
Abstract
Sphingosine-1-phosphate (S1P) and its receptors have been implicated in functions of Langerhans cells and atopic dermatitis. In this study, we investigated the roles of S1P receptor type 2 (S1P2) in a mouse model of atopic dermatitis, which was induced by topical application of 2,4-dinitrochlorobenzene (DNCB) on ventral skin on D0, followed by repeated DNCB challenge on both ears from D7 to D49. Wild-type mice with atopic dermatitis displayed severe inflammation and mast cell accumulation in ear tissues and elevated IgE levels in serum. Furthermore, the mice showed significantly increased sizes of draining lymph nodes, high levels of inflammatory cytokines (IL-4, IL-13, IL-17, and IFN-γ) in the ears and lymph nodes and high levels of chemokines CCL17 and CCL22 in ears. Administration of JTE-013, a selective antagonist of S1P2 (3 mg/kg, i.p, from D19 to D49) before DNCB challenge significantly suppressed DNCB-induced atopic responses in ears and lymph nodes. JTE-013 administration also significantly decreased the lymph nodes sizes, the levels of inflammatory cytokines (IL-4, IL-13, IL-17, and IFN-γ) in the ears and lymph nodes, and the levels of chemokines CCL17 and CCL22 in ears. Furthermore, the inflammatory responses of atopic dermatitis were greatly ameliorated in S1pr2 gene-deficient mice. As CCL17 and CCL22 are CCR4 ligands, acting as Th2-attracting chemokines, we investigated CCL17 and CCL22 expression in bone marrow-derived dendritic cells (BMDCs) from wild-type and S1pr2 gene-deficient mice. Addition of IL-4 (10 ng/mL) markedly increased the levels of CCL17 and CCL22, but IL-4-induced CCL17 and CCL22 expression was significantly blunted in BMDCs from S1pr2 gene-deficient mice. Furthermore, pretreatment with JTE-013 (1−30 μM) dose-dependently suppressed this induction in BMDCs from wild-type mice. Our results demonstrate that blockage of S1P2 ameliorates not only DNCB-induced atopic dermatitis symptoms but also Th2 cell-attracting capacity of dendritic cells, suggesting S1P2 as a potential therapeutic target for atopic dermatitis.
Similar content being viewed by others


Introduction
Atopic dermatitis (AD) is a chronic, pruritic inflammatory skin disorder with a complex etiology and heterogeneous presentation [1]. AD is the most common skin disease, and can significantly compromise quality of life owing to associated emotional distress, sleep disruption, and social awkwardness [2]. AD affects up to 20% of children worldwide and can predispose infants and children to other atopic diseases, including allergic asthma, allergic keratoconjunctivitis, allergic rhinitis, and food allergy, a process termed atopic march [1]. Decreased concentration and enhanced metabolism of sphingosine-1-phosphate (S1P) have been reported in the skin lesions of dogs suffering from AD [3].
S1P is a specific ligand for five G protein-coupled receptors, S1P1-5 [4]. S1P is synthesized from sphingosine via sphingosine kinases 1 and 2 and is degraded irreversibly by sphingosine lyase [5, 6]. S1P and S1P receptors have been implicated in the development of AD-like skin lesions in NC/Nga mice [7, 8] and animal skin allograft rejection [9]. S1P levels in plasma are elevated in patients severely affected with psoriasis, another immune-mediated chronic inflammatory skin disorder [10, 11]. Previously, antiproliferative and anti-inflammatory effects of S1P were reported in mouse models of psoriasis and Th17 differentiation [12, 13]. Shin et al. [14] recently revealed the importance of S1P generated from sphingosine kinase 2 in Th17 differentiation of naive CD4+ T cells and in the pathogenesis of psoriasis. Topical application of S1P and FTY720, an S1P modulator, attenuates allergic contact dermatitis reactions [15, 16]. Topical administration of S1P diminished imiquimod-induced ear swelling and epidermal thickness in the ear [12]. In heterozygous S1P lyase-deficient mice, mild hyperplasia with orthokeratotic hyperkeratosis developed in the skin [17]. S1P modulates antigen capture by murine and human Langerhans cells via S1P2 [7, 18]. However, there has been no preclinical study on S1P2 in atopic responses. In the current study, we used a murine 2,4-dinitrochlorobenzene (DNCB)-induced AD model to investigate the roles of S1P2 in vivo and examined overall atopic responses in S1pr2 gene-deficient mice and in JTE-013, a specific S1P2 antagonist-treated mice.
Materials and methods
Materials
JTE‐013 was purchased from Cayman Chemicals (Ann Arbor, MI). 1-Chloro-2,4-dinitrobenzene (DNCB) and olive oil were obtained from Sigma‐Aldrich (St. Louis, MO). Recombinant mouse GM‐CSF and IL‐4 were purchased from Shenandoah Biotechnology (Warminster, PA, USA).
Animals
Three S1pr2 heterozygous mice were kindly provided by Richard Proia at the NIH [19]. The mice had a mixed C57BL/6 and 129Sv background. They had been backcrossed to Balb/c mice for eight generations [20]. S1pr2 wild-type littermates (WT) and knockout (KO) mice were housed in the Laboratory Animal facility at Pusan National University and provided with standard laboratory chow and water ad libitum. The mice were housed in standard plastic cages (two mice per cage) with sawdust as bedding and maintained under controlled conditions, with the temperature at 22–24 °C, humidity at 60% ± 5%, and alternating light/dark cycles (lights were on between 7:00 h and 19:00 h). The animal protocol used in this study was reviewed and approved by the Pusan National University–Institutional Animal Care Committee (PNU–IACUC) with respect to procedural ethics and scientific care.
Induction of atopic dermatitis in Balb/c mice and JTE-013 administration
Following a simple randomization procedure, 7-week-old male S1P2 WT Balb/c mice and S1P2-KO mice were divided into five groups (n = 6): a PBS-treated control S1P2 WT group, a DNCB-treated S1P2 WT mouse group, a PBS-treated control S1P2-KO group, a DNCB-treated S1P2-KO mouse group, and a JTE-013/DNCB-treated S1P2 WT group. To induce experimental AD, the ventral skin was shaved, and 300 μL of 1% DNCB in acetone/olive oil (3:1) was applied to the ventral skin on day 0. Starting on day 7, the mice were challenged by application of 200 μL of 0.3% DNCB to the ears every other day for as many as 42 days. From day 19 until the completion of the experiment, the JTE-013/DNCB-treated S1P2 WT group was administered JTE-013 (3 mg/kg body weight) intraperitoneal (i.p.) injection 30 min before challenge. The mice were sacrificed on day 49.
Histologic analysis and mast cell count in skin samples
After the mice were sacrificed on day 49, ear tissues were fixed in 10% formalin, embedded in paraffin and sectioned (8-μm-thick sections). The sections were stained with hematoxylin and eosin (H&E) or toluidine blue. For H&E staining, the sections were washed in running tap water for 5 min, counterstained with hematoxylin solution for 90 s, washed again in running tap water, dehydrated, and mounted on coverslips with Permount.
For the detection of mast cells by toluidine blue staining, the sections were washed in running tap water, stained with toluidine blue solution for 2 min, rinsed in distilled water, dehydrated, and mounted on coverslips with Permount.
Measurement of total serum IgE levels
The IgE levels in mouse serum were determined using ELISA kits (eBioscience, San Diego, CA). Briefly, 96-well plates (NUNC, USA) were coated overnight at 4 °C with capture antibodies for IgE. Following washing, the plates were blocked for 2 h at room temperature with blocking buffer. Then, serum was added to each well, and the plates were incubated for 2 h at room temperature. Pretitrated biotin-conjugated detection antibodies for IgE were then added and incubated for 1 h at room temperature. Pretitrated streptavidin-HRP was added and incubated for 30 min at room temperature, and the plates were incubated with substrate solution for 15 min at room temperature. Stop solution was then added, and the absorbance was read at 450 nm.
Reverse transcriptase-PCR
To assess the expression levels of inflammatory markers in the ears of the mice by RT-PCR, first-strand cDNA was first synthesized from total RNA isolated from skin tissues using TRIzol reagent (Invitrogen, Waltham, MA, USA). The synthesized cDNA products, primers for each gene, and Promega Go-Taq DNA polymerase (Madison, WI, USA) were used for PCR. Specific primers and PCR conditions were previously described [21]. Aliquots (7 μL) were electrophoresed in 1.2% agarose gels and stained with StaySafeTM nucleic acid gel stain (Real Biotech Corporation, Taipei, Taiwan). The intensity of each PCR product was quantified by using ImageJ software (NIH, Bethesda, MD, USA) and normalized to GAPDH levels.
Generation and cell culture of bone marrow-derived dendritic cells (BMDCs)
Dendritic cells were generated from the bone marrow cells of S1P2 WT or KO mice, as previously described by Lutz et al. [22]. In brief, bone marrow cells were flushed from the mouse femurs and tibias using sterile PBS and placed in RPMI-1640 medium containing 10% FBS, 100 units/mL penicillin, 50 μg/mL streptomycin, recombinant mouse GM-CSF (20 ng/mL) and 50 μM 2-mercaptoethanol. On day 3, fresh medium containing GM-CSF was added. On day 7, BMDCs were collected, and the CD11c+ cells were sorted using anti-mouse CD11c microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany).
Western blotting
BMDCs were harvested and resuspended in RIPA lysis buffer (GenDEPOT, Baker, USA). The concentrations of proteins were determined using a BCA protein assay (Thermo Scientific, Rockford, IL, USA). Proteins (30 μg) were resolved by 10% SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to nitrocellulose membranes, which were incubated with specific primary antibodies recognizing β-actin and p-Stat6 and then incubated with HRP-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA, USA). The light intensities were determined using an enhanced chemiluminescence system (Pierce Biotechnology Inc., Rockford, IL, USA).
Statistics
The results are expressed as the means ± standard error of the mean (SEM) based on six evaluations of the animal experiments and as the means ± SEs based on three evaluations for the BMDC experiments. The significance of the differences was determined by analysis of variance (ANOVA) and Tukey’s multiple comparison test. Significance was accepted for P values < 0.05; asterisk indicates a significant difference compared to the PBS-treated group or untreated controls; hash indicates a significant difference compared to the DNCB-treated group. The analyses were performed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
DNCB-induced atopic dermatitis on the ears was suppressed in the S1P2-KO mice
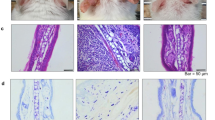
S1P2 has been suggested to mediate mast cell degranulation [23, 24] and to modulate the functions of Langerhans cells and dendritic cells; that is, it regulates antigen capture, maturation, migration, and cytokine production [7, 18, 21]. To investigate the roles of S1P2 in overall atopic responses, we used in vivo DNCB-induced AD models consisting of S1P2 WT and S1P2-KO Balb/c mice (Fig. 1a). DNCB induced AD on the ears of the S1P2 WT mice, as confirmed by extensive hypertrophy of the epidermis, as visualized with H&E staining, compared with the ear epidermis of the mice in the PBS-treated control group (Fig. 1b). However, for the S1P2-KO mice, DNCB induced less severe AD on the ears than it did in the WT mice (Fig. 1b). Serum IgE levels were measured to examine the immunological effect of DNCB administration. Hyperproduction of IgE was observed in the DNCB-treated WT mice (Fig. 1c). However, in S1P2-KO mice, DNCB induced an increase in serum IgE levels, but this increase was significantly lower than that in the WT mice (Fig. 1c). Infiltration of mast cells into the dermis was measured using toluidine blue staining. The number of mast cells was significantly increased by DNCB treatment in both the S1P2 WT and KO mice (Fig. 1d, e). However, the number of mast cells in the S1P2-KO mice was significantly lower than it was in the S1P2 WT mice (Fig. 1d, e). Furthermore, the levels of inflammatory cytokines IL-4, IL-13, IL-17A, and INF-γ were measured because AD is thought to be regulated not only by the Th2 response but also by Th1 and Th17 responses [25,26,27]. The mRNA levels of the four cytokines were significantly elevated in the ear tissues after AD induction in the S1P2 WT mice (Fig. 2). However, the increases in cytokine mRNA levels were significantly lower in the S1P2-KO mice than they were in the WT mice (Fig. 2). In summary, hypertrophy and an increased infiltration of mast cells were found in the epidermis, as was increased IgE levels in the serum, and the increased cytokine levels were substantially lower in the S1P2-KO mice than they were in the WT mice.

a Experimental protocol. A murine model of DNCB‐induced atopic dermatitis was established through DNCB sensitization on day 0 and with repeated DNCB challenge on days 7–49. S1P2 WT and KO mice were treated with PBS or DNCB. b Ear tissue sections were stained with H&E for histological analysis. c Blood was collected on day 49 of the experiment. Serum levels of IgE were measured by ELISA. Toluidine blue staining of the skin obtained from DNCB-treated S1P2 WT and KO mice was used to identify mast cells. d Sections of the ear samples from the mice in which atopic dermatitis was induced were stained with toluidine blue, and e the number of mast cells (dark violet‐stained cells) was counted using a light microscope. The results are presented as the means ± SEM (n = 6). ***P < 0.001. ###P < 0.001.

RT-PCR analyses of the Th2 cytokines (IL-4 and IL-13), Th17 cytokine (IL-17A), and Th1 cytokine (INF-γ) were performed using mRNA isolated from mouse ear tissues. a mRNA levels are expressed as ratios with respect to GAPDH mRNA. Representative RT-PCR images are shown in b. The results are presented as the means ± SEM (n = 6). *P < 0.01, ***P < 0.001. ###P < 0.001.
DNCB-induced atopic responses in lymph nodes are suppressed in the S1P2-KO mice
Next, we investigated lymph node size and cytokine production in the mouse lymph nodes. DNCB induced an increase in the size of draining lymph nodes; however, the increase was significantly lower in the S1P2-KO mice than it was in the S1P2 WT mice (Fig. 3). The levels of IL-4, IL-13, IL-17A, and INF-γ were also measured in the mouse lymph nodes. The mRNA levels of the four cytokines were significantly elevated in the lymph nodes after AD induction in the S1P2 WT mice (Fig. 4). However, the increases in IL-4, IL-13, IL-17A, and INF-γ were significantly lower in the S1P2-KO mice than they were in the WT mice (Fig. 4). In summary, lymph node size and inflammatory cytokine levels in the lymph nodes were largely suppressed in the S1P2-KO mice compared to the size and levels in the WT mice.

a The lymph nodes were photographed to record morphological changes. b The weight of lymph nodes was measured. The results are presented as the means ± SEM (n = 6). ***P < 0.001. ###P < 0.001.

RT-PCR analyses of the Th2 cytokines (IL-4 and IL-13), Th17 cytokine (IL-17A), and Th1 cytokine (INF-γ) were performed using mRNA isolated from mouse lymph node tissues. a mRNA levels are expressed as ratios with respect to GAPDH mRNA. Representative RT-PCR images are shown in b. The results are presented as the means ± SEM (n = 6). *P < 0.01, ***P < 0.001. ###P < 0.001.
JTE-013 inhibits DNCB-induced atopic responses on the mouse ears and in the lymph nodes
The functions of S1P2 in both the atopy induction phase (antigen sensitization) and elicitation phase were blocked in the S1P2-KO mice. To identify the therapeutic potential of S1P2 in the elicitation phase, we treated S1P2 WT mice with JTE-013 after induction of AD (Fig. 5a). The extensive hypertrophy of the epidermis, as induced by DNCB, which had been observed on the ears of the S1P2 WT mice, was significantly suppressed by the JTE-013 treatment, as shown in Fig. 5b. The elevated serum IgE levels were also inhibited by the JTE-013 treatment, as shown in Fig. 5c. Similarly, the increased infiltration of mast cells into the dermis was suppressed by the JTE-013 treatment, as shown in Fig. 5d, e. In addition, the increased levels of IL-4, IL-13, IL-17, and INF-γ in the ear tissues were significantly reduced by the JTE-013 treatment (Fig. 5f).

a Experimental protocol. A murine model of DNCB‐induced atopic dermatitis was established through DNCB sensitization on day 0 and repeated DNCB challenges on days 7–49. S1P2 WT mice were treated with either PBS or DNCB, and JTE‐013 (an S1P2 antagonist) was administered by i.p. injection 30 min before the DNCB challenge. b Ear tissue sections were stained with H&E for histological analysis. c Blood was collected on day 49 of the experiment. Serum levels of IgE were measured by ELISA. Toluidine blue staining of skin obtained from DNCB-treated S1P2 WT mice was used to identify mast cells. d Sections of ear samples from mice in which atopic dermatitis was induced were stained with toluidine blue, and e the number of mast cells (dark violet‐stained cells) was counted using a light microscope. RT-PCR analyses for Th2 cytokines (IL-4 and IL-13), Th17 cytokine (IL-17A), and Th1 cytokine (INF-γ) were performed using mRNA isolated from mouse ear tissues. f mRNA levels are also expressed as ratios with respect to GAPDH mRNA. The results are presented as the means ± SEM (n = 6). ***P < 0.001. ##P < 0.01, ###P < 0.001.
The increase in lymph node size was attenuated by the JTE-013 treatment (Fig. 6a, b), and the increased mRNA levels of the four cytokines in the lymph nodes were also significantly suppressed following JTE-013 treatment (Fig. 6c). In summary, histologic and immunologic manifestations of DNCB-induced AD in the ears and lymph nodes were largely suppressed by the administration of JTE-013.

a The lymph nodes were photographed to record morphological changes. b the Weight of lymph nodes was measured. RT-PCR analyses of the Th2 cytokines (IL-4 and IL-13), Th17 cytokines (IL-17A), and Th1 cytokines (INF-γ) were performed using mRNA isolated from mouse lymph node tissues. c mRNA levels are expressed as ratios with respect to GAPDH mRNA. The results are presented as the means ± SEM (n = 6). ***P < 0.001. ##P < 0.01, ###P < 0.001.
Effects of S1P2 deficiency and JTE-013 administration on CCL17 and CCL22 levels in ear samples and bone marrow-derived dendritic cells
The therapeutic efficacy of JTE-013 in the elicitation phase is mediated by S1P2 in several cell types, such as mast cells, Langerhans cells, and dendritic cells. That is, the suppressive effects of JTE-013 and S1P2 deficiency on mast cell degranulation and the pro-allergic functions of S1P2 in Langerhans cells and dendritic cells have been previously reported, and these effects included antigen capture, maturation, migration, and cytokine production [7, 18, 21, 23, 24]. S1P2 was not expressed in the T cell lineage, and thus, T cells are presumed to not be involved [28]. Recently, elevated levels of CCL17 and CCL22 in the skin of AD patients were reported to be positively correlated with disease severity [29,30,31]. In addition, CCR4, a major trafficking receptor for Th2 cells, is the receptor for CCL17 and CCL22 and is expressed on Th2 cells infiltrated in the skin lesions of AD patients [30, 32]. Thus, we focused on the S1P2-mediated regulation of chemokine CCL17 and CCL22 levels in the skin. As shown in Fig. 7a, b, DNCB treatment induced an increase in the mRNA levels of CCL17 and CCL22 in the ears, and the levels were lower in the S1P2-KO mice than they were in the S1P2 WT mice. Furthermore, JTE-013 treatment suppressed the expression of both of these chemokines in the S1P2 WT mice (Fig. 7c, d). This function of S1P2 on the expression of CCL17 and CCL22 was confirmed in vitro in BMDCs following treatment with IL-4 (Fig. 8). In the presence of JTE-013, IL-4-induced induction of CCL17 and CCL22 was inhibited in a concentration-dependent manner (Fig. 8a, b), and this inhibition was also observed in the BMDCs obtained from the S1P2-KO mice (Fig. 8c, d). As phosphorylation of stat6 is a key step in IL-4 signaling, we examined the phosphorylation of stat6. As shown in Fig. 9, IL-4 increased the phospho-stat6 levels in the BMDCs; however, in the presence of JTE-013, stat6 phosphorylation was inhibited in a concentration-dependent manner, and this inhibition was also observed in the BMDCs from the S1P2-KO mice. Taken together, the results strongly suggest that S1P2 has an important role in the expression of CCL17 and CCL22 in dendritic cells by interrupting IL-4 and stat6 signaling.

RT-PCR analyses of CCL17 and CCL22 were performed using mRNA isolated from the ear tissues of the PBS- or DNCB-treated S1P2 WT and KO mice (a, b). RT-PCR analyses of CCL17 and CCL22 were performed using mRNA isolated from ear tissues of the PBS-, DNCB-, or JTE-013/DNCB-treated mice (c, d). mRNA levels are expressed as ratios with respect to GAPDH mRNA (b, d). Representative RT-PCR images are shown in a, c. The results are presented as the means ± SEM (n = 6). ***P < 0.001. #P < 0.05, ###P < 0.001.

a, b Immature S1P2 WT DCs were pretreated with vehicle or the indicated concentrations of JTE‐013 1 h before IL-4 administration and allowed to mature in the presence of 10 ng/mL IL-4 for 24 h. mRNA levels are expressed as ratios with respect to GAPDH mRNA (a). Representative RT-PCR images are shown in b. c, d Immature S1P2 WT BMDCs or KO BMDCs were treated with 10 ng/mL IL-4 for 24 h. The mRNA expression levels of CCL17 and CCL22 were determined by RT‐PCR. The expression levels of CCL17 and CCL22 were determined by RT-PCR using GAPDH as an internal reference (c). Representative RT-PCR images are shown in d. The results shown were obtained from three independent experiments presented as the means ± SEs (n = 3). **P < 0.05, ***P < 0.001. #P < 0.05, ##P < 0.01, ###P < 0.001.

a, b Immature S1P2 WT BMDCs or KO BMDCs were treated with 10 ng/mL IL-4 for 24 h. c, d Immature S1P2 WT DCs were pretreated with vehicle or the indicated concentrations of JTE‐013 1 h before IL-4 administration and allowed to mature in the presence of 10 ng/mL IL-4 for 24 h to determine the phospho-stat6 levels. Western blotting was conducted on cell lysates. Relative protein levels compared with β-actin are shown in histograms (a, c). Representative Western blots are shown in b, d. The results shown are obtained from three independent experiments presented as the means ± SEs (n = 3). *P < 0.05. #P < 0.05.
Discussion
In previous studies, S1P receptors were implicated in the development of AD-like skin lesions in NC/Nga mice, as FTY720 prevented spontaneous dermatitis and impaired the initiation of contact hypersensitivity [8, 16, 33, 34]. In addition, elevated S1P plasma levels were observed in patients with severe psoriasis [10, 11], and the importance of S1P in the pathogenesis of psoriasis in relation to Th17 differentiation was found [12,13,14]. In particular, the topical application of S1P and FTY720 attenuated allergic contact dermatitis reactions [15, 16] and diminished imiquimod-induced psoriasis [12]. However, specific S1P receptors have not been investigated. In the current study, for the first time, the functions of S1P2 in AD were elucidated using S1P2-deficient mice and JTE-013 treatment. Three main S1P2 functions were found in DNCB-induced AD. First, S1P2 exacerbated inflammatory AD in vivo, which is a novel finding. Second, a blockade of S1P2 in the elicitation phase could be used as a potential therapeutic strategy for the treatment of AD, as the effectiveness was shown with JTE-013 treatment. Third, S1P2 activation in dendritic cells was involved in the expression of CCL17 and CCL22, which are Th2-attracting chemokines in the skin.
The in vivo anti-dermatitis effects of JTE-013 administration and S1P2 deficiency may be caused not only by the inhibition of S1P2 in dendritic cells and mast cells [7, 18, 21, 23, 24] but also partly by the inhibition of S1P2 in other cell types, such as keratinocytes [35, 36]. The pharmacological and genetic inhibition of S1P lyase decreased keratinocyte proliferation and induced differentiation [37]. In a previous study, we found that JTE-013 treatment caused a decrease in IL-4+ T cells in draining lymph nodes in an ovalbumin-induced allergic asthma model [21]. Although the S1P2 receptor was not expressed in the T cell lineage [28], further investigation into T cells, such as Th17 differentiation, may be necessary [14]. The in vivo efficacy of the JTE-013 treatment and S1P2 deficiency could have been due to the following: (1) the accumulation of mast cells was suppressed in the epidermis, (2) the overproduction of inflammatory cytokines (IL-4, IL-13, IL-17A, and INF-γ) was suppressed in the epidermis and lymph nodes, and (3) the expression of CCL17 and CCL22 was suppressed in the epidermis and dendritic cells.
References
Davidson WF, Leung DYM, Beck LA, Berin CM, Boguniewicz M, Busse WW, et al. Report from the National Institute of Allergy and Infectious Diseases workshop on “Atopic dermatitis and the atopic march: Mechanisms and interventions”. J Allergy Clin Immunol. 2019;143:894–913.
Leung DY, Guttman-Yassky E. Assessing the current treatment of atopic dermatitis: Unmet needs. J Allergy Clin Immunol. 2017;139:S47–8.
Baumer W, Rossbach K, Mischke R, Reines I, Langbein-Detsch I, Luth A, et al. Decreased concentration and enhanced metabolism of sphingosine-1-phosphate in lesional skin of dogs with atopic dermatitis: disturbed sphingosine-1-phosphate homeostasis in atopic dermatitis. J Invest Dermatol. 2011;131:266–8.
Park SJ, Im DS. Sphingosine 1-phosphate receptor modulators and drug discovery. Biomol Ther. 2017;25:80–90.
Leong WI, Saba JD. S1P metabolism in cancer and other pathological conditions. Biochimie. 2010;92:716–23.
Spiegel S, Milstien S. Functions of the multifaceted family of sphingosine kinases and some close relatives. J Biol Chem. 2007;282:2125–9.
Bock S, Pfalzgraff A, Weindl G. Sphingosine 1-phospate differentially modulates maturation and function of human Langerhans-like cells. J Dermatol Sci. 2016;82:9–17.
Kohno T, Tsuji T, Hirayama K, Watabe K, Matsumoto A, Kohno T, et al. A novel immunomodulator, FTY720, prevents spontaneous dermatitis in NC/Nga mice. Biol Pharm Bull. 2004;27:1392–6.
Yanagawa Y, Hoshino Y, Kataoka H, Kawaguchi T, Ohtsuki M, Sugahara K, et al. FTY720, a novel immunosuppressant, prolongs rat skin allograft survival by decreasing T-cell infiltration into grafts. Transpl Proc. 1999;31:1227–9.
Checa A, Xu N, Sar DG, Haeggstrom JZ, Stahle M, Wheelock CE. Circulating levels of sphingosine-1-phosphate are elevated in severe, but not mild psoriasis and are unresponsive to anti-TNF-α treatment. Sci Rep. 2015;5:12017.
Mysliwiec H, Baran A, Harasim-Symbor E, Choromanska B, Mysliwiec P, Milewska AJ, et al. Increase in circulating sphingosine-1-phosphate and decrease in ceramide levels in psoriatic patients. Arch Dermatol Res. 2017;309:79–86.
Schaper K, Dickhaut J, Japtok L, Kietzmann M, Mischke R, Kleuser B, et al. Sphingosine-1-phosphate exhibits anti-proliferative and anti-inflammatory effects in mouse models of psoriasis. J Dermatol Sci. 2013;71:29–36.
Liao JJ, Huang MC, Goetzl EJ. Cutting edge: alternative signaling of Th17 cell development by sphingosine 1-phosphate. J Immunol. 2007;178:5425–8.
Shin SH, Cho KA, Hahn S, Lee Y, Kim YH, Woo SY, et al. Inhibiting Sphingosine Kinase 2 Derived-sphingosine-1-phosphate ameliorates psoriasis-like skin disease via blocking Th17 differentiation of naive CD4 T lymphocytes in mice. Acta Derm Venereol. 2019;99:594–601.
Reines I, Kietzmann M, Mischke R, Tschernig T, Luth A, Kleuser B, et al. Topical application of sphingosine-1-phosphate and FTY720 attenuate allergic contact dermatitis reaction through inhibition of dendritic cell migration. J Invest Dermatol. 2009;129:1954–62.
Nakashima D, Kabashima K, Sakabe J, Sugita K, Kobayashi T, Yoshiki R, et al. Impaired initiation of contact hypersensitivity by FTY720. J Invest Dermatol. 2008;128:2833–41.
Schumann J, Grevot A, Ledieu D, Wolf A, Schubart A, Piaia A, et al. Reduced activity of sphingosine-1-phosphate lyase induces podocyte-related glomerular proteinuria, skin irritation, and platelet activation. Toxicol Pathol. 2015;43:694–703.
Japtok L, Schaper K, Baumer W, Radeke HH, Jeong SK, Kleuser B. Sphingosine 1-phosphate modulates antigen capture by murine Langerhans cells via the S1P2 receptor subtype. PLoS ONE. 2012;7:e49427.
Kono M, Belyantseva IA, Skoura A, Frolenkov GI, Starost MF, Dreier JL, et al. Deafness and stria vascularis defects in S1P2 receptor-null mice. J Biol Chem. 2007;282:10690–6.
Park SJ, Im DS. Deficiency of sphingosine-1-phosphate receptor 2 (S1P2) attenuates bleomycin-induced pulmonary fibrosis. Biomol Ther. 2018;27:318–26.
Park SJ, Im DS. Blockage of sphingosine-1-phosphate receptor 2 attenuates allergic asthma in mice. Br J Pharmacol. 2019;176:938–49.
Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92.
Olivera A, Dillahunt SE, Rivera J. Interrogation of sphingosine-1-phosphate receptor 2 function in vivo reveals a prominent role in the recovery from IgE and IgG-mediated anaphylaxis with minimal effect on its onset. Immunol Lett. 2013;150:89–96.
Oskeritzian CA, Price MM, Hait NC, Kapitonov D, Falanga YT, Morales JK, et al. Essential roles of sphingosine-1-phosphate receptor 2 in human mast cell activation, anaphylaxis, and pulmonary edema. J Exp Med. 2010;207:465–74.
Kim JY, Jeong MS, Park MK, Lee MK, Seo SJ. Time-dependent progression from the acute to chronic phases in atopic dermatitis induced by epicutaneous allergen stimulation in NC/Nga mice. Exp Dermatol. 2014;23:53–7.
Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. 2008;128:2625–30.
Muraro A, Lemanske RF Jr., Hellings PW, Akdis CA, Bieber T, Casale TB, et al. Precision medicine in patients with allergic diseases: Airway diseases and atopic dermatitis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol. 2016;137:1347–58.
Sawicka E, Zuany-Amorim C, Manlius C, Trifilieff A, Brinkmann V, Kemeny DM, et al. Inhibition of Th1- and Th2-mediated airway inflammation by the sphingosine 1-phosphate receptor agonist FTY720. J Immunol. 2003;171:6206–14.
Matsuo K, Hatanaka S, Kimura Y, Hara Y, Nishiwaki K, Quan YS, et al. A CCR4 antagonist ameliorates atopic dermatitis-like skin lesions induced by dibutyl phthalate and a hydrogel patch containing ovalbumin. Biomed Pharmacother. 2019;109:1437–44.
Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7.
Stutte S, Quast T, Gerbitzki N, Savinko T, Novak N, Reifenberger J, et al. Requirement of CCL17 for CCR7- and CXCR4-dependent migration of cutaneous dendritic cells. Proc Natl Acad Sci USA. 2010;107:8736–41.
Homey B, Steinhoff M, Ruzicka T, Leung DY. Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol. 2006;118:178–89.
Tsuji T, Yoshida Y, Iwatsuki R, Inoue M, Fujita T, Kohno T. Therapeutic approach to steroid-resistant dermatitis using novel immunomodulator FTY720 (Fingolimod) in combination with betamethasone ointment in NC/Nga mice. Biol Pharm Bull. 2012;35:1314–9.
Tsuji T, Okuno S, Kuroda A, Hamazaki J, Chikami T, Sakurai S, et al. Therapeutic approach to mite-induced intractable dermatitis using novel immunomodulator FTY720 ointment (fingolimod) in NC/Nga mice. Allergol Int. 2016;65:172–9.
Kim DS, Kim SY, Kleuser B, Schafer-Korting M, Kim KH, Park KC. Sphingosine-1-phosphate inhibits human keratinocyte proliferation via Akt/protein kinase B inactivation. Cell Signal. 2004;16:89–95.
Jeong SK, Kim YI, Shin KO, Kim BW, Lee SH, Jeon JE, et al. Sphingosine kinase 1 activation enhances epidermal innate immunity through sphingosine-1-phosphate stimulation of cathelicidin production. J Dermatol Sci. 2015;79:229–34.
Jeon S, Song J, Lee D, Kim GT, Park SH, Shin DY, et al. Inhibition of sphingosine 1-phosphate lyase activates human keratinocyte differentiation and attenuates psoriasis in mice. J Lipid Res. 2020;61:20–32.
Acknowledgements
This research was supported by the Basic Science Research Program of the Korean National Research Foundation funded by the Korean Ministry of Education, Science and Technology (NRF-2019R1A2C1005523).
Author information
Authors and Affiliations
Contributions
SJP and DSI designed the experiments. SJP performed the experiments and analyzed the data. SJP and DSI wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Park, SJ., Im, DS. Blockage of sphingosine-1-phosphate receptor 2 attenuates 2,4-dinitrochlorobenzene-induced atopic dermatitis in mice. Acta Pharmacol Sin 41, 1487–1496 (2020). https://doi.org/10.1038/s41401-020-0412-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0412-8
Keywords
This article is cited by
-
Human umbilical cord mesenchymal stem cell treatment alleviates symptoms in an atopic dermatitis-like mouse model
Stem Cell Research & Therapy (2023)
-
Pathogenic sphingosine 1-phosphate pathway in psoriasis: a critical review of its pathogenic significance and potential as a therapeutic target
Lipids in Health and Disease (2023)
-
Inhibitory Effect of Oroxylin A in a Mouse Model of Atopic Dermatitis
Inflammation (2023)
-
Atopic dermatitis: an expanding therapeutic pipeline for a complex disease
Nature Reviews Drug Discovery (2022)
