
Abstract
We have recently developed an enzyme-directed immunostimulant (EDI) prodrug motif, which is metabolized to active immunostimulant by cancer cells and, following drug efflux, activates nearby immune cells, resulting in immunogenicity. In this study, we synthesized several EDI prodrugs featuring an imidazoquinoline immunostimulant resiquimod (a Toll-like receptor 7/8 agonist) covalently modified with glycosidase enzyme-directing groups selected from substrates of β-glucuronidase, α-mannosidase, or β-galactosidase. We compared the glycosidase-dependent immunogenicity elicited by each EDI in RAW-Blue macrophages following conversion to active immunostimulant by complementary glycosidase. At a cellular level, we examined EDI metabolism across three cancer cell lines (B16 melanoma, TC2 prostate, and 4T1 breast cancer). Comparing the relative immunogenicity elicited by each EDI/cancer cell combination, we found that B16 cells produced the highest EDI prodrug immunogenicity, achieving >95% of that elicited by unmodified resiquimod, followed by TC2 and 4T1 cells (40% and 30%, respectively). Immunogenicity elicited was comparable for a given cell type and independent of the glycosidase substrate in the EDIs or differences in functional glycosidase activity between cell lines. Measuring drug efflux of the immunostimulant payload and efflux protein expression revealed that EDI/cancer cell-mediated immunogenicity was governed by efflux potential of the cancer cells. We determined that, following EDI conversion, immunostimulant efflux occurred through both P-glycoprotein-dependent and P-glycoprotein-independent transport mechanisms. Overall, this study highlights the broad ability of EDIs to couple immunogenicity to the metabolism of many cancers that exhibit drug efflux and suggests that designing future generations of EDIs with immunostimulant payloads that are optimized for drug efflux could be particularly beneficial.
Similar content being viewed by others

Introduction
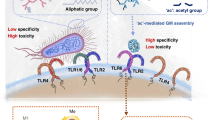
Directed enzyme prodrug therapy (DEPT) is a drug delivery technique that uses enzyme-directed chemotherapeutic prodrugs to enhance tissue-specific drug accumulation and limit off-target cytotoxicity via enzymatic conversion of prodrug to active drug [1,2,3]. However, DEPT is complicated by the bystander effect; a phenomenon whereby the drug payload, once liberated by complementary enzyme, can diffuse to interact with adjacent off-target bystander cells in local proximity [4]. The bystander effect is particularly problematic in cancer due to the expression of transport proteins, including members of the adenosine-binding cassette (ABC)-transporter superfamily, which traffic small-molecule xenobiotics from within cancer cells to the extracellular space via a myriad of active transport processes collectively termed drug efflux [5]. Building on these findings, our group proposed a strategy termed bystander-assisted immunotherapy (BAIT) that extends the concept of DEPT to immunostimulant payloads by activating nearby immune cells via the bystander effect (Fig. 1a). To this end, we developed an enzyme-directed immunostimulant (EDI) prodrug motif, which is metabolized by cancer cells, resulting in immunogenicity that is enhanced by drug efflux and the bystander effect [6, 7]. In first-generation EDIs, the immunostimulant imiquimod [8] was chosen for its synthetic simplicity rather than potency, and the enzyme-directing groups were specifically matched to cancer cell model systems that overexpressed complementary enzyme and transport proteins required for BAIT.

a The mechanism of action underlying BAIT: (i) An enzyme-directed immunostimulant (EDI) prodrug is taken up by cancer cells, and (ii) enzymes within cancer cells metabolize EDI prodrug to active immunostimulant. (iii) The active immunostimulant is effluxed from within cancer cells to the extracellular space. (iv) Effluxed immunostimulant activates bystander immune cells, which (v) initiate an immune response in local proximity to the cancer cells. b Overview of first-generation EDIs (IMQ-Gal and IMQ-Man) and the EDIs developed in this work, EDI (7), (10), and (13). Each EDI was tested for conversion to immunostimulant by exogenous enzyme or by cancer cell metabolism followed by drug efflux. In this work, we determine the effect of using different enzyme substrates in EDIs across cancer cell lines of varied expression of complementary enzyme.
The present study builds on our previous work by comparing the performance of a small catalog of more potent EDIs across multiple enzyme-directing groups and cancer cell lines without a priori matching to complementary enzyme expression. For the immunostimulant payload, we use the imidazoquinoline immunostimulant resiquimod (RSQ), an agonist of innate immune cell Toll-like receptors (TLRs) 7 and 8 featuring established anticancer efficacy [9, 10], nanomolar potency [11], and a well-defined structure–activity relationship [12]. For enzyme-directing groups in our EDI catalog, we selected glycosidase-labile substrates for their general ability to pair with the Warburg effect in cancer cells, which favor glycolysis [13, 14]. Specifically, we selected β-glucuronidase (β-glu) [15], α-mannosidase (α-man) [16,17,18], and β-galactosidase (β-gal) [19, 20], because we envisioned that the established glycosidase expression and functional activity across many cancer types [21, 22] would make these glycosidase-directed immunostimulants broadly applicable [23, 24]. Among these glycosidases, β-glu is unique because it is localized intracellularly in healthy cells but found extracellularly in tumor and necrotic tissues, although it remains unclear whether extracellular β-glu is derived from cancer cells themselves or introduced through other sources such as tumor-infiltrating lymphocytes [3, 25, 26]. Each glycosidase has been employed as an enzyme target, either in DEPT [27, 28] or BAIT [6, 7], but there have been few direct comparisons of different enzyme-directing groups in a single enzyme-directed prodrug system [29] and, with the exception of the present study, none that compare EDIs. As such, we were interested in comparing EDIs targeted to different glycosidases endogenously expressed across several cancer cell lines. The cancer types chosen for this study were melanoma, prostate cancer, and breast cancer, because they are among the top five most frequently diagnosed cancers in the United States [30]. In addition, it has been established that imidazoquinolines exhibit antitumor efficacy in mouse tumor models of the corresponding cancer cell lines, specifically for the B16 melanoma [31], TRAMP prostate [32], and 4T1 breast [33] cancers used in this study. The specific activities of the target glycosidases have been reported for some of these cell lines [34, 35]; however, established expression of a particular glycosidase was not used as a selection criterion. Rather, we anticipated that differences in glycosidase activity and drug efflux potential between cancer cell lines would provide insight into which factors affect the cancer-mediated immunogenicity of EDIs in BAIT (Fig. 1b).
Materials and methods
Synthesis of EDIs
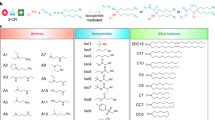
Intermediates (8) and (11) were synthesized according to literature procedure [6, 7]. EDIs (7), (10), and (13) were synthesized as detailed in Fig. 2 and Supplementary Information.

a Base-catalyzed elimination of the methyl ester-protected glucuronide scaffold. b Synthesis of β-glucuronidase-directed EDI (7). (i) (a) Ac2O, pyridine, DCM, rt, 3.5 h; (b) 33% (w/w) HBr/AcOH, DCM, rt, 3 h; (ii) 4-hydroxy-3-nitrobenzaldehyde, Ag2O, MeCN, rt, 4 h; (iii) NaBH4, 6:1 CHCl3/2-propanol, 0 °C to rt, 5 h; (iv) 4-nitrophenyl chloroformate, pyridine, DCM, 0 °C, 2 h; (v) resiquimod, DIPEA, THF, microwave: 90 °C, 1 h; (vi) (a) Pd(PPh3)4, aniline, DCM, rt, 3 h; (b) NaOMe, 1:1 THF/MeOH, 0 °C, 3 h. c Synthesis of α-mannosidase-directed EDI (10). (vii) resiquimod, DIPEA, THF, microwave: 90 °C, 1.5 h; (viii) NaOMe, MeOH, 0 °C to rt, 1.5 h. d Synthesis of β-galactosidase-directed EDI (13). (ix) resiquimod, DIPEA, THF, microwave: 90 °C, 2.5 h; (x) NaOMe, MeOH, 0 °C to rt, 2.5 h.
Standardization of glycosidase enzyme stock solutions
β-Glu, β-gal, and α-man were filtered through sterile 0.2-µm filters and titrated in RAW-Blue (RB) assays to ensure non-pyrogenicity at the indicated working concentrations. For comparison of glycosidases, we standardized our definition of glycosidase activity such that 1 U is defined as 1.0 µmol/min of liberated 4-nitrophenol at 37 °C from a 1 mM solution of corresponding 4-nitrophenyl β-D-glucuronide (β-glu substrate), 4-nitrophenyl β-D-galactopyranoside (β-gal substrate), or 4-nitrophenyl α-D-mannopyranoside (α-man substrate). For β-glu and β-gal, activity was measured at pH 7.4 in Dulbecco’s phosphate-buffered saline (PBS). For α-man, a pH 4.6 acetate buffer was used to match the enzyme’s optimal pH. In this way, enzyme stock solutions were standardized at 1 U/mL, which were diluted to provide the working solutions used in subsequent assays.
Cell culture
Cell lines of B16-F10 (B16) melanoma, TRAMP-C2 (TC2) prostate cancer, 4T1-Luc2 (4T1) breast cancer, and RB macrophages were cultured according to the manufacturer’s instructions. RB passages 5–18 were used for all experiments. Because the NF-κB reporter system drifts with passage number, only raw absorbance values within individual figures are comparable. For detailed cell culture procedures and media formulations, see Supplementary Information.
RB assay
Unless otherwise noted, the complete cell media used for this and all following cell experiments is comprised of Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose, 2 mM L-glutamine, and 100 U/mL PenStrep, supplemented with 10% heat inactivated fetal bovine serum (HI-FBS). Activation of RB cells (Figs. 3 and 5, Supplementary Figs. S1 and S2) was performed similar to the manufacturer’s instructions with some modifications. Briefly, RB cells were seeded in 96-well plates (1 × 105 cells/well) in 180 µL of complete media. Test solutions (RSQ, enzymatic conversion experiments, and cancer cell supernatants) were filtered through 0.2-µm filters, and 20 µL of each solution was administered to the respective wells to achieve the indicated concentrations at a final volume of 200 µL/well. Plates were incubated for 18 h before measuring NF-κB transcription via secreted embryonic alkaline phosphatase (SEAP) detection protocol.

The indicated EDI (10 µM) was incubated with complementary enzyme (red bars/circles) or buffer alone (blue bars/squares) and conversion to RSQ was quantified by LC-MS (left axis). Immunogenicity via NF-κB transcription was assessed at each time interval via RAW-Blue assay (right axis) for a EDI (7) and β-glucuronidase (β-glu, 0.1 mU/mL, PBS, pH 7.4); b EDI (10) and α-mannosidase (α-man, 5 mU/mL, acetate buffer, pH 4.6), and c EDI (13) and β-galactosidase (β-gal, 5 mU/mL, pH 7.4). Samples were incubated (37 °C) for the indicated time intervals before analysis. Error bars are the standard deviation of mean values from triplicate measurements. P < 0.001 for all LC-MS samples with enzyme compared to samples without enzyme at >6 h incubation time. For RB assay, *P < 0.05, **P < 0.01, and ***P < 0.001 for EDIs incubated with enzyme compared to EDIs incubated with PBS or acetate buffer alone as indicated.
SEAP detection protocol
SEAP detection media (1.168 mg/mL 5-bromo-4-chloro-3’-indolylphosphate p-toluidine salt in 1 M aqueous diethanolamine) was used to measure SEAP for all RB Assays. To an optically transparent bottomed 96-well plate, 180 µL of SEAP detection media was added to each well. Next, 20 µL of supernatant from the RB assays above was added to each well, and the plates were incubated for the amount of time necessary to achieve significant positive control absorbance (1–6 h). Plates directly compared within each experiment were incubated for the same amount of time. Following incubation, the absorbance of each well was measured at 620 nm. Experiments were performed in triplicate, and where indicated in the text, a blank was subtracted from the values reported. Where indicated, absorbance values were normalized using the absorbance from RSQ-positive control as the maximum value and absorbance from the media blank as the minimum value.
Liquid chromatography–mass spectrometry (LC-MS) quantification protocol
LC-MS/MS chromatography was performed using a Kinetex® C18 column (100 mm × 2.1 mm, 2.6 μm). Enzyme reactions were separated using a gradient of 0.05% formic acid and 0.2% acetic acid in H2O (Mobile Phase A) and 90% acetonitrile, 9.9% H2O, and 0.1% formic acid (Mobile Phase B). Quantification of EDI metabolites was performed using an API 4000 Q-Trap MS system in positive mode relative to phenacetin internal standard according to published protocol [36].
Combined LC-MS/RB assay
To affect enzymatic hydrolysis of (7), 180 µL of a working solution of β-glu (0.11 mU/mL in PBS) was added to wells in a 96-well plate alongside wells that contained only PBS (no enzyme). Next, 20 µL of (7, 100 µM in PBS) was added to each well and incubated at 37 °C. To affect the enzymatic hydrolysis of (10), 180 µL of a working solution of α-man (5.6 mU/mL in acetate buffer, pH 4.6) was added to wells in a 96-well plate alongside wells that contained only acetate buffer (no enzyme). Next, 20 µL of (10, 100 µM in acetate buffer) was added to each well and incubated at 37 °C. To affect enzymatic hydrolysis of (13), 180 µL of a working solution of β-gal (5.6 mU/mL in PBS) was added alongside wells that contained only PBS (no enzyme). Next, 20 µL of (13, 100 µM in PBS) was added to each well and incubated at 37 °C. The 96-well plates were incubated at 37 °C for the time intervals indicated. At each time interval, assay solution (1 µL) was removed, diluted to 40 µL with PBS, and 20 µL of this diluted assay solution was used in the RB assay protocol (25 nM final total concentration of EDI+RSQ). The remaining assay solution was quenched by addition of 65 µL of 1 M formic acid containing 25 µM phenacetin (LC-MS internal standard) and analyzed for corresponding EDI and RSQ by LC-MS.
Cell-mediated conversion of reporter glycosidase substrates
Cancer cells or RB macrophages were seeded at 5 × 105 cells/well in 24-well plates in complete cell media (900 μL) using DMEM without phenolphthalein. The plates were incubated at 37 °C for 6 h. Next, 100 μL of stock glycosidase substrate solution (5 mM; 4-nitrophenyl β-D-glucuronide, 4-nitrophenyl α-D-mannopyranoside, or 4-nitrophenyl β-D-galactopyranoside) was added to the homogenous cell suspensions, resulting in a 0.5 mM substrate concentration in each well. Plates were incubated (37 °C), and at the indicated time intervals (Fig. 4a), samples were collected and centrifuged (300 × g for 5 min). Supernatants were transferred to an optically transparent bottomed 96-well plate and ultraviolet–visible light (UV-Vis) was used to measure the absorbance of 4-nitrophenol (405 nm) liberated at each time interval. This was performed for all cell lines with each glycosidase substrate in triplicate.

a Each glycoside reporter (1 mM) was incubated with cancer cells before measuring liberated 4-nitrophenol via absorbance (405 nm) at the indicated time intervals. b 4-Nitrophenyl β-glucuronide was incubated with cancer cell lysate before measuring liberated 4-nitrophenol via absorbance (405 nm) at the indicated time intervals. Error bars in a and b represent the standard deviation from the mean for triplicate measurements. c P-gp expression was measured by Western blot in 1 WT MDCK, 2 MDR MDCK, 3 TC2 (2.5 × 105–5 × 105 cells), 4 TC2 (1.5 × 107–2 × 107 cells), 5 B16 (2.5 × 105–5 × 105 cells), 6 B16 (1.5 × 107–2 × 107 cells), 7 4T1 (2.5 × 105–5 × 105 cells), or 8 4T1 (1.5 × 107–2 × 107 cells). d Histograms of cancer cell Rh123 fluorescence (x-axis) measured by flow cytometry (λex = 507 nm, λem = 529 nm). Cancer cells were incubated with Rh123 (1 µM) and subsequently with RSQ (1 or 100 µM) or PSC 833 P-gp inhibitor (1 µM). Red dashed lines are used as a reference to cells loaded with Rh123 alone. Histograms are representative of experiments performed in hexaplet. e Mean fluorescence intensity of Rh123 retention normalized with respect to cells loaded with Rh123 alone. Error bars are the standard deviation of normalized mean values. **P < 0.01, ***P < 0.001, and ns = no significance for the indicated sample/cell type compared to cells loaded with Rh123 alone.
Cancer cell lysate conversion of 4-nitrophenyl β-D-glucuronide
Cancer cells (5 × 105) were centrifuged (300 × g for 5 min), and the supernatant was discarded. The cells were resuspended in 1 mL of cold lysis buffer (5 mM EDTA, 150 mM NaCl, 50 mM Tris-HCl, 1% Triton X-100, pH 8.0). Samples were incubated on ice for 20 min and then centrifuged (5580 × g for 10 min). Aliquots of supernatant (180 µL) were transferred to an optically transparent bottomed 96-well plate, and 4-nitrophenyl β-D-glucuronide (20 µL, 10 mM) was added. Samples were incubated at 37 °C for the indicated time intervals (Fig. 4b). The absorbance of the supernatant was analyzed by UV-Vis to measure the absorbance of liberated 4-nitrophenol (405 nm) at each time interval. This experiment was performed in triplicate for all three cancer cell lines.
P-glycoprotein (P-gp) expression in cancer cell lines
Total protein concentration for standardized plasma membrane preparations of Madin-Darby canine kidney (MDCK) and multidrug-resistant (MDR) MDCK cells was determined using a Pierce BCA Protein Assay Kit according to the manufacturer’s instructions. Total protein (30 μg) isolated from MDCK or MDR MDCK cells was used as a positive control. Similar preparations were made from B16, TC2, and 4T1 cells at low (2.5 × 105–5 × 105) and high (1.5 × 107–2 × 107) cell counts. The samples were separated on a 4%–15% Tris–HCl gradient polyacrylamide gel and electrotransferred to polyvinylidene fluoride membrane. The membrane was incubated in blocking buffer (5% nonfat dry milk in PBS with 0.1% Tween 20) for 1 h at room temperature. The membrane was then incubated with P-gp detection antibody C219 (Thermo Fisher) overnight at 4 °C, washed (0.1% Tween 20 in PBS), and incubated with alkaline phosphatase-conjugated secondary antibody (Promega) for 30 min at 25 °C. The membrane underwent a final wash before staining using Immune Star AP substrate (Bio-Rad) according to the manufacturer’s instructions (Fig. 4c).
Cancer cell efflux assay
Cancer cells were seeded in 6-well plates (2 × 105 cells/well) in 1 mL of complete media and allowed to adhere by incubating at 37 °C for 12 h (~70% confluency). Cells were then loaded with 1 μM rhodamine 123 (Rh123) followed by incubation at 37 °C for 1 h. Next, cells were loaded with RSQ (1 or 100 μM), 1 μM of P-gp inhibitor PSC 833 (positive control), or complete media (negative control). Cells were then incubated (30 min at 37 °C) before washing with PBS (1 mL), trypsinizing with Trypsin-EDTA solution, and neutralizing using Trypsin neutralizing solution (5% HI-FBS in PBS). The resulting cell suspensions were centrifuged (300 × g for 5 min), and cell pellets were suspended in 300 μL FACS solution (PBS, 2% HI-FBS, 1 mM EDTA, 0.1% NaN3) before analysis by flow cytometry to measure intracellular Rh123. Gates for each cell population were set (Supplementary Fig. S4), and 5000 events were collected inside the gate (Fig. 4d). This experiment was performed in hexaplet for all three cancer cell lines. Mean fluorescence intensity from each experiment was normalized relative to cells treated with Rh123 alone (Fig. 4e).
Cancer-mediated immunogenicity of EDIs
Cancer cells were seeded at 5 × 105 cells/well in 24-well plates in complete cell media and incubated at 37 °C for 6 h. RSQ or EDIs (7, 10, or 13) were added to wells containing either cancer cells or cancer cell media to yield a final concentration of 250 nM, and samples were incubated at 37 °C for 48 h. The cancer cell supernatant (20 µL) was then transferred to a 96-well plate containing 180 µL of RB cells and quantified using the RB assay and SEAP detection protocols above. Immunogenicity of EDIs (measured as absorbance at 620 nm) was normalized by using RSQ as the maximum and cancer cell media as the minimum absorbance values (Fig. 5).

a Schematic of the workflow for experiments testing the cancer-mediated immunogenicity of EDIs. b RSQ (red, 250 nM) or EDIs (250 nM) were incubated with cancer cells or cancer cell media alone (48 h, 37 °C). Supernatant immunogenicity of each sample was assessed via NF-κB transcription in a RB assay. Error bars are the standard deviation of mean values for experiments performed in triplicate and normalized with respect to the RSQ control and cell media blanks: *P < 0.05 and **P < 0.01 for experiments compared to EDI incubated with media alone.
Statistical analyses
All data are presented as mean ± SEM. All experiments were performed at least three times. Statistical significance for all comparisons was done using a two-tailed t test assuming unequal variances.
Results
Synthesis of EDI prodrugs
We first set out to synthesize EDIs that could be converted to RSQ immunostimulant by complementary β-glu, α-man, or β-gal enzyme. Initial attempts at synthesizing β-glucuronide EDI (7) from methyl bromo-2,3,4-triacetylglucuronate were met with difficulty due to the proclivity of this substrate to undergo 4,5 elimination under even mild basic conditions (Fig. 2a). To circumvent this, synthesis of (7) began with the preparation of allyl-D-glucuronate (1) from D-(+)-glucuronic acid according to literature procedure (Fig. 2b) [37]. This was followed by peracetylation and anomeric bromination to yield protected glycosyl donor (2). Koenigs–Knorr O-glycosylation with 4-hydroxy-3-nitrobenzaldehyde afforded (3) with clean control over the anomeric stereochemistry, followed by reduction with NaBH4 to provide (4). The alcohol (4) was treated with 4-nitrophenyl chloroformate to yield activated carbonate (5), followed by microwave-assisted carbamoylation with RSQ to afford (6). Deprotection of (6) required two sequential steps: (1) Tsuji–Trost deallylation followed by (2) Zemplén deacetylation to provide (7) via a route that effectively circumvented elimination. Overall, the synthesis of (7) was achieved in 8 linear steps with a total yield of 2.4% from allyl-D-glucuronate (1). Synthesis of the α-mannopyranoside (10) and β-galactopyranoside (13) EDIs began with preparation of activated 4-nitrophenyl carbonates, (8) and (11) respectively, according to previously reported chemistry [6, 7]. Microwave-assisted carbamoylation between these compounds and RSQ cleanly afforded the protected EDIs (9) and (12). Zemplén deacetylation provided EDIs (10) and (13) in 2 steps from reported intermediates with yields of 48% and 26%, respectively (Fig. 2c, d).
EDIs are metabolized to RSQ by complementary exogenous glycosidase
Following the synthesis of each EDI, we tested to see whether these compounds could liberate RSQ selectively in the presence of complementary glycosidase. At a molecular level, glycosidase-mediated conversion of EDIs to RSQ was monitored by LC-MS. In addition, we measured the immunogenicity of the reaction metabolites via stimulation of RB macrophages, a reporter immune cell line known to express TLRs 7/8. Activation of TLRs 7/8 leads to NF-κB transcription that, in RB cells, is linked to SEAP production, which can be quantified by a colorimetric assay [38]. Prior to incubation with EDI, the amount of glycosidase used was optimized (via titration in RB assays) to eliminate any background immunogenicity caused by the enzyme. Thus β-glu was used at a final activity of 0.1 mU/mL, while α-man and β-gal were used at 5 mU/mL (for standardization of enzyme activity, see Materials and methods). Minimal conversion of each EDI to RSQ occurred over 48 h in PBS (pH 7.4) or acetate buffer (pH 4.6) without enzyme, and EDIs without enzyme did not activate RB cells. However, in the presence of complementary glycosidase, all EDIs were converted to RSQ in quantities that could activate RB cells within 20 h (Fig. 3 and Supplementary Fig. S1). Additional experiments with mismatched glycosidases and EDIs were also performed (Supplementary Fig. S2). While most mismatched combinations exhibited diminished immunogenicity, it was found that β-glu-directed EDI (7) could also elicit an immune response in the presence of β-gal. To ensure that our β-gal stock was not contaminated with β-glu, we tested it with the highly sensitive and specific fluorometric β-glu substrate, 4-methylumbelliferyl β-glucuronide. This experiment revealed no β-glu activity in our β-gal (Supplementary Fig. S3).
Cancer cell lines feature varied levels of glycosidase activity
To determine how the functional glycosidase activities in each cancer cell line affects cancer-mediated immunogenicity from EDI prodrugs, we measured the relative enzyme activity in each cancer cell line using reporter molecules for functional β-glu, α-man, and β-gal (4-nitrophenyl β-glucuronide, α-mannopyranoside, and β-galactopyranoside). Of the three glycosidase reporters, the α-man substrate liberated the most 4-nitrophenol in every cell line after 48 h (Fig. 4a). Relative to the α-man substrate, the β-gal substrate liberated 40% ± 4%, 45% ± 2%, and 56% ± 3% of 4-nitrophenol in B16, TC2, and 4T1 cell lines, respectively. The β-glu substrate exhibited minimal conversion to 4-nitrophenol in all cancer cell lines (11% ± 0.5%, 0% ± 0.5%, and 5% ± 0.6% relative to α-man substrate for B16, TC2, and 4T1 cell lines, respectively). We envisioned that the minimal conversion of β-glu substrate in cancer cells could either be due to impaired uptake or due to a lack of functional β-glu. To test this, cancer cells were lysed and β-glu substrate was incubated with the cell lysate (Fig. 4b). In this case, enhanced liberation of 4-nitrophenol was observed, indicating the presence of intracellular β-glu and supporting our hypothesis that cell entry is hindered for the β-glu substrate in cancer cells.
Cancer cell lines efflux RSQ through multiple pathways
In order to investigate the efflux of RSQ from cancer cells, we first looked for P-gp expression in each cancer cell line by Western blot (Fig. 4c) [39]. P-gp was detected in TC2 and B16 cells but not in 4T1 cells. We then measured retention of Rh123 (P-gp substrate) in cancer cells alone or in the presence of RSQ or PSC 833 (P-gp inhibitor) by flow cytometry (Fig. 4d, Supplementary Fig. S5). Interestingly, PSC 833 caused a slight but statistically significant (P < 0.01) decrease in Rh123 retention for B16 cells; however, Rh123 retention was increased 2-fold and 1.5-fold in the presence of inhibitor in TC2 and 4T1 cells, respectively (Fig. 4e). We then monitored the impact of RSQ on the efflux of Rh123. At low loading concentrations of RSQ (1 µM), a modest decrease in Rh123 fluorescence was observed in all cell lines, indicating that RSQ is not a competitive substrate for Rh123 efflux at equimolar concentrations in any cancer cell line tested. However, at higher RSQ loading (100 µM), an increase in Rh123 retention was observed in TC2 cells but not in B16 or 4T1 cells.
Cancer cells elicit immunogenicity from EDIs via BAIT
The performance of each EDI in a BAIT model system was compared by treating cancer cells with EDIs (7, 10, and 13) and measuring the immunogenicity of the cell supernatant in RB macrophages (Fig. 5a). EDIs exhibited minimal immunogenicity in cancer cell media alone. Each cancer cell line caused varied degrees of immune cell activation from the EDIs (Fig. 5b). B16 cells provided greater immunogenicity than the other cell lines with every EDI. TC2 and 4T1 cells provided comparable but lower levels of immunogenicity from each EDI despite the large differences in functional glycosidase activity as measured with the 4-nitrophenol reporter substrates. Interestingly, no differences in immunogenicity were observed between different EDIs in any given cancer cell line.
Discussion
Following synthesis of EDIs (7), (10), and (13), we determined, via RB assay, that attachment of enzyme-directing groups abrogated immune cell activation and that addition of exogenous complementary glycosidase restored immunogenicity. This assay, in tandem with LC-MS, demonstrates that immunogenicity results from hydrolysis of the EDI enzyme substrates to liberate free RSQ immunostimulant. In addition, we demonstrated that EDIs are specifically metabolized to RSQ by their complementary glycosidase, except for the β-glu EDI (7) that was also metabolized by β-gal. Because both β-gal and β-glu are sourced from Escherichia coli, we initially suspected that our β-gal stock could have residual β-glu activity. However, testing our β-gal stock with a fluorometric β-glu substrate proved that there was no cross-contamination. Therefore, we concluded that (7) is a substrate for both β-glu and β-gal, likely stemming from active site homology between the two enzymes [40].
After exploring enzymatic hydrolysis and subsequent immune cell activation of the EDIs, we next measured the relative functional glycosidase activity in the chosen cancer cell lines to determine whether EDI immunogenicity correlates to in vitro glycosidase activity. We chose the 4-nitrophenyl glycoside reporter substrates because the 4-nitrophenol reporter molecule is conserved between each substrate, and therefore efflux of the 4-nitrophenol reporter would be identical between glycosidase substrates in a given cell line. We envisioned that this experiment would lend insight into the behavior of each EDI as it pertains to relative conversion by functional enzyme in each cancer cell type. Across all cancer cell lines, the α-man substrate was converted to the greatest extent, while conversion of the β-glu substrate was negligible. Based on previous reports, we suspected that the β-glu substrate may have difficulty permeating the cells due to the negatively charged carboxylate moiety, thereby obscuring the true amount of functional intracellular β-glu [26]. Elevated conversion of the β-glu substrate by cancer cell lysate confirmed the presence of intracellular β-glu and supports the hypothesis that the glucuronide moiety hinders cellular uptake of the 4-nitrophenyl β-glucuronide reporter molecule. Taken together, these experiments demonstrated that choice of enzyme-directing group affects the conversion rate of enzyme-directed substrates in each cell type. This also suggested that (7) would elicit relatively low cancer-mediated immunogenicity and (10) should outperform the other EDIs due to the increased activity of α-man in each cell line.
After comparing functional enzyme activity across cancer cell lines, we next examined the efflux of RSQ from cancer cells, as the RSQ payload is the product of enzymatic conversion common to EDIs (7), (10), and (13). It is well established that cancer cells use multiple drug efflux pathways to transport drugs from within cells to the extracellular space, including ABC transport proteins P-gp [41], breast cancer resistance protein 1 [42], and multi-drug resistance protein 1 [43]. Our group previously demonstrated that the imidazoquinoline, imiquimod, competes for efflux with the known P-gp substrate, Rh123 [44] in drug-resistant rat prostate cancer cells [7]. Although both imiquimod and RSQ are of the imidazoquinoline family, slight structural modifications can greatly impact P-gp-mediated efflux [45], and RSQ efflux from cancer cells has never been reported. Thus we were interested in determining whether RSQ was a substrate for the previously characterized efflux potential of B16 [46], TC2 [47], and 4T1 [48] cancer cells. Specifically, we were interested in testing RSQ competition with P-gp-mediated efflux of Rh123 due to the broad substrate scope and prevalence of P-gp in many types of cancer [49]. Competition experiments with Rh123 suggested that RSQ is effluxed via P-gp-mediated routes in TC2 cells because RSQ causes more intracellular Rh123 to be retained. Efflux occurs through P-gp-independent routes in B16 cells and possibly P-gp-dependent or independent routes in 4T1 cells, where P-gp expression was too low to be detected by Western blot but efflux was sensitive to addition of P-gp inhibitor. Together, these results suggest that RSQ is a competitive substrate for P-gp-mediated Rh123 efflux, particularly in TC2 cells, but that RSQ efflux occurs through P-gp-independent routes in B16 and 4T1 cells.
With functional glycosidase activity and RSQ efflux potential of each cancer cell line characterized, we next tested cancer cell-mediated immunogenicity of each EDI to determine how enzyme activity and efflux potential correlates to immunogenicity in BAIT. Of each cancer cell/EDI combination, B16 cells elicited the greatest immunogenicity from all EDIs compared to other cancer cell lines. On the other hand, the immunogenicity elicited by TC2 and 4T1 cells was substantially lower, despite the observation that 4T1 cells featured the greatest α-man and β-gal activities of any cell lines tested. Overall, these results demonstrate that the cancer-mediated immunogenicity of EDIs in these cancer cell lines appears not to directly correlate with the functional activity of each glycosidase. In addition, although RSQ competes with Rh123 efflux in P-gp-expressing TC2 cells, the P-gp-mediated RSQ efflux experiments do not correlate with the observed immunogenicity of EDIs across cancer cell lines.
Overall, we report the preparation and in vitro testing of a small catalog of EDIs featuring the potent immunogenic payload RSQ. We demonstrated that these EDIs feature abrogated immunogenicity until they are metabolized to immunogenic RSQ in the presence of complementary glycosidase. We used 4-nitrophenyl glycoside reporter substrates to assess the functional activity of complementary glycosidases across multiple model cancer cell lines, thereby providing insight into how EDIs are metabolized to immunostimulants. The maximum substrate hydrolysis by cancer cells was observed using 4-nitrophenyl α-mannopyranoside, suggesting that (10) should be metabolized the fastest and therefore elicit the greatest immunogenicity within each cancer cell line. Conversely, the negligible conversion of 4-nitrophenyl β-glucuronide by each cancer cell line suggested that (7) would exhibit impaired cellular uptake and limited cancer-mediated immunogenicity. However, immunogenicity among all EDIs was comparable for a given cell type. Competitive efflux experiments demonstrated that RSQ competes with Rh123 for P-gp-mediated efflux in TC2 cells, with P-gp expression confirmed by Western Blot. In contrast, no competition with Rh123 efflux from B16 cells was observed. Competition between Rh123 and RSQ in 4T1 cells was not significant, likely due to low levels of P-gp expression. Taken together with the observed cancer-mediated immunogenicity of EDIs when used in BAIT, we conclude that RSQ undergoes efflux through both P-gp-mediated and P-gp-independent routes. More work is needed to characterize all possible routes of efflux, as RSQ could be a substrate for several of the many highly promiscuous transport proteins implicated in drug efflux. However, these results do suggest that efflux of the immunogenic payload is the rate-limiting step in BAIT. Therefore, the efflux potential of the target cancer cells, as well as the parent immunostimulant, may be the most important factors to consider when designing the next generation of EDIs. This also suggests that BAIT may be best suited to the treatment of drug-resistant cancers that commonly overexpress drug efflux proteins, and in the future we plan to generate new EDIs that are optimized to exploit drug efflux.
References
Mauger AB, Burke PJ, Somani HH, Friedlos F, Knox RJ. Self-immolative prodrugs: candidates for antibody-directed enzyme prodrug therapy in conjunction with a nitroreductase enzyme. J Med Chem. 1994;37:3452–8.
Florent JC, Dong X, Gaudel G, Mitaku S, Monneret C, Gesson JP, et al. Prodrugs of anthracyclines for use in antibody-directed enzyme prodrug therapy. J Med Chem. 1998;41:3572–81.
Bosslet K, Straub R, Blumrich M, Czech J, Gerken M, Sperker B, et al. Elucidation of the mechanism enabling tumor selective prodrug monotherapy. Cancer Res. 1998;58:1195–201.
Wei MX, Tamiya T, Rhee RJ, Breakefield XO, Chiocca EA. Diffusible cytotoxic metabolites contribute to the in vitro bystander effect associated with the cyclophosphamide/cytochrome P450 2B1 cancer gene therapy paradigm. Clin Cancer Res. 1995;1:1171–7.
Sun YL, Patel A, Kumar P, Chen ZS. Role of ABC transporters in cancer chemotherapy. Chin J Cancer. 2012;31:51–7.
Hantho JD, Strayer TA, Nielsen AE, Mancini RJ. An enzyme-directed imidazoquinoline for cancer immunotherapy. ChemMedChem. 2016;11:2496–500.
Burt AJ, Hantho JD, Nielsen AE, Mancini RJ. An enzyme-directed imidazoquinoline activated by drug resistance. Biochemistry. 2018;57:2184–8.
PubChem. Imiquimod. 2020. https://pubchem.ncbi.nlm.nih.gov/compound/57469.
Rook AH, Gelfand JM, Gelfand JC, Wysocka M, Troxel AB, Benoit B, et al. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood. 2015;126:1452–61.
Nishii N, Tachinami H, Kondo Y, Xia Y, Kashima Y, Ohno T, et al. Systemic administration of a TLR7 agonist attenuates regulatory T cells by dendritic cell modification and overcomes resistance to PD-L1 blockade therapy. Oncotarget. 2018;9:13301–12.
Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499.
Schiaffo CE, Shi C, Xiong Z, Olin M, Ohlfest JR, Aldrich CC, et al. Structure–activity relationship analysis of imidazoquinolines with Toll-like receptors 7 and 8 selectivity and enhanced cytokine induction. J Med Chem. 2014;57:339–47.
Warburg O. On the origin of cancer cells. Science. 1956;123:309–14.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95.
de Graaf M, Boven E, Scheeren HW, Haisma HJ, Pinedo HM. Beta-glucuronidase-mediated drug release. Curr Pharmacol Des. 2002;8:1391–403.
He L, Fan C, Kapoor A, Ingram AJ, Rybak AP, Austin RC, et al. α-Mannosidase 2C1 attenuates PTEN function in prostate cancer cells. Nat Commun. 2011;2:307.
Yue W, Jin YL, Shi GX, Liu Y, Gao Y, Zhao FT, et al. Suppression of 6A8 α-mannosidase gene expression reduced the potentiality of growth and metastasis of human nasopharyngeal carcinoma. Int J Cancer. 2004;108:189–95.
Legler K, Rosprim R, Karius T, Eylmann K, Rossberg M, Wirtz RM, et al. Reduced mannosidase MAN1A1 expression leads to aberrant N-glycosylation and impaired survival in breast cancer. Br J Cancer. 2018;118:847–56.
Chatterjee SK, Bhattacharya M, Barlow JJ. Glycosyltransferase and glycosidase activities in ovarian cancer patients. Cancer Res. 1979;39:1943–51.
Wagner J, Damaschke N, Yang B, Truong M, Guenther C, McCormick J, et al. Overexpression of the novel senescence marker β-galactosidase (GLB1) in prostate cancer predicts reduced PSA recurrence. PLoS ONE. 2015;10:e0124366.
Beratis NG, Kaperonis A, Eliopoulou MI, Kourounis G, Tzingounis VA. Increased activity of lysosomal enzymes in the peritoneal fluid of patients with gynecologic cancers and pelvic inflammatory disease. J Cancer Res Clin Oncol. 2005;131:371–6.
Sloane BF, Dunn JR, Honn KV. Lysosomal Cathepsin B. Correlation with metastatic potential. Science. 1981;212:1151–3.
Bernacki RJ, Niedbala MJ, Korytnyk W. Glycosidases in cancer and invasion. Cancer Metastasis Rev. 1985;4:81–101.
The Human Protein Atlas. The human pathology proteome. 2020. https://www.proteinatlas.org/humanproteome/pathology.
Juan TY, Roffler SR, Hou HS, Huang SM, Chen KC, Leu YL, et al. Antiangiogenesis targeting tumor microenvironment synergizes glucuronide prodrug antitumor activity. Clin Cancer Res. 2009;15:4600–11.
Chen KC, Schmuck K, Tietze LF, Roffler SR. Selective cancer therapy by extracellular activation of a highly potent glycosidic duocarmycin analogue. Mol Pharmacol. 2013;10:1773–82.
Grinda M, Clarhaut J, Renoux B, Tranoy-Opalinski I, Papot S. A self-immolative dendritic glucuronide prodrug of doxorubicin. MedChemComm. 2012;3:68–70.
Legigan T, Clarhaut J, Tranoy-Opalinski I, Monvoisin A, Renoux B, Thomas M, et al. The first generation of β-galactosidase-responsive prodrugs designed for the selective treatment of solid tumors in prodrug monotherapy. Angew Chem Int Ed. 2012;51:11606–10.
Tietze LF, Schuster HJ, Krewer B, Schuberth I. Synthesis and biological studies of different duocarmycin based glycosidic prodrugs for their use in the antibody-directed enzyme prodrug therapy. J Med Chem. 2009;52:537–43.
Weir HK, Thompson TD, Soman A, Møller B, Leadbetter S. The past, present, and future of cancer incidence in the United States: 1975 through 2020: predicting cancer incidence to 2020. Cancer. 2015;121:1827–37.
Dumitru CD, Antonysamy MA, Tomai MA, Lipson KE. Potentiation of the antitumor effects of imidazoquinoline immune response modifiers by cyclophosphamide. Cancer Biol Ther. 2010;10:155–65.
Han JH, Lee J, Jeon SJ, Choi ES, Cho SD, Kim BY, et al. In vitro and in vivo growth inhibition of prostate cancer by the small molecule imiquimod. Int J Oncol. 2013;42:2087–93.
Broomfield SA, van der Most RG, Prosser AC, Mahendran S, Tovey MG, Smyth MJ, et al. Locally administered TLR7 agonists drive systemic antitumor immune responses that are enhanced by anti-CD40 immunotherapy. J Immunol. 2009;182:5217–24.
Bosmann HB, Bieber GF, Brown AE, Case KR, Gersten DM, Kimmerer TW, et al. Biochemical parameters correlated with tumour cell implantation. Nature. 1973;246:487–9.
Liu G, Khanna V, Kirtane A, Grill A, Panyam J. Chemopreventive efficacy of oral curcumin: a prodrug hypothesis. FASEB J. 2019;33:9453–65.
Abbasi A, Paragas EM, Joswig-Jones CA, Rodgers JT, Jones JP. Time course of aldehyde oxidase and why it is nonlinear. Drug Metab Dispos. 2019;47:473–83.
El Alaoui A, Schmidt F, Monneret C, Florent JC. Protecting groups for glucuronic acid: application to the synthesis of new paclitaxel (taxol) derivatives. J Org Chem. 2006;71:9628–36.
InvivoGen. NF-κB reporter raw 264.7 murine macrophages. 2020. https://www.invivogen.com/raw-blue.
Mealey KL, Dassanayake S, Burke NS. Establishment of a cell line for assessing drugs as canine P-glycoprotein substrates: proof of principle. J Vet Pharmacol Ther. 2017;40:545–51.
Islam MR, Tomatsu S, Shah GN, Grubb JH, Jain S, Sly WS. Active site residues of human β-glucuronidase: evidence for GLU 540 as the nucleophile and GLU 451 as the acid-base residue. J Biol Chem. 1999;274:23451–5.
Juliano RL, Ling VA. Surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta Biomembr. 1976;455:152–62.
Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95:15665–70.
Cole SPC. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem. 2014;289:30880–8.
Efferth T, Löhrke H, Volm M. Reciprocal correlation between expression of P-glycoprotein and accumulation of rhodamine 123 in human tumors. Anticancer Res. 1989;9:1633–7.
Hitchcock SA. Structural modifications that alter the P-glycoprotein efflux properties of compounds. J Med Chem. 2012;55:4877–95.
He X, Wang J, Dou J, Yu F, Cai K, Li X, et al. Antitumor efficacy induced by a B16F10 tumor cell vaccine treated with mitoxantrone alone or in combination with reserpine and verapamil in mice. Exp Ther Med. 2011;2:911–6.
Kammerer R, Buchner A, Palluch P, Pongratz T, Oboukhovskij K, Beyer W, et al. Induction of immune mediators in glioma and prostate cancer cells by non-lethal photodynamic therapy. PLoS ONE. 2011;6:e21834.
Bao L, Haque A, Jackson K, Hazari S, Moroz K, Jetly R, et al. Increased expression of P-glycoprotein is associated with doxorubicin chemoresistance in the metastatic 4T1 breast cancer model. Am J Pathol. 2011;178:838–52.
Alvarez M, Paull K, Monks A, Hose C, Lee JS, Weinstein J, et al. Generation of a drug resistance profile by quantitation of Mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J Clin Invest. 1995;95:2205–14.
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number 1R01CA234115. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank Professor Jeff Jones for assistance with LC-MS analysis of EDI conversion, as well as Professor Katrina Mealey and Darren Schnider for expert assistance with characterizing drug efflux from cancer cells and standardized plasma membrane preparations of MDCK and MDR MDCK cells. The authors also thank Professor Darrell Irvine for donating the 4T1 cells. All NMR characterization of synthetic intermediates and EDIs was made possible through use of the Washington State University NMR Center with equipment supported by NIH RR0631401, RR12948, NSF CHE-9115282, and DBI-9604689, the Murdock Charitable Trust, and private donors Don and Marianna Matteson.
Author information
Authors and Affiliations
Contributions
ATR synthesized EDIs and conducted all enzyme kinetics experiments. AJP performed 4-nitrophenyl glycoside reporter assays and the reported efflux experiments in each cell line. MD and LKO performed the RB stimulation experiments. AA performed all LC-MS experiments and quantitative analysis related to EDI enzymatic conversion. AJB and ATH assisted with synthesis of EDIs. AEN contributed preliminary Rh123/RSQ competition data and assisted with NMR characterization. RJM conceived of the project and helped in the overall experimental design. All authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors are inventors of United States provisional patent application 63/002,924 which relates to the prodrugs reported in this work.
Supplementary information
Rights and permissions
About this article

Cite this article
Ryan, A.T., Pulukuri, A.J., Davaritouchaee, M. et al. Comparing the immunogenicity of glycosidase-directed resiquimod prodrugs mediated by cancer cell metabolism. Acta Pharmacol Sin 41, 995–1004 (2020). https://doi.org/10.1038/s41401-020-0432-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0432-4
Keywords
This article is cited by
-
Engineered nanomedicines block the PD-1/PD-L1 axis for potentiated cancer immunotherapy
Acta Pharmacologica Sinica (2022)
-
Nanomedicine and cancer immunotherapy
Acta Pharmacologica Sinica (2020)
