
Abstract
P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) are involved in intestinal barrier. Short-chain fatty acids (SCFAs) play important roles in maintaining intestinal barrier. In this study we explored how SCFAs affected the expression and function of intestinal P-gp and BCRP in rats. Rats received 150 mM acetate, propionate or butyrate in drinking water for 4 weeks. In SCFA-treated rats, the expression and function of intestinal P-gp were decreased, but those of intestinal BCRP were increased; intestinal p-p65 was also decreased, which was positively related to P-gp protein expression. Among the three SCFAs tested, butyrate exhibited the strongest induction or inhibitory effect, followed by propionate and acetate. Similar results were observed in mouse primary enterocytes and Caco-2 cells treated with acetate (5 mM), propionate (2 mM), or butyrate (1 mM). In Caco-2 cells, addition of butyrate, vorinostat, and valproate (two classic HDAC inhibitors), Bay117082 (selective inhibitor of NF-κB activation) or NF-κB p65 silencing significantly decreased the expression of P-gp and the level of phosphorylated p65 (p-p65). Furthermore, butyrate attenuated the expression of P-gp and p-p65 induced by TNF-α (NF-κB activator) and theophylline (HDAC activator). However, vorinostat, valproate, Bay117082, TNF-α or p65 silencing hardly affected BCRP protein expression. But GW9662 (selective PPARγ antagonist) or PPARγ silencing abolished BCRP induction by butyrate and troglitazone (PPARγ agonist). SCFAs-treated rats showed higher intestinal protein expression of PPARγ, which was positively related to BCRP protein expression. Butyrate increased plasma exposure of fexofenadine but decreased that of rosuvastatin following oral dose to rats. In conclusion, SCFAs exert opposite effects on the expression and function of intestinal P-gp and BCRP; butyrate downregulated P-gp expression and function possibly via inhibiting HDAC/NF-κB pathways; butyrate induced BCRP expression and function partly via PPARγ activation.
Similar content being viewed by others

Introduction
P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), two key ATP-binding cassette (ABC) efflux transporters expressed in intestinal epithelial cells, participate in the construction of the intestinal barrier. Both P-gp and BCRP extrude their substrates back into the intestinal lumen, maintaining low systemic exposure to many oral drugs by limiting their absorption. Factors such as food, comedication, and physical condition affect the expression and function of the two transporters. Food and beverages can influence the absorption of coadministered drugs by modulating intestinal transporters, among which the inhibitory effects of grapefruit juice on P-gp has been well described [1]. The gut mucosa of patients with ulcerative colitis has significantly lower expression of BCRP and P-gp [2], but P-gp expression in the colonic mucosa of patients with steroid-refractory ulcerative colitis is significantly higher [3]. The higher dose requirement of tacrolimus in active Crohn’s disease may be related to the high expression of intestinal P-gp [4]. Our previous studies have illustrated that diabetes impairs the functions and expression levels of intestinal P-gp [5, 6] and BCRP [6, 7], altering the plasma exposure of protoberberine alkaloids, glibenclamide, and atorvastatin following oral dosing. Moreover, P-gp has been reported to reduce the adhesion of some bacteria to enterocytes, contributing to the resistance of potential bacterial toxicity and reducing the risk of gastrointestinal disorder [8].
Short-chain fatty acids (SCFAs), mainly including acetate, propionate, and butyrate, are bacterial fermentation products derived from soluble dietary fiber in the colon. A growing body of evidence has demonstrated key roles of SCFAs in maintaining the intestinal barrier by stabilizing specific transcription factors, facilitating tight junction assembly and mucin secretion [9,10,11]. The amount and abundance of fecal SCFAs vary with several factors, such as dietary habits, composition of gut microbes, and health status. High fiber diets are often characterized by increased amounts of fecal SCFAs [12]. Clinical trials have revealed that obesity is associated with increased amounts of total fecal SCFAs [13,14,15]. The influence of diabetes on intestinal SCFA levels is still controversial. Fecal SCFA levels in type 1 diabetic (T1D) patients have been reported to be comparable to those in healthy subjects [16, 17]. In another study, T1D subjects showed lower fecal propionate and butyrate levels as well as total SCFA levels compared with healthy controls [18]. We recently reported that diabetic rats induced by a combination of a high-fat diet and a low dose of streptozocin exhibited higher concentrations of intestinal SCFAs than control rats [19].
SCFAs, acting as histone deacetylase (HDAC) inhibitors, also show anti-intestinal inflammatory effects, partly by inhibiting nuclear factor κB (NF-κB) [20]. NF-κB is considered to be a crucial modulator of P-gp expression [21], indicating that SCFAs may regulate P-gp by inhibiting the NF-κB pathway or HDACs. We also reported that SCFAs, especially butyrate, impaired the function and expression of intestinal monocarboxylate transporter 6 by activating peroxisome proliferator-activated receptor gamma (PPARγ) [19]. Moreover, the PPARγ agonists pioglitazone and rosiglitazone have been reported to induce the expression of BCRP [22, 23]. These results indicate that SCFAs, which are modulated by dietary habits and health status, may regulate the expression/function of intestinal P-gp and BCRP, leading to alterations in the pharmacokinetics, efficacy, and toxic effects of their substrate drugs.
The aims of the study were: (1) to investigate alterations in the functions and expression levels of intestinal P-gp and BCRP by administration of SCFAs; (2) to explore whether these alterations were associated with inhibition of the NF-κB pathway or activation of the PPARγ pathway; and (3) to document the impact of these alterations on the pharmacokinetics of substrate drugs of P-gp and BCRP following oral dosing.
Materials and methods
Chemicals and reagents
Rhodamine 123 (Rho123), fexofenadine hydrochloride, and prazosin hydrochloride were purchased from J&K Scientific Ltd. (Shanghai, China). Rosuvastatin calcium, troglitazone, and vorinostat were obtained from Aladdin Co. Ltd. (Shanghai, China). GW9662, Bay117082, and valproate sodium were from MedChemExpress (New Jersey, USA). Recombinant human tumor necrosis factor alpha (TNF-α) was obtained from R&D Systems (Minnesota, USA). Reagents for cell culture fetal bovine serum (FBS), Dulbecco’s modified Eagle's medium (DMEM), insulin-transferrin-selenium (ITS), and nonessential amino acids (NEAAs) were from Gibco (Thermo Fisher Scientific, USA). RIPA lysis buffer and BCA protein assay kits were from Beyotime Institute of Biotechnology (Nanjing, China). Antibodies specific for ABCB1 (P-gp), ABCG2 (BCRP), NF-кB p65, and p-NF-кB p65 were purchased from Cell Signaling Technology (Danvers, USA). Antibodies against IκBα, p-IκBα, and PPARγ were obtained from Wanlei Biotechnology (Shenyang, China). Antibodies against β-actin and lamin B1 as well as anti-rabbit and anti-mouse IgG-conjugated horseradish peroxidase were purchased from Bioworld (Louis Park, USA). All other agents were of commercial grade.
Animals and treatments
Male Sprague–Dawley rats, body weight 160–180 g, and male BALB/c mice (8–12 weeks) were obtained from Sino-British SIPPR/BK Lab Animal Ltd. (Shanghai, China). Animals were housed in a controlled environment (ambient temperature of 24 ± 1 °C; relative humidity of 50% ± 5%) under a 12 h light-dark cycle with commercial standard rodent chow. The animal experiments were carried out in accordance with the guidelines on the Care and Use of Animals developed by the National Advisory Committee for Laboratory Animal Research. All animal experiments were approved by the Animal Ethics Committee of China Pharmaceutical University.
Rats acclimated for 3 days and were randomly divided into four groups: control rats, acetate rats, propionate rats and butyrate rats. They received ordinary water or water containing 150 mM sodium acetate, sodium propionate, or sodium butyrate, respectively. The concentrations of SCFAs in drinking water were determined from previous reports [19, 24]. Following 4 weeks of treatment, the experimental rats were used for the subsequent experiments.
Effects of SCFAs on the functions of intestinal P-gp and BCRP in rats
In situ single-pass perfusion was performed to evaluate the functions of intestinal P-gp and BCRP using intestinal excretion of Rho123 (a P-gp substrate) and prazosin (a BCRP substrate) according to methods described previously [25, 26]. In brief, the experimental rats, fasted overnight, were anaesthetized by pentobarbital sodium (60 mg/kg in 0.9% saline, ip). The jejunum (~10 cm) was isolated and cannulas were inserted into both ends. The isolated intestinal segment was perfused with Krebs–Henseleit buffer with a constant infusion pump at a rate of 0.2 mL/min. Then, the rats intravenously received Rho123 (0.2 mg/kg) and prazosin (0.5 mg/kg) via the tail vein. Effluent samples were collected at 30-min intervals via the distal cannula for 120 min. Blood samples were simultaneously collected into heparinized microtubes via the suborbital vein at 2, 5, 15, 30, 60, and 120 min after dosing. The rats were sacrificed at the end of the perfusion, and the areas (A) of the perfused intestinal segments were measured. The jejunum was quickly collected to measure the expression of the indicated proteins. The concentrations of Rho123 and prazosin in the perfusates and plasma samples were measured by high-performance liquid chromatography (HPLC). The intestinal excretion clearance (Peff) was calculated as follows. Peff = M/(AUC120 × A), where M is the total amount excreted via the intestinal wall and AUC120 is the area under plasma concentration-time curve from 0 to 120 min following the intravenous dosing.
Isolation and culture of primary intestinal epithelial cells from mice
Intestinal epithelial cells from mice were isolated according to a previous method [27]. The isolated cells were resuspended in DMEM and seeded at a density of 1 × 106 cells/well in 24-well culture plates precoated with rat tail collagen. Following a 12-h incubation in DMEM supplemented with 1% nonessential amino acids, ITS and 20% FBS in a humidified incubator of 5% CO2 at 37 °C, the medium was replaced with fresh medium containing sodium acetate (5 mM), propionate (2 mM), or butyrate (1 mM) and maintained for another 48 h. The concentrations of SCFAs in the cell experiment were determined according to previous reports [19, 28] and preliminary data. The MTT assay showed that these agents had no impact on cell viability at the tested concentrations.
Caco-2 cell culture and drug treatment
Human epithelial colorectal adenocarcinoma cells (Caco-2 cells) were obtained from Cell Bank, Chinese Academy of Sciences (Shanghai, China). Cells were seeded in 24-well plates at a density of 2 × 105 cells/well in DMEM supplemented with 20% FBS, 1% nonessential amino acids and antibiotics, and cultured in a humidified incubator of 5% CO2 at 37 °C. The cells were incubated with different concentrations of sodium acetate (0.5, 1.0, 2.0, 5.0, and 10.0 mM), propionate (0.2, 0.5, 1.0, 2.0, and 5.0 mM) or butyrate (0.1, 0.2, 0.5, 1.0, and 2.0 mM) for 48 h. Expression of P-gp and BCRP in Caco-2 cells was documented following treatment with the NF-κB inhibitor Bay117082 (2 μM), the HDAC inhibitor valproate sodium (5 mM), vorinostat (1 μM), or the PPARγ agonist troglitazone (5 μM). Caco-2 cells were also incubated with butyrate (1 mM) in the presence of the HDAC activator theophylline (5 μM), the NF-κB activator TNF-α (10 ng/mL), or the selective PPARγ antagonist GW9662 (5 μM). The MTT assay demonstrated that these agents had no cytotoxicity at the tested concentrations.
P-gp and BCRP functions in cells
The functions of P-gp and BCRP were documented using the cellular uptake of P-gp and BCRP substrates. The cultured cells were washed twice with warm Hank’s balanced salt solution (HBSS, pH 7.4) and preincubated in 0.5 mL of HBSS (pH 7.4) at 37 °C for 5 min. The uptake reactions were initiated by adding 0.5 mL of HBSS containing Rho123 (100 nM for Caco-2 cells and 200 nM for primarily cultured intestinal epithelial cells) or prazosin (200 nM). Following 120 min of incubation at 37 °C, uptake reactions were terminated by removing HBSS and washing the cells three times with ice-cold HBSS. Further, 120-min uptake experiments with quinidine (a P-gp substrate) or rosuvastatin (a BCRP substrate) were performed to confirm the altered functions of P-gp and BCRP in Caco-2 cells. The intracellular amounts of Rho123, quinidine, and prazosin were measured by HPLC. The intracellular amount of rosuvastatin was measured by high-performance liquid chromatography-mass spectrometry (HPLC-MS).
Small interfering RNA-mediated knockdown in Caco-2 cells
Caco-2 cells were transfected with siRNA specific for NF-κB p65 or PPARγ using Lipofectamine 3000 according to the manufacturer’s instructions. After incubation for 12 h, the transfection medium was replaced. Cells transfected with p65 siRNA were cultured for another 36 h. Cells transfected with PPARγ siRNA were further exposed to butyrate (1 mM) or troglitazone (5 μM) for 48 h. The siRNA sequences for NF-κB p65 were 5′-CCUGAGCACCAUCAACUAU-3′ (forward) and 5′-AUAGUUGAUGGUGCUCAGG-3′ (reverse). The siRNA sequences for PPARγ were 5′-CCAAGUUUGAGUUUGCUGU-3′ (forward) and 5′-ACAGCAAACUCAAACUUGG-3′ (reverse). Knockdown efficiencies of the NF-κB p65 and PPARγ proteins were confirmed by Western blot.
Western blot analysis and PCR analysis
The protein levels of the indicated proteins in rat jejunum and cells were measured by Western blot analysis. Briefly, samples from the jejunum and cells were lysed in RIPA lysis buffer containing 1 mM PMSF in an appropriate proportion. The nuclear proteins from rat intestines and Caco-2 cells were extracted to determine NF-κB nuclear translocation by a nuclear extract kit (KeyGen Biotech, Nanjing) according to the manufacturer’s instructions. The protein concentrations of tissue and cell lysates were measured by a BCA protein assay kit. Equal amounts of proteins were subjected to SDS-PAGE and transferred to nitrocellulose membranes (Pall Life Sciences, USA). Blots were blocked in 5% nonfat dry milk dissolved in Tris-buffered saline-0.1% Tween 20 (TBST) for 90 min. The membranes were then incubated with primary antibodies overnight at 4 °C, washed and then incubated with horseradish peroxidase-conjugated secondary anti-rabbit antibody for 90 min. The membranes were washed three times with TBST, and then the protein bands were visualized by incubating the membranes with a chemiluminescence (ECL) reagent prior to exposure to the gel imaging system (Tanon Science & Technology Co. Ltd., Shanghai). The densitometry was analyzed by a gel imaging system (Tanon 5200 Multi). The primary antibodies used were antibodies against P-gp (1:1000 dilution), BCRP (1:1000 dilution), PPARγ (1:500 dilution), NF-кB p65 (1:1000 dilution), p-NF-кB p65 (1:1000 dilution), IκBα (1:1000 dilution), p-IκBα (1:500 dilution), β-actin (1:3000 dilution) and lamin B1 (1:1000 dilution).
The mRNA levels of P-gp and BCRP in Caco-2 cells were assessed using qPCR according to our previous method [19]. Total mRNA was extracted using RNAiso Plus (Takara, Shiga, Japan). ReverTra Ace® master mix (Toyobo, Osaka, Japan) was used for the synthesis of cDNA. qPCR was performed on Roche lightcycler 96 (Penzberg, Germany) using Toyobo Thunderbird® SYBR mix. Primers used for targeted genes were ABCB1 (P-gp), 5′-ACAGAGGGGATGGTCAGTGT-3′ (forward) and 5′-TCACGGCCATAGCGAATGTT-3′ (reverse); ABCG2 (BCRP), 5′-AACCTGGTCTCAACGCCATC-3′ (forward) and 5′-GTCGCGGTGCTCCATTTATC-3′ (reverse); β-actin, 5′-AAGAGCTACGAGCTGCCTGAC-3′ (forward) and 5′-TCCTGCTTGCTGATCCACAT-3′ (reverse). RNA expression levels of the targeted genes were calculated using the 2-ΔΔCT method and normalized to the expression of β-actin.
Effects of butyrate on the pharmacokinetics of fexofenadine and rosuvastatin following oral administration
The clinical significance of the altered P-gp and BCRP by SCFAs was investigated by documenting the pharmacokinetics of their substrates fexofenadine and rosuvastatin following oral administration to rats. Rats were randomly divided into a control group and a butyrate group. They received ordinary water or water containing 150 mM butyrate sodium for 4 weeks. Then, the experimental rats (weighing ~350 g) were fasted overnight and orally received fexofenadine (10 mg/kg) and rosuvastatin (5 mg/kg). Blood samples (250 μL) were collected under light ether anesthesia at 5, 10, 20, 30, 45, 60, 120, 240, 480, and 720 min after dosing. Plasma samples were obtained by centrifugation and stored at −80 °C until analysis. Plasma concentrations of fexofenadine and rosuvastatin were measured using a validated HPLC-MS method. Pharmacokinetic parameters were individually estimated using noncompartmental analysis on Phoenix WinNonlin 8.1 (Pharsight, St Louis, MO, USA).
Drug assays
Quantification of Rho123 and prazosin was performed using an HPLC system (Shimadzu, Japan) with terazosin serving as the internal standard following liquid–liquid extraction with ethyl acetate. Chromatographic separation was performed on a YMC-Triart C18 column at a flow rate of 1.0 mL/min. The mobile phases were 10 mM ammonium acetate-0.9% acetic acid aqueous solution and acetonitrile. The wavelength was set to excitation at 485 nm and emission at 546 nm for Rho123 and excitation at 250 nm and emission at 390 nm for prazosin. The ranges of calibration curves were 0.78–100.00 ng/mL for Rho123 and 3.12–400.00 ng/mL for prazosin in biological samples.
Quantification of fexofenadine and rosuvastatin was performed using an HPLC-MS system (Shimadzu, Japan) with phenacetin as an internal standard following liquid–liquid extraction with ethyl acetate acidified with formic acid. Chromatographic separation was performed on a YMC-Triart C18 column at a flow rate of 0.2 mL/min. The mobile phases were 0.1% formic acid in water and acetonitrile. The mass spectrometer was operated in positive mode to detect the respective [M + H]+ ions; m/z 502.4 for fexofenadine, 482.3 for rosuvastatin and 180.3 for phenacetin. The ranges of calibration curves were 1.95–250.00 ng/mL for fexofenadine in plasma and rosuvastatin in plasma and cells.
Statistical analysis
Data are presented as means ± S.D. (standard deviation). One-way ANOVA was used for multiple group comparisons followed by Bonferroni’s post hoc test. Statistical significance was considered at the level of P < 0.05.
Results
Effects of SCFAs on the protein expression levels and functions of intestinal P-gp and BCRP in rats
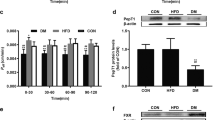
The protein expression levels and functions of intestinal P-gp and BCRP were measured in SCFA-treated rats. The results showed that SCFAs oppositely regulated the protein expression of intestinal P-gp and BCRP, significantly decreasing P-gp but inducing BCRP protein expression in the intestines of rats (Fig. 1a). In accordance with the altered expression of proteins, SCFAs significantly attenuated intestinal excretion of Rho123 but increased that of prazosin following intravenous dosing (Fig. 1b). The areas under the plasma concentration-time curves (AUCs) of both Rho123 and prazosin were not affected (Fig. 1c–e). These results indicated that SCFAs impaired the function of P-gp and enhanced the function of BCRP in the intestines of rats. Among the three tested SCFAs, butyrate exhibited the strongest induction or inhibitory effect, followed by propionate and acetate.

SCFA-treated rats drank water containing 150 mM sodium acetate, sodium propionate or sodium butyrate for 4 weeks. a Levels of P-gp and BCRP proteins in the intestines of rats; b Intestinal excretion of Rho123 (iv 0.2 mg/kg) and prazosin (iv 0.5 mg/kg); c Plasma concentration-time profiles of Rho123 (iv 0.2 mg/kg); d Plasma concentration-time profiles of prazosin (iv 0.5 mg/kg); e Area under the plasma concentration-time curve values (AUCs) of Rho123 and prazosin in rats. Protein expression of (f) NF-кB p65 and phosphorylated p65, g IкBα and phosphorylated IкBα, and h nuclear NF-кB p65 in the intestines of SCFA-treated rats. Data are expressed as means ± S.D. (n = 5). *P < 0.05, **P < 0.01 vs control (Con) rats.
Effects of SCFAs on the protein expression and function of P-gp and BCRP in primarily cultured mouse intestinal epithelial cells
The effects of SCFAs on the protein expression levels and functions of P-gp and BCRP were also documented using primarily cultured intestinal epithelial cells from mice. In line with the in vivo results, 48 h of incubation with SCFAs decreased P-gp protein expression and increased BCRP protein expression (Fig. 2a). The strongest inhibition or induction was observed in cells treated with butyrate. The expression of P-gp protein in cells incubated with butyrate decreased to ~40% that of the control cells, while the level of BCRP protein more than doubled. The functions of P-gp and BCRP were characterized by uptake of their substrates Rho123 and prazosin, respectively (Fig. 2b). Consistent with the altered protein expression of P-gp and BCRP, 48 h of incubation with butyrate or propionate remarkably enhanced the cellular uptake of Rho123 and decreased the cellular uptake of prazosin. Incubation with acetate showed a similar trend, but the result was not significant.

SCFAs (acetate 5 mM, propionate 2 mM, or butyrate 1 mM) were administered to primary intestinal epithelial cells for 48 h and Caco-2 cells for 72 h. a Levels of P-gp and BCRP proteins in primary intestinal epithelial cells incubated with SCFAs; b Uptake of the P-gp substrate Rho123 and the BCRP substrate prazosin in primary intestinal epithelial cells incubated with SCFAs; c Uptake of P-gp substrates (Rho123 and quinidine) and (d) BCRP substrates (prazosin and rosuvastatin) in Caco-2 cells incubated with SCFAs; e Relative mRNA expression of P-gp and BCRP in Caco-2 cells; Levels of P-gp and BCRP proteins in Caco-2 cells incubated with (f) Ac, (g) Pro, and (h) But. Data are expressed as means ± S.D. (n = 6). *P < 0.05, **P < 0.01 vs control (Con) group.
Effects of SCFAs on the expression and function of P-gp and BCRP in Caco-2 cells
The functions of P-gp and BCRP in Caco-2 cells treated with acetate (5 mM), propionate (2 mM) or butyrate (1 mM) were evaluated using the cellular uptake of two specific substrates (Rho123 and quinidine for P-gp; prazosin and rosuvastatin for BCRP). In agreement with the findings in the primarily cultured intestinal epithelial cells of mice, SCFAs increased the uptake of both Rho123 and quinidine but decreased the uptake of both prazosin and rosuvastatin in Caco-2 cells (Fig. 2c, d), demonstrating the impairment of P-gp function and enhancement of BCRP function. Consistently, SCFAs reduced the mRNA levels of P-gp but increased the mRNA levels of BCRP (Fig. 2e). Concentration-dependent inhibition and induction of the targeted proteins were observed (Fig. 2f–h). Butyrate showed the strongest induction or inhibitory effect.
Butyrate impaired the protein expression of P-gp by inhibiting histone deacetylase
Butyrate was selected for further investigation of the pathways involved in alterations in the functions and expression of P-gp and BCRP by SCFAs. As butyrate is a typical HDAC inhibitor [29], the roles of HDAC inhibition were investigated. Two additional classic HDAC inhibitors, vorinostat and valproate, were employed to mimic the effects of butyrate [30]. The results showed that butyrate, vorinostat, and valproic acid inhibited the expression of the P-gp protein (Fig. 3a). However, unlike butyrate, vorinostat and valproic acid had no impact on the expression of the BCRP protein. The protein expression levels of total NF-κB p65, phosphorylated NF-κB p65 (p-p65), inhibitor of nuclear factor kappa B alpha (IκBα), and phosphorylated IκBα (p-IκBα) were simultaneously measured. The results demonstrated that the three tested HDAC inhibitors significantly inhibited the phosphorylation of IκBα (Fig. 3c) and decreased the levels of both p65 and p-p65 (Fig. 3b), but the ratio of p-p65 to p65 remained unaltered. The roles of HDAC inhibition in butyrate-mediated impairment of P-gp protein expression were further documented using the HDAC activator theophylline [31, 32]. The results showed that theophylline induced the expression of the P-gp protein, accompanied by increases in the levels of NF-κB p65, p-65, and p-IκBα (Fig. 3d–f). The theophylline-mediated inductions were almost reversed by butyrate.

Effects of butyrate (But, 1 mM), valproate (Vpa, 5 mM) and vorinostat (Vor, 1 μM) on the expression of (a) P-gp and BCRP, (b) NF-кB p65 and phosphorylated p65 (p-p65), and (c) IкBα and phosphorylated IкBα (p-IкBα); Effects of butyrate (But, 1 mM) on the induction of (d) P-gp, (e) NF-кB p65 and p-p65, and (f) IкBα and p-IкBα protein expression by theophylline (5 μM). Data are expressed as means ± S.D. (n = 5). **P < 0.01 vs control (Con) group. ##P < 0.01 vs cells treated with theophylline only.
Butyrate impaired the protein expression of P-gp by inhibiting the NF-кB pathway
NF-κB is considered to be a transcriptional factor of P-gp [33, 34]. Bay117082 (a selective inhibitor of NF-κB activation) [35] and NF-κB p65 silencing were used to investigate the roles of the NF-κB pathway in the expression of P-gp and BCRP proteins. The results showed that Bay117082 and NF-κB p65 silencing markedly decreased the protein expression of P-gp without affecting the protein expression of BCRP (Fig. 4a, e), demonstrating that inhibition of the NF-κB pathway downregulated the protein expression of P-gp. Further study showed that Bay117082 prevented the phosphorylation of p65, leading to a decrease in the level of p-p65 without affecting the expression of total NF-κB p65 protein (Fig. 4b), resulting in a low ratio of p-p65 to total p65. Unlike Bay117082, butyrate decreased the expression of total NF-κB p65 protein and p-p65 protein (Fig. 4b) without altering the ratio of p-p65 to NF-κB p65. Moreover, Bay117082 and butyrate significantly lowered the level of nuclear p65 protein (Fig. 4d), inferring impairment of NF-κB nuclear translocation. Butyrate also significantly reduced the level of p-IκBα but did not affect the total protein level of IκBα (Fig. 4c).

Expression of (a) P-gp and BCRP, (b) NF-кB p65 and p-p65, and (c) IκBα and p-IκBα in Caco-2 cells coadministered with TNF-α (10 ng/mL) and But (1 mM) or Bay (2 μM) for 72 h; (d) Levels of nuclear NF-кB p65 protein in Caco-2 cells incubated with But or Bay. Effects of NF-кB p65 silencing on (e) P-gp and BCRP and (f) NF-кB p65 protein expression; correlation analysis for protein levels of p-p65 and P-gp (g) in Caco-2 cells co-administered with TNF-α and But or Bay and (h) in the intestines of SCFA-treated rats. Data are expressed as means ± S.D. (n = 5). **P < 0.01 vs control (Con) cells, ##P < 0.01 vs cells treated with TNF-α only.
The role of the NF-κB pathway in the impaired expression of P-gp protein by butyrate was further investigated in Caco-2 cells using TNF-α as an NF-κB activator [36]. As expected, TNF-α activated the NF-κB pathway and significantly induced the protein expression of P-gp (Fig. 4a), which was reflected by increases in p-p65, p65 and p-IκBα levels (Fig. 4b, c). Butyrate attenuated the increased levels of P-gp, p-p65, p65, and p-IκBα induced by TNF-α. Treatment with Bay117082 (Fig. 4a–c) reversed the increases in the levels of P-gp, p-p65, and p-IκBα induced by TNF-α.
The levels of p65, p-p65, IκBα, and p-IκBα in the intestines of SCFA-treated rats were also measured. In line with the in vitro findings, SCFAs decreased the protein expression levels of p-IκBα, p-p65, p65, and nuclear p65 (Fig. 1f–h), which paralleled the decreased expression of P-gp. Correlation analysis showed a positive correlation between NF-κB activation and P-gp expression (Fig. 4g, h).
BCRP expression was not affected by Bay117082, vorinostat, valproic acid, TNF-α, or NF-κB p65 silencing (Figs. 3a and 4a, e), suggesting that SCFA-mediated upregulation of BCRP was independent of histone deacetylase inhibition and the NF-κB pathway.
Butyrate increased BCRP protein expression in a PPARγ-dependent manner
The roles of PPARγ on the induction of BCRP by butyrate were investigated in Caco-2 cells treated with the PPARγ agonist troglitazone and the selective PPARγ antagonist GW9662. The results showed that butyrate and troglitazone increased the expression of the BCRP protein, which was attenuated by GW9662 (Fig. 5a). Further study showed that butyrate itself induced the expression of the PPARγ protein in a concentration-dependent manner (Fig. 5b). The roles of PPARγ in the induction of BCRP by butyrate were verified using silencing PPARγ with siRNA transfection. The expression of PPARγ in silenced cells successfully decreased (Fig. 5f) and the protein level was only 38.4% that of negative cells. Silencing PPARγ almost abolished the upregulation of the BCRP protein by butyrate and troglitazone (Fig. 5d). Consistent with the in vitro results, acetate, propionate, and butyrate also induced PPARγ expression in the intestines of rats (Fig. 5c). Obvious positive correlations were found between BCRP protein levels and PPARγ protein levels in both butyrate-treated Caco-2 cells and the intestines of SCFA-treated rats (Fig. 5g, h), confirming the involvement of PPARγ in the induction of BCRP protein by butyrate.

a Effects of butyrate sodium (But, 1 mM), the PPARγ agonist troglitazone (Tgz, 5 μM) and the PPARγ antagonist GW9662 (GW, 5 μM) on the protein expression of P-gp and BCRP. b Butyrate concentration-dependently induced the expression of the PPARγ protein in Caco-2 cells. c Effects of acetate (Ac), propionate (Pro) and butyrate (But) on the protein expression of the PPARγ in the intestines of SCFA-treated rats. Effects of But and Tgz on the protein expression of (d) BCRP and (e) P-gp in Caco-2 cells following PPARγ silencing. f Expression of the PPARγ protein was downregulated by PPARγ silencing with specific siRNA. Correlation analysis for protein levels of PPARγ and BCRP (g) in Caco-2 cells incubated with But or (h) in the intestines of SCFA-treated rats. Data are expressed as means ± S.D. (n = 5). *P < 0.05, **P < 0.01 vs control (Con) group; ^^P < 0.01 vs But or Tgz group; ##P < 0.01 vs But or Tgz group in negative control silenced cells.
Troglitazone slightly decreased the expression of the P-gp protein, but GW9662 could not restore the impaired expression of the P-gp protein by butyrate or troglitazone (Fig. 5a). In addition, PPARγ silencing had no impact on the decreased expression of P-gp by butyrate or troglitazone (Fig. 5e), inferring that butyrate and troglitazone inhibited the expression of P-gp via pathways independent of PPARγ.
Effects of butyrate on the pharmacokinetics of fexofenadine and rosuvastatin in rats
To study the clinical significance of the alterations in intestinal P-gp and BCRP, the pharmacokinetic behaviors of two clinical substrates recommended by the FDA (fexofenadine for P-gp and rosuvastatin for BCRP) were documented after oral administration to butyrate-treated rats (Fig. 6). Consistent with previous reports [37, 38], a double peak of fexofenadine in the plasma of rats was observed following oral administration, and the first peak (Cmax1) and the secondary peak (Cmax2) occurred at ~15 and 240 min, respectively. In line with the altered expression levels and functions of intestinal P-gp and BCRP, butyrate increased plasma exposure to fexofenadine in rats. The estimated AUC0–12h and Cmax1 of fexofenadine in butyrate-treated rats were 215.0 ± 22.0 h·ng/mL and 48.7 ± 3.5 ng/mL, respectively, which were significantly higher (P < 0.05) than these parameters (180.0 ± 18.4 h·ng/mL for AUC0–12h and 40.8 ± 3.6 ng/mL for Cmax1) in control rats. Compared with control rats (23.5 ± 3.6 ng/mL), the Cmax2 (26.3 ± 3.4 ng/mL) for fexofenadine in butyrate-treated rats showed an increasing trend, but no significance was observed. In contrast, butyrate significantly decreased oral plasma exposure to rosuvastatin in rats. The AUC0–12h (57.1 ± 7.0 h·ng/mL) and Cmax (29.9 ± 4.4 ng/mL) in butyrate-treated rats were significantly (P < 0.05) decreased to 62.3% and 72.6% of those (91.6 ± 14.5 h·ng/mL for AUC0–12h and 41.2 ± 6.2 ng/mL for Cmax) in control rats, respectively. No significant differences were found for the Tmax and t1/2 values between the two groups following oral administration of both fexofenadine and rosuvastatin to rats. These alterations in the pharmacokinetic profiles may largely result from the altered oral absorption affected by P-gp and BCRP in the intestine.

a Plasma concentration-time profiles of fexofenadine following oral administration of 10 mg/kg; b Plasma concentration-time profiles of rosuvastatin following oral administration of 5 mg/kg. Data are expressed as means ± S.D. (n = 5).
Discussion
The main findings were that the SCFAs oppositely regulated expression levels and functions of P-gp and BCRP. SCFAs downregulated the expression of P-gp protein but increased the expression of BCRP protein in the intestines of rats. In line with altered protein expression, SCFAs decreased intestinal efflux of Rho123 and increased that of prazosin following intravenous dosing without altering their plasma exposure. Butyrate showed the strongest inhibitory or induction potency, followed by propionate and acetate. These findings were confirmed in primary mouse epithelial cells and Caco-2 cells. In line with protein expression, the three SCFAs also induced mRNA expression of BCRP but downregulated mRNA expression of P-gp, implying that SCFAs oppositely affected the transcriptional activity of P-gp and BCRP.
The mechanisms by which SCFAs lead to opposite alterations in expression levels and functions of intestinal P-gp and BCRP were investigated using butyrate as the representative SCFA in Caco-2 cells. Biological responses of butyrate on host cells generally result from inhibition of HDACs [39] or activation of G protein-coupled receptors (GPCRs, such as GPR41 and GPR43) [39, 40]. GPR43 has been identified in the rat and human colon [41] as well as in Caco-2 cells [42]. First, we determined the contributions of GPR43 to butyrate-induced alterations in the expression of P-gp and BCRP using GLPG0974, an antagonist of GPR43 [43, 44]. In contrast to our expectations, GLPG0974 could not attenuate butyrate-induced alterations in the expression of P-gp and BCRP (data not shown), suggesting that these alterations were independent of GPR43. Then, we investigated whether butyrate altered the expression of P-gp and BCRP by inhibiting HDACs in Caco-2 cells. We found that all three HDAC inhibitors (butyrate, valproate, and vorinostat) significantly decreased P-gp expression. Butyrate almost abolished the induction of P-gp expression by the HDAC activator theophylline. These results suggested that HDACs possibly participate in the regulation of P-gp expression.
The NF-κB family is a critical nuclear factor that regulates a variety of genes, including P-gp [34]. In the inactive form, NF-κB dimers combine with inhibitory molecules referred to as IκB proteins such as IκBα. Once phosphorylated, the IκB protein is ubiquitinated and degraded from the complex, allowing NF-κB to translocate into the nucleus and bind with DNA sequences of target genes [45]. Several reports have shown that HDAC activity and expression are associated with NF-κB activation [46, 47]. Therefore, we hypothesized that butyrate inhibited P-gp expression via the HDAC/NF-κB pathway. It was observed that the expression of NF-κB p65 was downregulated by HDAC inhibitors but upregulated by theophylline. Although total expression of IκBα remained unchanged, the p-IκBα level was reduced by HDAC inhibitors but elevated by theophylline. We deduced that butyrate, acting as an HDAC inhibitor, inhibited the activation of NF-κB. The roles of the NF-κB pathway in P-gp expression were further investigated. Bay117082 (an inhibitor of IκBα phosphorylation) was used as a positive control, and TNF-α was used as an NF-κB activator. As expected, butyrate and Bay117082 significantly attenuated both basal and TNF-α-induced activation of NF-κB, accompanied by decreased protein expression of P-gp. In vivo data also showed that the expression of the P-gp protein was positively related to the level of p-p65. Moreover, NF-κB p65 silencing markedly diminished the protein expression of P-gp in Caco-2 cells. It has been reported that NF-κB transactivates an mdr1 reporter plasmid in HCT15 cells [34]. Two NF-κB binding sites, (a) CCTTTCGGGG located in the first intron of the MDR1 promoter [34] and (b) −6092GGGAATTCT located upstream from the MDR1 transcription start site [21], were identified separately by two research teams. These results further confirmed that the inhibition of the HDAC/NF-κB pathways was involved in the downregulation of intestinal P-gp expression by butyrate. Acetate and propionate, although to a lesser degree, have also been reported to be HDAC inhibitors [48,49,50] and NF-κB inhibitors [51, 52].
Although the results from several reports [53,54,55] were contrary to ours, some reports supported our findings. For example, Encarnacao et al. reported that butyrate impaired the expression of P-gp in the LS1034 cell line [56]. Cummins et al reported that butyrate did not affect the expression and function of P-gp in Caco-2 cells [57]. Among 4 colon carcinoma cell lines (SW620, HCT-15, DLD-1, and LS180), a major increase in MDR1 mRNA and P-gp protein expression by butyrate was only observed in SW620 cells [58]. Butyrate and trichostatin A (TSA, another HDAC inhibitor) were reported to induce P-gp expression in small-cell lung carcinoma drug-sensitive cells but strongly decrease P-gp expression in etoposide-resistant cells [59]. Similarly, TSA increased the expression and function of P-gp in K562 cells but showed a decrease in K562/ADR cells [60]. These results indicated that the discrepancies in the effects of butyrate on P-gp expression seemed to result from basal levels of P-gp, cell type and incubation time, which require further investigation.
Notably, vorinostat, valproic acid, Bay117082, TNF-α, and NF-κB silencing did not affect the expression of BCRP, suggesting that butyrate-mediated induction of BCRP was independent of HDAC inhibition or NF-κB pathway inhibition. The PPARγ agonists pioglitazone and rosiglitazone have been reported to induce the expression of BCRP [22, 23]. Butyrate has been reported to activate PPARγ [61, 62]. These results suggested that butyrate induced the expression of BCRP possibly by activating PPARγ. A series of studies were designed to confirm this deduction. In line with previous reports, butyrate concentration-dependently induced the expression of the PPARγ protein [19, 63]. Although a report showed that propionate did not affect PPARγ expression [63], propionate and acetate at higher concentrations have been reported to stimulate PPARγ activation [62, 64]. The induction of BCRP protein expression by butyrate or troglitazone was attenuated by the PPARγ antagonist GW9662 and PPARγ silencing. In vivo data demonstrated that acetate, propionate, and butyrate-induced PPARγ expression in the intestines of rats, which was positively correlated with the protein expression of intestinal BCRP. PPARγ has been reported to directly regulate the transcription of BCRP. A conserved enhancer region containing three functional PPAR response elements (PPAREs) was identified upstream from the human ABCG2 gene [65]. All these results demonstrated that butyrate-induced BCRP expression by activating the PPARγ pathway. Neither GW9662 nor PPARγ silencing restored the impairment of P-gp expression, further confirming that the regulation of P-gp expression by butyrate was independent of the PPARγ pathway. Surprisingly, troglitazone also decreased the expression of the P-gp protein, which could not be restored by GW9662 and PPARγ silencing. Troglitazone has been reported to inhibit HDACs [66] or the NF-κB pathway [67, 68], which may partly explain the above finding.
To evaluate the clinical significance of butyrate-induced alterations in the expression and function of intestinal P-gp and BCRP, pharmacokinetic profiles of both the P-gp substrate fexofenadine and the BCRP substrate rosuvastatin were documented following oral administration to butyrate-treated rats. Butyrate treatment significantly increased oral plasma exposure to fexofenadine and decreased that of rosuvastatin, which was consistent with the alterations in the expression levels and functions of intestinal P-gp and BCRP. The results indicated that the SCFA-induced alterations in expression levels and functions of intestinal P-gp and BCRP affected the pharmacokinetics of oral drugs. Butyrate was reported to enhance the anticancer effects of the P-gp substrate irinotecan, accompanied by decreased P-gp expression [56]. In vivo data also demonstrated the therapeutic potential for the use of butyrate in combination with irinotecan in chemoresistant tumors [56]. Dietary fiber generally increases the amounts of intestinal SCFAs [12], indicating that food–drug interactions should be cautioned due to SCFA-mediated alterations in expression levels and functions of intestinal P-gp and BCRP. Moreover, intestinal excretion of uric acid is mainly mediated by BCRP [69]. Dietary fibers suppress the elevation of serum uric acid induced by dietary RNA and increase its excretion in the feces of rats [70]. A clinical report also showed that the intake of dietary fiber was inversely associated with the risk of hyperuricemia [71]. In addition, health status may alter the expression of intestinal P-gp and BCRP, which may be attributed to alterations in the intestinal contents of SCFAs. For instance, we once reported that diabetes significantly increased SCFA contents in the intestines of rats [19]. This may be a reason that diabetes impairs the function and expression of P-gp but induces the function and expression of BCRP in the intestines of rats, leading to remarkably decreased plasma exposure of atorvastatin following oral dosing to rats [6]. Another example is obesity, which is characterized by increased fecal SCFAs [13,14,15], decreases intestinal P-gp expression and increases the oral bioavailability of P-gp substrate drugs [72]. Our study provides new insight into the relationship between bacterial metabolite SCFAs and transporters and suggests that SCFA-mediated dosage adjustment should be considered in specific populations. The clinical significance of SCFA-induced alterations in the expression and function of intestinal P-gp and BCRP requires further investigation.
In conclusion, SCFAs oppositely regulated the expression levels and functions of intestinal P-gp and BCRP via different mechanisms. Inhibition of the HDAC/NF-κB pathways was involved in butyrate-induced downregulation of P-gp expression. Butyrate induced the expression and function of BCRP partly through PPARγ activation. These findings would aid in understanding the effects of intestinal SCFAs on the pharmacokinetics of oral drugs.
References
Nakanishi T, Tamai I. Interaction of drug or food with drug transporters in intestine and liver. Curr Drug Metab. 2015;16:753–64.
Gutmann H, Hruz P, Zimmermann C, Straumann A, Terracciano L, Hammann F, et al. Breast cancer resistance protein and P-glycoprotein expression in patients with newly diagnosed and therapy-refractory ulcerative colitis compared with healthy controls. Digestion. 2008;78:154–62.
Sambuelli AM, Negreira SM, Gil AH, Huernos SP, Goncalves S, Toro MA, et al. Multidrug resistance gene (MDR-1) expression in the colonic mucosa of patients with refractory ulcerative colitis. Acta Gastroenterol Latinoam. 2006;36:23–32.
Buchman AL, Paine MF, Wallin A, Ludington SS. A higher dose requirement of tacrolimus in active Crohn’s disease may be related to a high intestinal P-glycoprotein content. Dig Dis Sci. 2005;50:2312–5.
Yu S, Yu Y, Liu L, Wang X, Lu S, Liang Y, et al. Increased plasma exposures of five protoberberine alkaloids from Coptidis Rhizoma in streptozotocin-induced diabetic rats: is P-GP involved? Planta Med. 2010;76:876–81.
Wang Z, Yang H, Xu J, Zhao K, Chen Y, Liang L, et al. Prediction of atorvastatin pharmacokinetics in high-fat diet and low-dose streptozotocin-induced diabetic rats using a semiphysiologically based pharmacokinetic model involving both enzymes and transporters. Drug Metab Dispos. 2019;47:1066–79.
Liu H, Liu L, Li J, Mei D, Duan R, Hu N, et al. Combined contributions of impaired hepatic CYP2C11 and intestinal breast cancer resistance ddition of butyrate, vorinostprotein activities and expression to increased oral glibenclamide exposure in rats with streptozotocin-induced diabetes mellitus. Drug Metab Dispos. 2012;40:1104–12.
Crowe A. The role of P-glycoprotein and breast cancer resistance protein (BCRP) in bacterial attachment to human gastrointestinal cells. J Crohn’s Colitis. 2011;5:531–42.
Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell host Microbe. 2015;17:662–71.
Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr. 2009;139:1619–25.
Jung TH, Park JH, Jeon WM, Han KS. Butyrate modulates bacterial adherence on LS174T human colorectal cells by stimulating mucin secretion and MAPK signaling pathway. Nutr Res Pr. 2015;9:343–9.
Tomova A, Bukovsky I, Rembert E, Yonas W, Alwarith J, Barnard ND, et al. The effects of vegetarian and vegan diets on gut microbiota. Front Nutr. 2019;6:47.
Rahat-Rozenbloom S, Fernandes J, Gloor GB, Wolever TM. Evidence for greater production of colonic short-chain fatty acids in overweight than lean humans. Int J Obes (Lond). 2014;38:1525–31.
Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18:190–5.
Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TM, Comelli EM. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes. 2014;4:e121.
de Groot PF, Belzer C, Aydin O, Levin E, Levels JH, Aalvink S, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS One. 2017;12:e0188475.
Samuelsson U, Ludvigsson J. The concentrations of short-chain fatty acids and other microflora-associated characteristics in faeces from children with newly diagnosed type 1 diabetes and control children and their family members. Diabet Med. 2004;21:64–7.
Lassenius MI, Fogarty CL, Blaut M, Haimila K, Riittinen L, Paju A, et al. Intestinal alkaline phosphatase at the crossroad of intestinal health and disease - a putative role in type 1 diabetes. J Intern Med. 2017;281:586–600.
Xu F, Zhu L, Qian C, Zhou J, Geng D, Li P, et al. Impairment of intestinal monocarboxylate transporter 6 function and expression in diabetic rats induced by combination of high-fat diet and low dose of streptozocin: involvement of butyrate-peroxisome proliferator-activated receptor-gamma activation. Drug Metab Dispos. 2019;47:556–66.
Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. 2011;22:849–55.
Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe S, Mills GB, et al. Induction of human MDR1 gene expression by 2-acetylaminofluorene is mediated by effectors of the phosphoinositide 3-kinase pathway that activate NF-kappaB signaling. Oncogene. 2002;21:1945–54.
Urakami-Takebayashi Y, Kuroda Y, Murata T, Miyazaki M, Nagai J. Pioglitazone induces hypoxia-inducible factor 1 activation in human renal proximal tubular epithelial cell line HK-2. Biochem Biophys Res Commun. 2018;503:1682–8.
Lin Y, Bircsak KM, Gorczyca L, Wen X, Aleksunes LM. Regulation of the placental BCRP transporter by PPAR gamma. J Biochem Mol Toxicol. 2017;31:e21880.
Lucas S, Omata Y, Hofmann J, Bottcher M, Iljazovic A, Sarter K, et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat Commun. 2018;9:55.
Mei D, Li J, Liu H, Liu L, Wang X, Guo H, et al. Induction of multidrug resistance-associated protein 2 in liver, intestine and kidney of streptozotocin-induced diabetic rats. Xenobiotica. 2012;42:709–18.
Moriguchi J, Kato R, Nakagawa M, Hirotani Y, Ijiri Y, Tanaka K. Effects of lipopolysaccharide on intestinal P-glycoprotein expression and activity. Eur J Pharm. 2007;565:220–4.
Di Claudio F, Muglia CI, Smaldini PL, Orsini Delgado ML, Trejo FM, Grigera JR, et al. Use of a collagen membrane to enhance the survival of primary intestinal epithelial cells. J Cell Physiol. 2017;232:2489–96.
Fukushima A, Aizaki Y, Sakuma K. Short-chain fatty acids induce intestinal transient receptor potential vanilloid type 6 expression in rats and Caco-2 cells. J Nutr. 2009;139:20–5.
Santini V, Gozzini A, Ferrari G. Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application. Curr Drug Metab. 2007;8:383–93.
Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126:455–67.
Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200:689–95.
Kadiyala CS, Zheng L, Du Y, Yohannes E, Kao HY, Miyagi M, et al. Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J Biol Chem. 2012;287:25869–80.
Li X, Mu P, Qiao H, Wen J, Deng Y. JNK-AKT-NF-kappaB controls P-glycoprotein expression to attenuate the cytotoxicity of deoxynivalenol in mammalian cells. Biochem Pharmacol. 2018;156:120–34.
Bentires-Alj M, Barbu V, Fillet M, Chariot A, Relic B, Jacobs N, et al. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene. 2003;22:90–7.
Montalbano AM, Albano GD, Bonanno A, Riccobono L, Di Sano C, Ferraro M, et al. Autocrine acetylcholine, induced by IL-17A via NFkappaB and ERK1/2 pathway activation, promotes MUC5AC and IL-8 synthesis in bronchial epithelial cells. Mediat Inflamm. 2016;2016:9063842.
Ferrari D, Speciale A, Cristani M, Fratantonio D, Molonia MS, Ranaldi G, et al. Cyanidin-3-O-glucoside inhibits NF-kB signalling in intestinal epithelial cells exposed to TNF-alpha and exerts protective effects via Nrf2 pathway activation. Toxicol Lett. 2016;264:51–8.
Pathak SM, Kumar AR, Musmade P, Udupa N. A simple and rapid high performance liquid chromatographic method with fluorescence detection for the estimation of fexofenadine in rat plasma–application to preclinical pharmacokinetics. Talanta. 2008;76:338–46.
Kamath AV, Yao M, Zhang Y, Chong S. Effect of fruit juices on the oral bioavailability of fexofenadine in rats. J Pharm Sci. 2005;94:233–9.
Liu H, Wang J, He T, Becker S, Zhang G, Li D, et al. Butyrate: a double-edged sword for health? Adv Nutr. 2018;9:21–9.
Bolognini D, Tobin AB, Milligan G, Moss CE. The pharmacology and function of receptors for short-chain fatty acids. Mol Pharmacol. 2016;89:388–98.
Tazoe H, Otomo Y, Karaki S, Kato I, Fukami Y, Terasaki M, et al. Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed Res. 2009;30:149–56.
D’Souza WN, Douangpanya J, Mu S, Jaeckel P, Zhang M, Maxwell JR, et al. Differing roles for short chain fatty acids and GPR43 agonism in the regulation of intestinal barrier function and immune responses. PLoS ONE. 2017;12:e0180190.
Pizzonero M, Dupont S, Babel M, Beaumont S, Bienvenu N, Blanque R, et al. Discovery and optimization of an azetidine chemical series as a free fatty acid receptor 2 (FFA2) antagonist: from hit to clinic. J Med Chem. 2014;57:10044–57.
Li M, van Esch B, Henricks PAJ, Folkerts G, Garssen J. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor alpha-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front Pharmacol. 2018;9:533.
Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annu Rev biophysics. 2013;42:443–68.
Lehmann A, Denkert C, Budczies J, Buckendahl A-C, Darb-Esfahani S, Noske A, et al. High class I HDAC activity and expression are associated with RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer. 2009;9:395.
Leus NG, Zwinderman MR, Dekker FJ. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-kappaB-mediated inflammation. Curr Opin Chem Biol. 2016;33:160–8.
Jin UH, Cheng Y, Park H, Davidson LA, Callaway ES, Chapkin RS, et al. Short chain fatty acids enhance aryl hydrocarbon (Ah) responsiveness in mouse colonocytes and caco-2 human colon cancer cells. Sci Rep. 2017;7:10163.
Soliman ML, Combs CK, Rosenberger TA. Modulation of inflammatory cytokines and mitogen-activated protein kinases by acetate in primary astrocytes. J Neuroimmune Pharmacol 2013;8:287–300.
Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008;19:587–93.
Liu T, Li J, Liu Y, Xiao N, Suo H, Xie K, et al. Short-chain fatty acids suppress lipopolysaccharide-induced production of nitric oxide and proinflammatory cytokines through inhibition of NF-kappaB pathway in RAW264.7 cells. Inflammation. 2012;35:1676–84.
Inan MS, Rasoulpour RJ, Yin L, Hubbard AK, Rosenberg DW, Giardina C. The luminal short-chain fatty acid butyrate modulates NF-kappaB activity in a human colonic epithelial cell line. Gastroenterology. 2000;118:724–34.
Zhao L, Bin S, He HL, Yang JM, Pu YC, Gao CH, et al. Sodium butyrate increases P-gp expression in lung cancer by upregulation of STAT3 and mRNA stabilization of ABCB1. Anticancer Drugs. 2018;29:227–33.
Pasvanis S, Tremblay S, Dumais N. High sodium butyrate levels induce MDR1 activation in colorectal cells: impact of 15-deoxy-delta(12,14)-prostaglandin J(2) on the resistance to saquinavir. Biochem Biophys Res Commun. 2012;418:609–15.
Yan JK, Gong ZZ, Zhang T, Cai W. Sodium butyrate attenuates soybean oil-based lipid emulsion-induced increase in intestinal permeability of lipopolysaccharide by modulation of P-glycoprotein in Caco-2 cells. Biochem Biophys Res Commun. 2017;482:791–5.
Encarnacao JC, Pires AS, Amaral RA, Goncalves TJ, Laranjo M, Casalta-Lopes JE, et al. Butyrate, a dietary fiber derivative that improves irinotecan effect in colon cancer cells. J Nutr Biochem. 2018;56:183–92.
Cummins CL, Mangravite LM, Benet LZ. Characterizing the expression of CYP3A4 and efflux transporters (P-gp, MRP1, and MRP2) in CYP3A4-transfected Caco-2 cells after induction with sodium butyrate and the phorbol ester 12-O-tetradecanoylphorbol-13-acetate. Pharmacol Res. 2001;18:1102–9.
Frommel TO, Coon JS, Tsuruo T, Roninson IB. Variable effects of sodium butyrate on the expression and function of the MDR1 (P-glycoprotein) gene in colon carcinoma cell lines. Int J Cancer. 1993;55:297–302.
El-Khoury V, Breuzard G, Fourre N, Dufer J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1) gene expression by a transcription-dependent mechanism in a drug-resistant small cell lung carcinoma cell line model. Br J Cancer. 2007;97:562–73.
Balaguer TM, Gomez-Martinez A, Garcia-Morales P, Lacueva J, Calpena R, Reverte LR, et al. Dual regulation of P-glycoprotein expression by trichostatin A in cancer cell lines. BMC Mol Biol. 2012;13:25.
Kinoshita M, Suzuki Y, Saito Y. Butyrate reduces colonic paracellular permeability by enhancing PPARgamma activation. Biochem Biophys Res Commun. 2002;293:827–31.
Alex S, Lange K, Amolo T, Grinstead JS, Haakonsson AK, Szalowska E, et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2013;33:1303–16.
Wachtershauser A, Loitsch SM, Stein J. PPAR-gamma is selectively upregulated in Caco-2 cells by butyrate. Biochem Biophys Res Commun. 2000;272:380–5.
Nepelska M, de Wouters T, Jacouton E, Béguet-Crespel F, Lapaque N, Doré J, et al. Commensal gut bacteria modulate phosphorylation-dependent PPARγ transcriptional activity in human intestinal epithelial cells. Sci Rep. 2017;7:43199.
Szatmari I, Vámosi G, Brazda P, Balint BL, Benko S, Széles L, et al. Peroxisome proliferator-activated receptor γ-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281:23812–23.
Davies GF, Ross AR, Arnason TG, Juurlink BH, Harkness TA. Troglitazone inhibits histone deacetylase activity in breast cancer cells. Cancer Lett. 2010;288:236–50.
Ban JO, Kwak DH, Oh JH, Park EJ, Cho MC, Song HS, et al. Suppression of NF-kappaB and GSK-3beta is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chem Biol Interact. 2010;188:75–85.
Ruan H, Pownall HJ, Lodish HF. Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-kappaB. J Biol Chem. 2003;278:28181–92.
Liu X. ABC family transporters. Adv Exp Med Biol. 2019;1141:13–100.
Koguchi T, Nakajima H, Wada M, Yamamoto Y, Innami S, Maekawa A, et al. Dietary fiber suppresses elevations of uric acid and allantoin in serum and urine induced by dietary RNA and increases its excretion to feces in rats. J Nutr Sci Vitaminol (Tokyo). 2002;48:184–93.
Sun Y, Sun J, Zhang P, Zhong F, Cai J, Ma A. Association of dietary fiber intake with hyperuricemia in U.S. adults. Food Funct. 2019;10:4932–40.
Murakami T, Bodor E, Bodor N. Modulation of expression/function of intestinal P-glycoprotein under disease states. Expert Opin Drug Metab Toxicol. 2020;16:59–78.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81573490, 81872930, and 81673505); “Cyan Blue”, “Six Talent Peaks” and “333” Project of Jiangsu Province, and “Double First-Class” university project (No. CPU2018GY22).
Author information
Authors and Affiliations
Contributions
Research design: QSX and XDL. Conduction of the experiments: QSX, JXZ, ML, PHL, LZ, LJ, MMJ, and XNL. Data analysis: QSX, ZJW, and XDL. Writing or revision of the manuscript: QSX, XDL, LL, ZJW, and LJ.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Xie, Qs., Zhang, Jx., Liu, M. et al. Short-chain fatty acids exert opposite effects on the expression and function of p-glycoprotein and breast cancer resistance protein in rat intestine. Acta Pharmacol Sin 42, 470–481 (2021). https://doi.org/10.1038/s41401-020-0402-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0402-x
Keyword
This article is cited by
-
Changes in the fecal microbiota of breast cancer patients based on 16S rRNA gene sequencing: a systematic review and meta-analysis
Clinical and Translational Oncology (2024)
-
Short-chain fatty acids in diseases
Cell Communication and Signaling (2023)
-
Correlation analysis of breast fibroadenoma and the intestinal flora based on 16S rRNA sequencing*
Oncology and Translational Medicine (2021)
