM homeostasis [4]. Cardiac fibrosis is a result of CF activation [1, 30]. Mechanical and neurohumoral stimuli can promote CF proliferation and induce the transformation of CFs into myofibroblasts, a critical event during fibrosis development [2, 31]. Myofibroblasts increase ECM synthesis, display enhanced migratory and contractile abilities, and express proinflammatory cytokines, which further exacerbate fibrosis [11]. Ang II, the principal component of the renin-angiotensin-aldosterone system, has been well demonstrated to be a key fibrotic stimulator [32]. Here, we observed that Ang II treatment led to cardiac fibrosis accompanied by elevated miR-99b-3p levels. In cultured rat CFs, forced expression of miR-99b-3p promoted ECM production, increased the level of α-SMA, a representative marker of transformed myofibroblasts, and facilitated the proliferation and migration of cells. Conversely, inhibition of miR-99b-3p could attenuate the fibrotic responses in CFs triggered by Ang II. In addition, the fibrotic phenotypes observed in Ang II-treated mice were significantly alleviated by miR-99b-3p antagomir injection. Together, these findings indicate that miR-99b-3p may be a mediator and interventional target for Ang II-induced cardiac fibrosis.
In this study, the miR-99b-3p level was found to be negatively correlated with GSK-3β expression, which was strongly reduced during cardiac fibrosis. GSK-3β was predicted to be a candidate target of miR-99b-3p. The amino acid and mRNA sequences of GSK-3β are relatively conserved among rats, mice, and humans, as indicated by the Basic Local Alignment Search Tool (Supplementary Tables S4 and S5). Moreover, there is a shared miR-99b-3p binding sequence in the GSK-3β 3′UTRs of these species. A dual-luciferase reporter assay demonstrated that miR-99b-3p inhibited luciferase activity in cells expressing the wild-type GSK-3β 3′UTR but not the mutant form. It is known that miRNAs exert their functions primarily by binding with the 3′UTRs of target genes, leading to mRNA degradation or translational silencing [33]. The qRT-PCR analysis data showed that the mRNA level of GSK-3β in CFs was not affected by miR-99b-3p. Nevertheless, GSK-3β protein expression was markedly reduced by the miR-99b-3p mimic, whereas it was upregulated following treatment with the miR-99b-3p inhibitor. These observations indicate that miR-99b-3p affects the expression of GSK-3β by repressing translation. Similar regulatory mechanisms have been observed for other miRNAs [34, 35]. For example, it was reported that miR-21 had no detectable influence on the mRNA level of Spry1 in CFs but resulted in strong inhibition of Spry1 protein expression [12]. The effect of miR-99b-3p on modulating GSK-3β was further supported by our in vivo experiments. In Ang II-infused mice, the protein level of GSK-3β in heart tissues was decreased, which could be restored by administration of a specific miR-99b-3p antagomir.
GSK-3β is a constitutively active serine/threonine kinase involved in a variety of cellular processes, including glucose metabolism, gene transcription, immune responses, proliferation, and apoptosis [36]. It has been implicated in the pathogenesis of cancer, diabetes, inflammation, and cardiovascular diseases [37]. Recently, accumulating evidence has revealed that GSK-3β also plays an important role in fibroblast biology and exerts an antifibrotic effect [19, 38]. Inhibition of GSK-3β has been proven to trigger extensive fibrotic responses in isolated CFs. Mice with fibroblast-specific knockout of GSK-3β suffered from progressive fibrogenesis and severe left ventricular dysfunction [39]. In contrast, pressure overload-induced fibrosis was ameliorated in the hearts of GSK-3β knock-in mice [40]. In addition, activation of GSK-3β with pharmacological reagents could protect against cardiac fibrosis both in vitro and in vivo [41]. In line with these studies, we found that transfection with GSK-3β siRNA resulted in fibrotic responses. More importantly, miR-99b-3p knockdown attenuated Ang II-induced ECM synthesis and CF activation, which could be strongly blunted by silencing GSK-3β. These results suggest that miR-99b-3p can promote cardiac fibrosis at least partially by suppressing GSK-3β.
To date, GSK-3β has been demonstrated to negatively regulate cardiac fibrosis, but the mechanism by which it suppresses fibrogenesis remains to be elucidated. Intriguingly, some previous studies indicate that there is crosstalk between GSK-3β and TGF-β/Smad3 in mediating fibrotic responses of the diseased heart [19, 38, 39]. The canonical TGF-β/Smad signaling pathway has been well recognized to play a predominant role in fibroblast activation and ECM production [42]. The binding of TGF-β to its membrane receptor leads to the activation of downstream Smad proteins by phosphorylation. Phosphorylated Smad2 and Smad3 trimerize with Smad4 and then translocate into the nucleus, where they act as transcription factors to induce the expression of fibrosis-related genes [43, 44]. A recent study showed that in the normal healthy heart, GSK-3β directly interacted with Smad3, thereby maintaining a low level of activity of Smad3. Inhibition or deletion of GSK-3β induced phosphorylation of Smad3 at the C-terminal Ser 423/425 residues, leading to enhanced transcriptional activity of Smad3 and eventually resulting in excessive fibrosis and adverse ventricular remodeling [39]. This mechanism was supported by the results that SIS3, a small molecule Smad3 inhibitor, could alleviate fibrotic changes and rescue cardiac function in GSK-3β knockout mice [19, 39]. Furthermore, the effect of GSK-3β was specific to Smad3, as GSK-3β deletion had no influence on Smad2 phosphorylation and activation [22]. In our present work, miR-99b-3p decreased GSK-3β protein expression and promoted the level of phosphorylated Smad3. In contrast, miR-99b-3p inhibition restored the Ang II-induced reduction in GSK-3β expression and simultaneously attenuated the increase in Smad3 phosphorylation. In addition, treatment with SIS3 alleviated miR-99b-3p-stimulated expression of fibrotic markers. Therefore, it may be speculated that the modulation of the GSK-3β/Smad3 cascade is responsible for miR-99b-3p-mediated cardiac fibrosis.
Although we have presented evidence concerning the participation of miR-99b-3p in Ang II-triggered cardiac fibrosis by targeting GSK-3β, there are still some obvious limitations in our study. First, the roles of miR-99b-3p in the pathogenesis of cardiac fibrosis should be further confirmed in vivo, especially in animals with cardiac-specific depletion or overexpression of miR-99b-3p. Changes in miR-99b-3p should also be detected in human samples to support potential antifibrotic intervention by modulating miR-99b-3p. To date, little is known about miR-99b-3p expression in the setting of cardiac disorders. A recent study reported that miR-99b-3p was markedly upregulated in aortic valves from patients with degenerative aortic stenosis [45], a common valvular heart disease accompanied by extensive cardiac fibrosis [46,47,48]. This finding may suggest a possible connection between elevated miR-99b-3p levels and cardiovascular abnormalities. However, there is still a lack of clinical investigations on miR-99b-3p expression in cardiac tissues. Second, it is not known whether the same conclusion can be drawn in the setting of cardiac fibrosis caused by stimuli other than Ang II, for example, by pressure overload, ischemia or diabetes. Moreover, a single miRNA may regulate a broad range of transcripts [49]. Several targets of miR-99b-3p have been identified previously, such as PCDH19 and HoxD3 [15, 16]. Thus, we cannot rule out the involvement of genes other than GSK-3β in miR-99b-3p-mediated fibrogenesis.
In summary, our present work revealed that miR-99b-3p promoted Ang II-induced cardiac fibrosis. The expression of miR-99b-3p was increased in cultured CFs after Ang II treatment and in the hearts of mice that received Ang II infusion. Overexpression of miR-99b-3p induced fibrotic responses in CFs. In contrast, miR-99b-3p inhibition protected against Ang II-mediated fibrosis both in vitro and in vivo. The profibrotic effects of miR-99b-3p could be at least partially attributed to its suppression of GSK-3β. These findings add new information about the biological functions of miR-99b-3p and suggest that modulation of miR-99b-3p may offer a potential treatment for cardiac fibrosis.
References
Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Asp Med. 2019;65:70–99.
Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis the fibroblast awakens. Circ Res. 2016;118:1021–40.
Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–74.
Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Disco. 2016;15:620–38.
Hinderer S, Schenke-Layland K. Cardiac fibrosis—a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. 2019;146:77–82.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14.
Zhou SS, Jin JP, Wang JQ, Zhang ZG, Freedman JH, Zheng Y, et al. miRNAS in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacol Sin. 2018;39:1073–84.
Boen JRA, Gevaert AB, De Keulenaer GW, Van Craenenbroeck EM, Segers VFM. The role of endothelial miRNAs in myocardial biology and disease. J Mol Cell Cardiol. 2020;138:75–87.
Ali Syeda Z, Langden SSS, Munkhzul C, Lee M, Song SJ. Regulatory mechanism of MicroRNA expression in cancer. Int J Mol Sci. 2020;21:1723.
Creemers EE, van Rooij E. Function and therapeutic potential of noncoding RNAs in cardiac fibrosis. Circ Res. 2016;118:108–18.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4.
Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, et al. MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation. 2016;133:291–301.
Zhang Y, Huang XR, Wei LH, Chung ACK, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol Ther. 2014;22:974–85.
Chang SE, Gao ZC, Yang Y, He K, Wang XF, Wang LM, et al. miR-99b-3p is induced by vitamin D3 and contributes to its antiproliferative effects in gastric cancer cells by targeting HoxD3. Biol Chem. 2019;400:1079–86.
Yao XB, Zhang HG, Liu YJ, Liu XM, Wang XH, Sun XF, et al. miR-99b-3p promotes hepatocellular carcinoma metastasis and proliferation by targeting protocadherin 19. Gene. 2019;698:141–9.
Hong HQ, Lu J, Fang XL, Zhang YH, Cai Y, Yuan J, et al. G3BP2 is involved in isoproterenol-induced cardiac hypertrophy through activating the NF-kappa B signaling pathway. Acta Pharmacol Sin. 2018;39:184–94.
Olson ER, Shamhart PE, Naugle JE, Meszaros JG. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase C delta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension. 2008;51:704–11.
Lal H, Ahmad F, Woodgett J, Force T. The GSK-3 family as therapeutic target for myocardial diseases. Circ Res. 2015;116:138–49.
Huang XR, Chung ACK, Yang FY, Yue WS, Deng CX, Lau CP, et al. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension. 2010;55:1165–71.
Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential role of smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–38.
Guo X, Ramirez A, Waddell DS, Li ZZ, Liu XD, Wang XF. Axin and GSK3-beta control Smad3 protein stability and modulate TGF-beta signaling. Genes Dev. 2008;22:106–20.
Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol Chem. 2009;284:19808–16.
Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta 1-induced extracellular matrix expression. Mol Pharmacol. 2006;69:597–607.
Pham TP, Kremer V, Boon RA. RNA-based therapeutics in cardiovascular disease. Curr Opin Cardiol. 2020;35:191–8.
Chandy M. A tangled tale of microRNA and cardiac fibrosis. Clin Sci. 2019;133:2217–20.
Pan ZW, Lu YJ, Yang BF. MicroRNAs: a novel class of potential therapeutic targets for cardiovascular diseases. Acta Pharmacol Sin. 2010;31:1–9.
Lu DC, Thum T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat Rev Cardiol. 2019;16:661–74.
Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M, et al. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J. 2018;39:2704–16.
Rockey DC, Bell PD, Hill JA. Fibrosis—a common pathway to organ injury and failure reply. N Engl J Med. 2015;373:96.
Leask A. Getting to the heart of the matter new insights into cardiac fibrosis. Circ Res. 2015;116:1269–76.
Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15–26.
Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–42.
Tay YMS, Tam WL, Ang YS, Gaughwin PM, Yang H, Wang WJ, et al. MicroRNA-134 modulates the differentiation of mouse embryonic stem cells, where it causes post-transcriptional attenuation of Nanog and LRH1. Stem Cells. 2008;26:17–29.
Zhang SB, Lin SY, Liu M, Liu CC, Ding HH, Sun Y, et al. CircAnks1a in the spinal cord regulates hypersensitivity in a rodent model of neuropathic pain. Nat Commun. 2019;10:4119.
Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–76.
Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–31.
Guo YJ, Gupte M, Umbarkar P, Singh AP, Sui JY, Force T, et al. Entanglement of GSK-3 beta, beta-catenin and TGF-beta 1 signaling network to regulate myocardial fibrosis. J Mol Cell Cardiol. 2017;110:109–20.
Lal H, Ahmad F, Zhou JB, Yu JE, Vagnozzi RJ, Guo YJ, et al. Cardiac fibroblast glycogen synthase kinase-3 beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation. 2014;130:419–30.
Matsuda T, Zhai P, Maejima Y, Hong C, Gao SM, Tian B, et al. Distinct roles of GSK-3 alpha and GSK-3 beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A. 2008;105:20900–5.
Zeng ZF, Wang QY, Yang XM, Ren YL, Jiao SH, Zhu QQ, et al. Qishen granule attenuates cardiac fibrosis by regulating TGF-beta /Smad3 and GSK-3 beta pathway. Phytomedicine 2019;62:152949.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–38.
Shi YG, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700.
Goumans MJ, ten Dijke P. TGF-beta signaling in control of cardiovascular function. Cold Spring Harb Perspect Biol. 2018;10:39.
Shi J, Liu H, Wang H, Kong XQ. MicroRNA expression signature in degenerative aortic stenosis. BioMed Res Int. 2016;2016:4682172.
Ramaraj R, Sorrell VL. Degenerative aortic stenosis. BMJ. 2008;336:550–5.
Krayenbuehl HP, Hess OM, Monrad ES, Schneider J, Mall G, Turina M. Left ventricular myocardial structure in aortic valve disease before, intermediate, and late after aortic valve replacement. Circulation. 1989;79:744–55.
Dweck MR, Joshi S, Murigu T, Alpendurada F, Jabbour A, Melina G, et al. Midwall fibrosis is an independent predictor of mortality in patients with aortic stenosis. J Am Coll Cardiol. 2011;58:1271–9.
Thum T. Noncoding RNAs and myocardial fibrosis. Nat Rev Cardiol. 2014;11:655–63.
Acknowledgements
This work was supported by grants from the National Major Special Projects for the Creation and Manufacture of New Drugs (2019ZX09301104), the National Engineering and Technology Research Center for New Drug Druggability Evaluation (Seed Program of Guangdong Province, 2017B090903004), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093), the Guangdong Basic and Applied Basic Research Foundation (2020A1515011512), and the Young Teacher Training Program of Sun Yat-sen University (18ykpy26).
Author information
Authors and Affiliations
Contributions
YHY and JTY conceived and designed the experiments. YHY, YHZ, XYB, JY, HZ, and LLZ performed the experiments. YQD and PXW analyzed the data. YHY and JTY wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Yu, Yh., Zhang, Yh., Ding, Yq. et al. MicroRNA-99b-3p promotes angiotensin II-induced cardiac fibrosis in mice by targeting GSK-3β. Acta Pharmacol Sin 42, 715–725 (2021). https://doi.org/10.1038/s41401-020-0498-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0498-z
Keywords
This article is cited by
-
MicroRNA delivery based on nanoparticles of cardiovascular diseases
Molecular and Cellular Biochemistry (2023)
