
Abstract
Cadmium (Cd) is an important environmental pollutant and long-term Cd exposure is closely related to autoimmune diseases, cancer, cardiovascular diseases (CVD), and hepatic dysfunction. Zinc (Zn) is an essential metal that plays key roles in protein structure, catalysis, and regulation of their function. Numerous studies have shown that Zn can reduce Cd toxicity; however, the underlying mechanisms have not been extensively explored. Preclinical studies have revealed direct competition for sarcolemmal uptake between these two metals. Multiple sarcolemmal transporters participate in Cd uptake, including Zn transporters, calcium channels, and DMT1 (divalent metal transporter 1). Zn also induces several protective mechanisms, including MT (metallothionein) induction and favorable redox homeostasis. This review summarizes current knowledge related to the role of Zn and metal transporters in reducing Cd toxicity and discusses potential future directions of related research.
Similar content being viewed by others

Introduction
Cadmium (Cd) is a nonessential heavy metal that induces adverse health effects in humans and other organisms [1]. It is now well accepted that Cd can accumulate in many organs, including the liver, kidney, heart, bone, pancreas, and testis, and it can adversely affect organ function and overall health. The most commonly used therapeutic strategy for heavy metal poisoning is chelation therapy, which promotes metal excretion. However, chelators for Cd toxicity have numerous safety and efficacy concerns [2,3,4]. The development of safe and efficient strategies to reduce Cd toxicity remains an area of ongoing research.
Dietary zinc (Zn) supplements are recognized for the important roles they play in the alleviation or prevention of Cd toxicity because Zn is readily available as a nutritional ingredient, affordable as a dietary supplement, and has not been described to have adverse side effects. Numerous studies have shown that Zn can reduce Cd toxicity. For example, Zn inhibits Cd accumulation and ROS production in Cd-treated HeLa cells and bovine aorta endothelial cells [5]. Zn supplementation improves the biochemical characteristics of the distal femur and femoral diaphysis in male rats chronically exposed to Cd [6]. Increased Zn consumption prevents alterations in lipid metabolism induced by Cd in male rats [7], and it reduces Cd-mediated hepatotoxicity in rats [8]. Zn influences several enzymes involved in DNA metabolism and inhibits apoptosis via its effect on transcription factors that are activated during apoptosis [9]. In general, the underlying mechanism of Zn protection as it relates to Cd toxicity could be divided into three activities: direct competition between these two metals, Zn-mediated MT induction, and Zn-mediated redox homeostasis. Zn and Cd have similar chemical structures and charges, and several studies have shown that Zn pretreatment reduces cellular Cd uptake; therefore, metal transporters are logical targets for further investigation [10,11,12]. Similarly, the roles of MT and redox homeostasis are important for reducing Cd toxicity. This review summarizes current knowledge related to the role of Zn and metal transporters in reducing Cd toxicity, discusses other relevant mechanisms, and presents potential future directions.
There is no agreement in the literature regarding how to treat Cd toxicity, and there are few human studies, which are typically anecdotal [13]. Available defense strategies against Cd are grouped according to their underlying mechanisms. These include antioxidant defense, mitochondrial protection, metal chelation, prevention of macromolecular damage, cytoskeletal rearrangements, hormetic response, co-exposure to other metals or trace elements, reduced uptake of Cd, removal of Cd, and altered toxicity of Cd by treatment with environmental factors [14]. Chelation therapy is commonly used clinically to treat metal toxicity [15]. Detoxification of Cd with EDTA and other chelators has been shown to be therapeutically beneficial in humans and animals [15]. However, chelation therapy is reported to have a number of safety and efficacy issues [16, 17]. Co-exposure to other metals or trace elements, such as Zn, is another possible therapeutic strategy. The mechanisms of Zn protection on Cd toxicity include direct competition between these two metals, Zn-mediated MT induction, and Zn-mediated redox homeostasis [14].
Zn mediated competition for sarcolemmal Cd uptake
Zn is an essential metal in biological systems. Zn was initially thought to cross cell membranes via iron channels; however, Zn transporters, some types of calcium channels, and DMT1 have been shown to mediate Zn uptake. Cd is a nonessential toxic heavy metal, and no Cd-specific transporters have been identified. Because Cd is chemically similar to Zn, both are positioned in Group 12 on the periodic table, both ions bind to identical macromolecular structures containing sulfur, oxygen, and/or nitrogen [18]. It is well established that Cd competes with Zn for transporter-mediated cellular entry [10,11,12, 19, 20]. For example, the Cd uptake rate is significantly lower after cellular exposure to Cd and Zn than it is after exposure to Cd alone (as demonstrated by Orchesella cincta, springtail insects) [11]. Zn supplementation counteracts acute Cd-induced nephrotoxicity in mice [21], and the Cd levels in cattle is inversely proportional to the levels of Zn. Another study revealed a protective role for Zn in counteracting Cd uptake and toxicity in the Madin-Darby bovine kidney epithelial cell line. The content of Cd in both the particulate and cytosolic fractions is decreased by Zn pretreatment in cultured vascular endothelial cells [22]. In Caco-2 cells, 200 μM Zn reduces Cd uptake by 55% [23]. Similarly, 30 µM Zn inhibits CM Cd uptake by 65% [10] and Zn reduces Cd uptake and accumulation in synoviocytes [24]. As discussed below, numerous sarcolemmal transporters and channels may be involved in Zn-mediated reduced Cd uptake, such as Zn transporters, calcium channels, and DMT1.
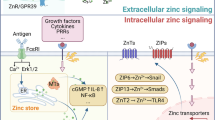
Zn transporters
The metal ion Zn is essential to all living cells and is present in both extracellular and intracellular fluids. Proteomic analysis suggests that nearly 10% of human proteins have the ability to bind Zn [25], and the average total cellular Zn content in mammalian cells is estimated to be between 100 and 250 μM [26]. Zn is a co-factor for many enzymes and is involved in forming and maintaining the correct three-dimensional conformation of proteins such as Zn-finger proteins. In addition to its structural and catalytic properties, labile or free Zn may function as a signaling ion exerting regulatory functions and as a second messenger to influence the activity of various enzymes and to control intracellular signaling pathways [27].
Because Zn is necessary for normal cellular function and is potentially toxic when in excess, homeostatic mechanisms have evolved to regulate the intracellular Zn concentration. The concentration of intracellular free Zn is regulated through the activity of Zn transporters and Zn-permeable ion channels and cysteine-rich metallothionein (MT) metal-binding proteins. Two families of eukaryotic Zn transporters have been identified: the ZIP (SLC39) and ZnT (SLC30) proteins [28]. Similar to Cd transporter (Table 1), Zn transporters have varied cellular distributions, transport activities, and tissue specificities (Table 2). ZIP proteins located in the sarcolemma increase Zn cytoplasmic concentration by importing Zn or by releasing Zn intracellularly vesicles. In contrast, ZnT proteins reduce the cytoplasmic Zn concentration by exporting Zn across the plasma membrane or sequestering Zn within intracellular vesicles.
Evidence for the role of Zn transporters in Zn uptake
Transfection of HEK293T cells with ZIP8 cDNA enhances Zn uptake by 40% [29]. ZIP8 siRNA-treated THP1 cells showed an increase in Zn content in the culture medium with a corresponding decrease within cells, suggesting that knockdown of ZIP8 reduced Zn intake [30]. ZIP14-mediated hepatic Zn accumulation was confirmed in ZIP14 KO mice and in HepG2 hepatocytes that were treated with ZIP14 siRNA [31].
Evidence for the role of Zn transporters in Cd uptake
In addition to Zn transport, both ZIP8 and ZIP14 Zn transporters have been shown to transport Cd [32]. ZIP8-mediated Cd uptake has been demonstrated by ZIP8 cRNA-injected Xenopus oocyte cultures [33] and ZIP14-mediated uptake of Cd can be competitively inhibited by Zn [34]. It is likely that other metal transporters can competitively transport both Zn and Cd, including some calcium channels and DMT1.
Evidence for the role of ZIP8 in Cd uptake
ZIP8 was originally identified in monocytes by screening cDNA transcripts that were highly induced by the Mycobacterium bovis cell wall; it was referred to as BCG-inducible gene in monocyte clone 103.10 (BIGM103) [35]. As a member of the SLC39A transporters, ZIP8 has a high affinity for divalent metal ions, including Zn2+, Fe2+, Mn2+, and the toxic heavy metal Cd. Experiments where ZIP8 capped RNAs were microinjected Xenopus oocyte cultures showed that the Km for Cd2+ is 0.48 μM, which is higher than that of Zn2+ and Fe2+ [36]. The transport characteristics of ZIP8 were further characterized using retroviral-mediated transfection of ZIP8 cDNA into mouse fetal fibroblast cultures (rvZIP8 cells), which revealed that ZIP8 is not a proton pump and is both HCO3− and energy dependent [36]. rvZIP8 cells exhibited a 10-fold increase in the rate of Cd influx and accumulation over that of control cells, and this uptake process was energy dependent and optimal at pH 7.5. To investigate Cd transport, Cd-resistant cell lines were established, including rat lung epithelial cells (LECs) [37] and Cd-resistant cells (A7 and B5), which were generated from MT-KO mouse cells [38]. These Cd-resistant cell lines displayed ZIP8 silencing and reduced cellular Cd content. ZIP8 knockdown has also been shown to be protective against Cd toxicity in a ZIP8-transfected HEK293 cell line [39] and in MT-KO mouse fibroblast cells [40]. In contrast, ZIP8 overexpression in cultured mouse fetal fibroblasts leads to a >10-fold increase in the rate of Cd influx and accumulation and a 30-fold increase in sensitivity to Cd-induced cell death [41]. The same result was observed in Xenopus oocytes [33]. Tumor necrosis factor-α (TNF-α) has been reported to induce ZIP8 expression in primary human lung epithelia and A549 cells, which then increases Cd uptake and cell death because of Cd-mediated apoptosis and necrosis [42]. The involvement of ZIP8 in Cd transport has been confirmed in multiple cell types; however, ZIP8 siRNA transfection does not alter Cd uptake in Caco-2 cells [43], suggesting that ZIP8-mediated Cd transport is cell type dependent.
In vivo studies revealed similar results. Cd treatment was found to cause acute renal failure and proximal tubular damage in mice overexpressing BTZIP8-3 but not in their nontransgenic littermates [44, 45]. Moreover, ZIP8 expression under normal conditions was found to be high in the liver, kidney, lung, and testis, which are all target organs of Cd toxicity.
Evidence for the role of ZIP14 in Cd uptake
Within ZIP protein family members, the amino acid sequences of ZIP8 and ZIP14 show the highest similarity, and ZIP14 shares a similar metal ion transport profile with ZIP8. However, ZIP14 has been investigated far less than ZIP8 and is presumed to play a less important role with respect to Cd-induced toxicity in the testis, kidney, and lung. ZIP14 was first identified as a Zn influx transporter in 2005 [46]. A role for ZIP14 in cellular Cd uptake was confirmed in mouse fetal fibroblasts and Madin-Darby canine kidney (MDCK) cells; further, ZIP14-mediated Cd uptake is proportional to cell toxicity, which can be competitively inhibited by Zn [34]. ZIP14 knockdown by siRNA transfection significantly reduces Cd uptake in proximal tubule cells [47]. Of interest, ZIP14 expression is downregulated in MT-KO Cd-resistant cells, which exhibit a decreased rate of Cd uptake [48]. Recent experiments have shown that transfection with a ZIP14 siRNA markedly decreases Cd uptake at lower Cd doses in Caco-2 cells [43].
Other Zn transporters involved in Cd transport
In addition to ZIP8 and ZIP14, several additional Zn transporters may be involved in Cd transport. HEK293 cells transduced with human ZIP2 reveal substrate selectivity in the following order: Zn2+ > Cd2+ ≥ Cu2+ > Co2+ [49]. Knockdown of ZnT3 in primary rat hippocampal neurons abolishes Zn-mediated protection from Cd-induced neurotoxicity, which indicates the involvement of ZnT3 in the mechanism of Cd-induced hippocampal neurotoxicity [50]. Another study indicated that downregulation of ZnT-1 by treatment with an siRNA resulted in regulation of the L-type Ca2+ channel, which could attenuate Cd2+ and Zn2+ permeation and alter Cd toxicity in rat hippocampal neurons [51]. While the role of Zn transporters in Cd uptake and toxicity has been well established using in vitro studies, further in vivo studies are needed to investigate the role of Zn transporters in whole organs and organisms.
The role of DMT1 in Cd toxicity
Evidence for the role of DMT1 as a Zn transporter
The divalent metal transporter 1 (DMT1), also known as NRAMP2, SLC11A2, and DCT1, is a member of the natural resistance-associated macrophage protein (Nramp) family. DMT1 was first identified in mice in 1995 [52]; it has 12 transmembrane domains, multiple membrane targeting motifs, one consensus transport motif, and two asparagine-linked glycosylation signals in an extra cytoplasmic loop. Both the N- and C-termini of the protein are located in the cytoplasm. DMT1 transports divalent cations in exchange for a single proton. DMT1 plays a crucial role in iron homeostasis and can mediate the transport of essential and toxic divalent metal ions, including Zn2+, Mn2+, Cu2+, Co2+, and Cd2+ [53]. The involvement of DMT1 in Zn transport has been shown in HEK293 cells [54] and Caco-2 cells transfected with DMT1 shRNA plasmids, which have lower Zn content compared to control cells [55]. These results confirm that DMT1 participates in active Zn transport.
Zn transported by DMT1 is likely cell type dependent, and DMT1 tends to have a lower affinity for Zn than many other metals. By using radiotracer assays and the metal-sensitive PhenGreen SK fluorophore in Xenopus oocytes, DMT1 overexpression in oocytes was shown to strongly stimulate Cd uptake, while there was only a fourfold increase in Zn uptake [56]. Recently, Shawki et al. [57] demonstrated that DMT1 is not required for the intestinal transport of Zn using a mouse model lacking intestinal DMT1. Thus, the role of DMT1 in Zn transport requires further in vitro and in vivo investigation.
Evidence for the role of DMT1 as Cd transporter
Overexpression of DMT1 in HEK293 cells was initially shown to increase time-dependent Cd uptake [58], and a follow-up study was able to increase DMT1 expression in transduced HEK293 cells by 50-fold over endogenous expression levels; the order for transport affinity in these cells was Mn2+ > Cd2+ > Fe2+ > Pb2+ ~ Co2+ ~ Ni2+ > Zn2+ [54]. Cd transport was significantly enhanced in human DMT1 RNA-injected oocytes [59] and in Caco-2 cells [60, 61], and it was suppressed in Caco-2 cells using DMT1 siRNA knockdown [62].
The role of calcium channels in Cd toxicity
Due to the similar divalent cationic properties of Ca2+, Cd2+, and Zn2+, ion channels are obvious candidates for Zn and Cd transport [63, 64]. Zn can permeate through Ca-transporting channels that belong to various families, including the voltage-gated calcium channels (VGCCs), NMDA receptors (which are primarily in neural tissues) [65], nicotinic acetylcholine receptors, and more recently, members of the transient receptor potential (TRP) superfamily [66]. The VGCC and TRP superfamilies have been more intensively studied than the others and deserve further discussion.
Evidence for the role of VGCCs as Zn transporters
In mammals, the VGCC family consists of 10 members that are divided into three subfamilies: Cav1, Cav2, and Cav3. The Cav1 subfamily members, more commonly known as L-type Ca2+ channels (LTCCs), are predominantly expressed in the heart, skeletal muscle, neurons, and endocrine cells. The Cav3 subfamily members (TTCCs) are expressed in the heart, liver, kidney, and in neurons [67]. Although VGCCs primarily transport Ca2+, they may also transport other divalent ions, including Zn [68] and Cd [69]. Zn can enter murine cortical neurons via L- and N-type voltage-gated Ca2+ channels [68], and L-type VGCC blockers (verapamil, nitrendipine, and nifedipine) inhibit Zn uptake in stimulated β-cells [70].
Evidence for the role of the TRP superfamily as Zn transporters
In mammals, the TRP superfamily includes 27 cation channels that are subdivided into six subfamilies based on sequence similarity: TRPA (ankyrin-rich protein), TRPC (canonical), TRPM (Melastin), TRPML (mucolipins), TRPP (Policystin), and TRPV (vanilloid). TRP is widely expressed in many tissues, including the intestine, kidney, placenta, and heart [71], and these channels are known to be the major pathways for Cd transport [72]. TRPV5 (also called epithelial Ca2+ channel, ECaC) and TRPV6 (also called CaT1) were reported in 1999 [73, 74] and are the two major Cd transporters in this superfamily. Several members of the TRP superfamily are Zn permeable, including TRPV5, TRPV6, TRPM3, TRPM6/7, TRPML1, and TRPA1 [66]. The overexpression of human TRPV6 in HEK293 and MCF-7 cells increases permeability for Zn2+, Cd2+, La3+, and Gd3+ [75], suggesting that TRPV6 is involved not only in Zn transport but also in the transport of other divalent heavy metal ions. HEK293 cells transfected with pTagRFP-C1-hTRPV5 or pTagRFP-C1-hTRPV6 exhibited increased Zn toxicity [72].
Evidence for the role of VGCCs as Cd transporters
The most important Ca2+ channels relevant to Cd transport are the VGCCs. While Cd entry through L-type channels in pituitary GH cells is very low relative to the levels of Ca2+ transport, this route of uptake is relevant to overall Cd toxicity, and Ca2+ channel blockers protect against Cd toxicity [76]. Increased Ca2+ in the renal tubular lumen decreases Cd accumulation in rabbit tubular cells, validating a role for Ca2+ transporters in renal Cd uptake [77]. Nimodipine, a VGCC antagonist, protects GH4C1 cells against Cd toxicity and increases the LD50 of CdCl2 from 15 to 45 μM, whereas the VGCC agonist BAY K8644 decreases the LD50 of CdCl2. These data indicate that transport of Cd through dihydropyridine-sensitive VGCCs is a major mechanism for Cd uptake and that pharmacologic VGCC blockade can provide protection against Cd toxicity [76]. In the hepatic human cell line WRL-68, the VGCC blockers nifedipine and verapamil inhibit Cd uptake by only 35%, suggesting that in these cells, VGCCs are not the predominant mechanism for cellular Cd entry [78]. In primary rat hepatocytes, diltiazem, verapamil, nifedipine, and nitrendipine inhibit Cd uptake with a 30% maximal decrease in Cd uptake [79]. In HeLa cells lacking VGCCs, Cd was unable to induce apoptosis or cellular toxicity [80], and mibefradil inhibition of T-type VGCCs reduces Cd uptake in a Cd-resistant cell line [81]. In cardiomyocytes, verapamil was shown to be the most effective Ca2+ channel antagonist, inhibiting Cd accumulation by 76%; the next most effective agents were nitrendipine (52%), diltiazem (50%), and nifedipine (29%) [10]. However, verapamil does not inhibit Cd uptake in Caco-2 cells, suggesting that there is VGCC-independent Cd uptake in these cells [23]. Nimodipine does not protect LLC-PK1 (kidney proximal tubule) or neuroblastoma cell lines from Cd toxicity [76], and it only provides partial protection in Chinese hamster ovary cells [80]. Thus, it appears that Cd uptake through Ca2+ channels varies with cell type and context, suggesting that VGCCs are involved in one pathway for Cd transport and toxicity, but there are other pathways.
Evidence for the role of the TRP superfamily as Cd transporters
The roles of TRPV6 and TRPV5 in Cd transport have been well characterized in vitro and in vivo. Mice fed a low-Ca diet showed higher intestinal mRNA levels of TRPV6 and exhibited a corresponding increase in hepatic and renal Cd accumulation, suggesting that TRPV6 may stimulate intestinal Cd absorption [82]. HEK293 cells transfected with pTagRFP-C1-hTRPV5 or pTagRFP-C1-hTRPV6 become sensitized to Cd [72], which can be inhibited by the nonspecific human TRPV6 inhibitor 2-aminoethoxydiphenyl borate (2-APB); these data support the idea that TRPV5 and TRPV6 have roles in controlling human cellular Cd uptake.
The role of metallothionein induction by Zn in reducing Cd toxicity
MTs are small cysteine-rich proteins capable of binding up to 7–12 metal ions per MT molecule; they were first described in 1957 as Cd-binding proteins in horse kidney cells [83]. MTs play an essential role in the cellular stress response, in maintaining the homeostasis of essential metals, and in the sequestration and detoxification of Cd and other heavy metals. In mammals, MTs are mainly cytoplasmic but can also be detected in lysosomes, mitochondria, and nuclei. Four MT isoforms, designated MT-1 to MT-4, have been identified, and MT-1 and MT-2 are the predominant isoforms that are expressed in most tissues [84]. MT-3 is constitutively expressed in the central nervous system, and MT-4 is primarily expressed in stratified squamous epithelium [84]. A broad range of cellular stressors, hormones, reactive oxygen species (ROS), and cytokines induce MT gene transcription [85]. Heavy metals rapidly induce MT-1 and MT-2 transcription via metal-regulatory transcription factor 1 (MTF-1) binding to metal-responsive elements (MREs) within promoter regions [86]. MTs are efficient scavengers of free radicals generated by oxidative stress [87], and MT-overexpressing and MT-KO mouse models provide validation of the role of MT in response to Cd exposure [88, 89]. Wild-type mice develop substantial Cd tolerance and a sevenfold higher LD50 than MT-KO mice [90]. Intrauterine Cd is toxic, and MT-KO pups exposed to Cd are growth restricted, confirming both a prenatal and postnatal role for MT as a major protein required for protection from Cd toxicity [91]. Repeated Cd dosing in MT-KO mice results in nephrotoxicity at one-tenth the dose required for nephrotoxicity in control mice [92], while MT overexpression protects against acute Cd lethality and hepatotoxicity [93]. In vitro engineered cardiac tissues (ECTs) generated using wild-type and MT-overexpressing neonatal cardiomyocytes display acute Cd toxicity and a partially protective role for cardiac-specific MT overexpression on Cd-induced apoptosis [88].
Cd toxicity can also be ameliorated by MT inducers, most notably Zn [92, 94, 95]. One mechanism for Zn-induced Cd tolerance is the Zn-mediated induction of MT synthesis [96]. Zn pretreatment increases MT expression and enhances resistance to Cd in MT-WT renal cells but not in Zn pretreated MT-1/MT-2 KO renal cells [97]; further, Zn protection from Cd toxicity via MT induction occurs in other tissues [95, 98]. The partial protective effect of Zn in MT-KO-derived ECTs confirms the presence of non-MT-mediated pathways in Zn-mediated protection from Cd toxicity, including direct competition for Cd and Zn for cellular uptake in bovine vascular endothelial cells [99], stellate cells [100], and murine neonatal ECTs [101]. Some animal models show a Zn-mediated protective effect related to Cd uptake and toxicity in the absence of MT induction [11] or in MT-KO mice [95]. MT induction may be directly related to Zn dosing with a dosing threshold required for MT induction [99]. Thus, the interdependence of MT and Zn on reducing Cd toxicity is likely dependent on the dose, sarcolemmal- and intracellular-specific mechanisms, and cell type.
Zn may reduce Cd toxicity by reducing ROS
ROS play an important role in Cd toxicity. The pathways involved in Cd-induced ROS are complex and multifactorial and have been well reviewed in the literature [102, 103]. The mechanisms of Cd-induced ROS can be summarized as follows. First, Cd liberates redox-active metals such as iron and copper from tightly regulated storage. Cd then displaces iron from ferritin, which in turn increases the concentrations of unbound iron ions. These free ions cause oxidative stress [104]. Second, Cd inhibits the electron transport chain, resulting in uncoupled electron flow and ROS formation [105]. Third, Cd depletes antioxidant scavengers. Glutathione (GSH) is a primary target of Cd that interacts with GSH by binding the cysteine thiolate anion. Reduced GSH activity further disturbs the cellular redox balance, which leads to enhanced production of ROS [106, 107]. Finally, Cd exposure suppresses antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) [105].
Zn is a potent antioxidant [108]. Zn plays an important role in reducing levels of ROS by several mechanisms. First, Zn acts as a co-factor for important enzymes that contribute to the proper functioning of the antioxidant defense system. Second, Zn promotes the stabilization of membranes and inhibits the enzyme nicotinamide adenine dinucleotide phosphate oxidase, a pro-oxidant enzyme. Finally, Zn can induce metallothionein synthesis, which is involved in the reduction of hydroxyl radicals (OH) and in the sequestration of the ROS produced under stress conditions [109].
Overall, ROS play an important role in Cd toxicity, and Zn has a major role in attenuating the increase in ROS [108], so it is reasonable that Zn could protect cells from Cd toxicity by suppressing ROS [110,111,112]. Zn upregulates antioxidant pathways (the Nrf2 pathway) and target genes (sod1, cat, and mt2) [113], and Zn-induced protection against Cd cytotoxicity in hepatic stellate cells occurs via maintenance of normal GSH, catalase, and GSH peroxidase activities [100]. In addition, similar results have been noted in PC12 cells [114]. The requirement in MT-KO mice of normal GSH status for Zn protection against Cd-induced renal damage may provide insight into MT-independent pathways for Zn-mediated Cd tolerance [115].
Conclusion
Zn is known to reduce Cd toxicity via direct competition for Cd uptake, induction of MT, and reduction of detrimental ROS. Various sarcolemmal transport systems have been identified to play a role in Cd accumulation, including Zn transporters, DMT1, and calcium channels, all of which may represent targets that could enhance protection from Cd. Many of the published studies on Cd toxicity and Zn countermeasures come from in vitro cell lines or small animal models. A broader range of cell types and more complex in vitro tissue culture systems as well as expanded in vivo studies are required to further confirm and characterize the contribution of cell- and organ-specific transport systems to Cd transport and Cd-induced toxicity and to test current FDA-approved and novel Cd countermeasures. Phase I and Phase II clinical trials are needed to determine the safety of Zn treatment in healthy individuals, which should be followed by limited cohort studies to determine the efficacy of Zn treatment in the reduction of Cd toxicity.
References
Zhang H, Reynolds M. Cadmium exposure in living organisms: a short review. Sci Total Environ. 2019;678:761–7.
Porru S, Alessio L. The use of chelating agents in occupational lead poisoning. Occup Med (Oxf, Engl). 1996;46:41–8.
Aposhian HV, Maiorino RM, Gonzalez-Ramirez D, Zuniga-Charles M, Xu Z, Hurlbut KM, et al. Mobilization of heavy metals by newer, therapeutically useful chelating agents. Toxicology. 1995;97:23–38.
Zhai Q, Narbad A, Chen W. Dietary strategies for the treatment of cadmium and lead toxicity. Nutrients. 2014;7:552–71.
Szuster-Ciesielska A, Stachura A, Slotwinska M, Kaminska T, Sniezko R, Paduch R, et al. The inhibitory effect of zinc on cadmium-induced cell apoptosis and reactive oxygen species (ROS) production in cell cultures. Toxicology. 2000;145:159–71.
Brzoska MM, Galazyn-Sidorczuk M, Rogalska J, Roszczenko A, Jurczuk M, Majewska K, et al. Beneficial effect of zinc supplementation on biomechanical properties of femoral distal end and femoral diaphysis of male rats chronically exposed to cadmium. Chem-Biol Interact. 2008;171:312–24.
Rogalska J, Brzoska MM, Roszczenko A, Moniuszko-Jakoniuk J. Enhanced zinc consumption prevents cadmium-induced alterations in lipid metabolism in male rats. Chem-Biol Interact. 2009;177:142–52.
Zhou J, Wu C, Tu J, Ling Y, Hu N, Zhang Y, et al. Assessment of cadmium-induced hepatotoxicity and protective effects of zinc against it using an improved cell-based biosensor. Sens Actuators A Phys. 2013;199:156–64.
Franklin RB, Costello LC. The important role of the apoptotic effects of zinc in the development of cancers. J Cell Biochem. 2010;106:750–7.
Limaye DA, Shaikh ZA. Cytotoxicity of cadmium and characteristics of its transport in cardiomyocytes. Toxicol Appl Pharmacol. 1999;154:59–66.
Sterenborg I, Vork NA, Verkade SK, van Gestel CA, van Straalen NM. Dietary zinc reduces uptake but not metallothionein binding and elimination of cadmium in the springtail, Orchesella cincta. Env Toxicol Chem. 2010;22:1167–71.
Zhang D, Liu J, Gao J, Shahzad M, Han Z, Wang Z, et al. Zinc supplementation protects against cadmium accumulation and cytotoxicity in madin-darby bovine kidney cells. PLoS ONE. 2014;9:e103427.
Bernhoft RA. Cadmium toxicity and treatment. Sci World J. 2013;2013:394652.
Sandbichler AM, Höckner M. Cadmium protection strategies—a hidden trade-off? Int J Mol Sci. 2016;17:139.
Kim J-J, Kim Y-S, Kumar V. Heavy metal toxicity: an update of chelating therapeutic strategies. J Trace Elem Med Biol. 2019;54:226–31.
Ding T, Luo JY, Yang SH, Yang MH. Recent research progress on natural medicines in treatment of cadmium toxicity. Zhongguo Zhong Yao Za Zhi. 2018;43:2006–13.
Gilman A, Philips FS. The treatment of acute cadmium intoxication in rabbits with 2,3-dimercaptopropanol (BAL) and other mercaptans. J Pharmacol Exp Ther. 1946;87:85–101.
Krężel A, Maret W. The biological inorganic chemistry of zinc ions. Arch Biochem Biophys. 2016;611:3–19.
Kaji T, Takata M, Hoshino T, Miyahara T, Kozuka H, Kurashige Y, et al. Role of zinc in protection against cadmium-induced toxicity in formation of embryonic chick bone in tissue culture. Toxicol Lett. 1988;44:219–27.
Pabis K, Gundacker C, Giacconi R, Basso A, Costarelli L, Piacenza F, et al. Zinc supplementation can reduce accumulation of cadmium in aged metallothionein transgenic mice. Chemosphere. 2018;211:855–60.
Tang W, Sadovic S, Shaikh ZA. Nephrotoxicity of cadmium-metallothionein: protection by zinc and role of glutathione. Toxicol Appl Pharmacol. 1998;151:276–82.
Kaji T, Mishima A, Koyanagi E, Yamamoto C, Sakamoto M, Kozuka H. Possible mechanism for zinc protection against cadmium cytotoxicity in cultured vascular endothelial cells. Toxicology. 1992;76:257–70.
Blais A, Lecoeur S, Milhaud G, Tome D, Kolf-Clauw M. Cadmium uptake and transepithelial transport in control and long-term exposed Caco-2 cells: the role of metallothionein. Toxicol Appl Pharmacol. 1999;160:76–85.
Bonaventura P, Lamboux A, Albarede F, Miossec P. Regulatory effects of zinc on cadmium-induced cytotoxicity in chronic inflammation. PLoS ONE. 2017;12:e0180879.
Guantario B, Capolupo A, Monti MC. Proteomic analysis of Zn depletion/repletion in the hormone-secreting thyroid follicular cell line FRTL-5. Nutrients. 2018;10. https://doi.org/10.3390/nu10121981.
Wu W, Bromberg PA, Samet JM. Zinc ions as effectors of environmental oxidative lung injury. Free Radic Biol Med. 2013;65:57–69.
Yamasaki S, Sakata-Sogawa K, Hasegawa A, Suzuki T, Kabu K, Sato E, et al. Zinc is a novel intracellular second messenger. J Cell Biol. 2007;177:637–45.
Ohashi W, Hara T, Takagishi T, Hase K, Fukada T. Maintenance of Intestinal epithelial homeostasis by zinc transporters. Dig Dis Sci. 2019;64:2404–15.
Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, et al. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem. 2012;287:34032–43.
Liu MJ, Bao S, Galvez-Peralta M, Pyle CJ, Rudawsky AC, Pavlovicz RE, et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Rep. 2013;3:386–400.
Kim MH, Aydemir TB, Kim J, Cousins RJ. Hepatic ZIP14-mediated zinc transport is required for adaptation to endoplasmic reticulum stress. Proc Natl Acad Sci USA. 2017;114:E5805–e14.
Thevenod F, Fels J. Channels, transporters and receptors for cadmium and cadmium complexes in eukaryotic cells: myths and facts. Biometals. 2019;32:469–89.
Liu Z, Li H, Soleimani M, Girijashanker K, Reed JM, He L, et al. Cd2+ versus Zn2+ uptake by the ZIP8 HCO3-dependent symporter: kinetics, electrogenicity and trafficking. Biochem Biophys Res Commun. 2008;365:814–20.
Girijashanker K, He L, Soleimani M, Reed JM, Li H, Liu Z, et al. Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter. Mol Pharmacol. 2008;73:1413–23.
Begum NA, Kobayashi M, Moriwaki Y, Matsumoto M, Toyoshima K, Seya T. Mycobacterium bovis BCG cell wall and lipopolysaccharide induce a novel gene, BIGM103, encoding a 7-TM protein: identification of a new protein family having Zn-transporter and Zn-metalloprotease signatures. Genomics. 2002;80:630–45.
He L, Girijashanker K, Dalton TP, Reed J, Li H, Soleimani M, et al. ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties. Mol Pharmacol. 2006;70:171–80.
Gao Y, Xu Y, Wu D, Yu F, Yang L, Yao Y, et al. Progressive silencing of the zinc transporter Zip8 (Slc39a8) in chronic cadmium-exposed lung epithelial cells. Acta Biochem Biophys Sin. 2017;49:444–9.
Yanagiya T, Imura N, Kondo Y, Himeno S. Reduced uptake and enhanced release of cadmium in cadmium-resistant metallothionein null fibroblasts. Life Sci. 1999;65:Pl177–82.
Martin P, Boulukos KE, Poggi MC, Pognonec P. Long-term extracellular signal-related kinase activation following cadmium intoxication is negatively regulated by a protein kinase C-dependent pathway affecting cadmium transport. FEBS J. 2009;276:1667–79.
Fujishiro H, Okugaki S, Kubota K, Fujiyama T, Miyataka H, Himeno S. The role of ZIP8 down-regulation in cadmium-resistant metallothionein-null cells. J Appl Toxicol. 2009;29:367–73.
Dalton TP, He L, Wang B, Miller ML, Jin L, Stringer KF, et al. Identification of mouse SLC39A8 as the transporter responsible for cadmium-induced toxicity in the testis. Proc Natl Acad Sci U S A. 2005;102:3401–6.
Napolitano JR, Liu MJ, Bao S, Crawford M, Nana-Sinkam P, Cormet-Boyaka E, et al. Cadmium-mediated toxicity of lung epithelia is enhanced through NF-kappaB-mediated transcriptional activation of the human zinc transporter ZIP8. Am J Physiol Lung Cell Mol Physiol. 2012;302:L909–18.
Fujishiro H, Hamao S, Tanaka R, Kambe T, Himeno S. Concentration-dependent roles of DMT1 and ZIP14 in cadmium absorption in Caco-2 cells. J Toxicol Sci. 2017;42:559–67.
Wang B, He L, Dong H, Dalton TP, Nebert DW. Generation of a Slc39a8 hypomorph mouse: markedly decreased ZIP8 Zn(2)(+)/(HCO(3)(−))(2) transporter expression. Biochem Biophys Res Commun. 2011;410:289–94.
Wang B, Schneider SN, Dragin N, Girijashanker K, Dalton TP, He L, et al. Enhanced cadmium-induced testicular necrosis and renal proximal tubule damage caused by gene-dose increase in a Slc39a8-transgenic mouse line. Am J Physiol Cell Physiol. 2007;292:C1523–35.
Taylor KM, Morgan HE, Johnson A, Nicholson RI. Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14. FEBS Lett. 2005;579:427–32.
Fujishiro H, Yano Y, Takada Y, Tanihara M, Himeno S. Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics. 2012;4:700–8.
Fujishiro H, Okugaki S, Nagao S, Satoh M, Himeno S. Characterization of gene expression profiles of metallothionein-null cadmium-resistant cells. Eisei Kagaku. 2006;52:292–9.
Franz MC, Pujol-Gimenez J. Reassessment of the transport mechanism of the human zinc transporter SLC39A2. Biochemistry. 2018;57:3976–86.
Ben Mimouna S, Le Charpentier T, Lebon S, Van Steenwinckel J, Messaoudi I, Gressens P. Involvement of the synapse-specific zinc transporter ZnT3 in cadmium-induced hippocampal neurotoxicity. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.28245.
Ohana E, Sekler I, Kaisman T, Kahn N, Cove J, Silverman WF, et al. Silencing of ZnT-1 expression enhances heavy metal influx and toxicity. J Mol Med (Berl). 2006;84:753–63.
Gruenheid S, Cellier M, Vidal S, Gros P. Identification and characterization of a second mouse Nramp gene. Genomics. 1995;25:514–25.
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–8.
Garrick MD, Singleton ST, Vargas F, Kuo HC, Zhao L, Knopfel M, et al. DMT1: which metals does it transport? Biol Res. 2006;39:79–85.
Espinoza A, Le Blanc S, Olivares M, Pizarro F, Ruz M, Arredondo M. Iron, copper, and zinc transport: inhibition of divalent metal transporter 1 (DMT1) and human copper transporter 1 (hCTR1) by shRNA. Biol Trace Elem Res. 2012;146:281–6.
Illing AC, Shawki A, Cunningham CL, Mackenzie B. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J Biol Chem. 2012;287:30485–96.
Shawki A, Anthony SR, Nose Y, Engevik MA, Niespodzany EJ, Barrientos T, et al. Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am J Physiol Gastrointest Liver Physiol. 2015;309:G635.
Olivi L, Sisk J, Bressler J. Involvement of DMT1 in uptake of Cd in MDCK cells: role of protein kinase C. Am J Physiol Cell Physiol. 2001;281:C793–800.
Okubo M, Yamada K, Hosoyamada M, Shibasaki T, Endou H. Cadmium transport by human Nramp 2 expressed in Xenopus laevis oocytes. Toxicol Appl Pharmacol. 2003;187:162–7.
Tallkvist J, Bowlus CL, Lonnerdal B. DMT1 gene expression and cadmium absorption in human absorptive enterocytes. Toxicol Lett. 2001;122:171–7.
Jumarie C, Campbell PG, Houde M, Denizeau F. Evidence for an intracellular barrier to cadmium transport through Caco-2 cell monolayers. J Cell Physiol. 1999;180:285–97.
Bannon DI, Abounader R, Lees PS, Bressler JP. Effect of DMT1 knockdown on iron, cadmium, and lead uptake in Caco-2 cells. Am J Physiol Cell Physiol. 2003;284:C44–50.
Palmiter RD, Findley SD. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995;14:639–49.
Inoue K, O’Bryant Z, Xiong ZG. Zinc-permeable ion channels: effects on intracellular zinc dynamics and potential physiological/pathophysiological significance. Curr Med Chem. 2015;22:1248–57.
Dolino DM, Chatterjee S, Maclean DM, Flatebo C, Bishop L, Shaikh SA, et al. The structure-energy landscape of NMDA receptor gating. Nat Chem Biol. 2017;13:1232–8.
Bouron A, Oberwinkler J. Contribution of calcium-conducting channels to the transport of zinc ions. Pflug Arch. 2014;466:381–7.
Gonzalez-Gutierrez G, Miranda-Laferte E, Contreras G, Neely A, Hidalgo P. Swapping the I-II intracellular linker between L-type CaV1.2 and R-type CaV2.3 high-voltage gated calcium channels exchanges activation attributes. Channels (Austin). 2010;4:42–50.
Kerchner GA, Canzoniero LM, Yu SP, Ling C, Choi DW. Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurons. J Physiol. 2000;528 Pt 1:39–52.
Fukuda J, Kawa K. Permeation of manganese, cadmium, zinc, and beryllium through calcium channels of an insect muscle membrane. Science. 1977;196:309–11.
Gyulkhandanyan AV, Lee SC, Bikopoulos G, Dai F, Wheeler MB. The Zn2+-transporting pathways in pancreatic beta-cells: a role for the L-type voltage-gated Ca2+ channel. J Biol Chem. 2006;281:9361–72.
Alonso-Carbajo L, Kecskes M, Jacobs G, Pironet A, Vennekens R. Muscling in on TRP channels in vascular smooth muscle cells and cardiomyocytes. Cell Calcium. 2017;66:48–61.
Kovacs G, Montalbetti N, Franz MC, Graeter S, Simonin A, Hediger MA. Human TRPV5 and TRPV6: key players in cadmium and zinc toxicity. Cell Calcium. 2013;54:276–86.
Peng JB, Chen XZ, Berger UV, Vassilev PM, Tsukaguchi H, Brown EM, et al. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J Biol Chem. 1999;274:22739–46.
Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, et al. Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem. 1999;274:8375–8.
Kovacs G, Danko T, Bergeron MJ, Balazs B, Suzuki Y, Zsembery A, et al. Heavy metal cations permeate the TRPV6 epithelial cation channel. Cell Calcium. 2011;49:43–55.
Hinkle PM, Kinsella PA, Osterhoudt KC. Cadmium uptake and toxicity via voltage-sensitive calcium channels. J Biol Chem. 1987;262:16333–7.
Wang Y, Zalups RK, Barfuss DW. Potential mechanisms involved in the absorptive transport of cadmium in isolated perfused rabbit renal proximal tubules. Toxicol Lett. 2010;193:61–8.
Souza V, Bucio L, Gutierrez-Ruiz MC. Cadmium uptake by a human hepatic cell line (WRL-68 cells). Toxicology. 1997;120:215–20.
Blazka ME, Shaikh ZA. Differences in cadmium and mercury uptakes by hepatocytes: role of calcium channels 1. Toxicol Appl Pharmacol. 1991;110:355–63.
Marchetti C. Role of calcium channels in heavy metal toxicity. ISRN Toxicol. 2013;2013:184360.
Leslie EM, Liu J, Klaassen CD, Waalkes MP. Acquired cadmium resistance in metallothionein-I/II(−/−) knockout cells: role of the T-type calcium channel Cacnalpha1G in cadmium uptake. Mol Pharmacol. 2006;69:629–39.
min KS, Ueda H, Tanaka K. Involvement of intestinal calcium transporter 1 and metallothionein in cadmium accumulation in the liver and kidney of mice fed a low-calcium diet. Toxicol Lett. 2008;176:85–92.
Margoshes M, Vallee BL. A cadmium protein from equine kidney cortex. J Am Chem Soc. 1957;79:4813–4.
Si M, Lang J. The roles of metallothioneins in carcinogenesis. J Hematol Oncol. 2018;11:107.
Ziller A, Fraissinet-Tachet L. Metallothionein diversity and distribution in the tree of life: a multifunctional protein. Metallomics. 2018;10:1549–59.
Ben Mimouna S, Boughammoura S, Chemek M, Haouas Z, Banni M, Messaoudi I. Disruption of the zinc metabolism in rat foetal brain after prenatal exposure to cadmium. Chem-Bio Interact. 2018;286:88–95.
Rios C, Santander I, Mendez-Armenta M. Metallothionein-I + II reduces oxidative damage and apoptosis after traumatic spinal cord injury in rats. Oxid Med Cell Longev. 2018:3265918. https://doi.org/10.1155/2018/3265918.
Yu H, Ye F, Yuan F, Cai L, Ji H, Keller BB. Neonatal murine engineered cardiac tissue toxicology model: impact of metallothionein overexpression on cadmium-induced injury. Toxicol Sci. 2018;165:499–511.
Akiyama M, Shinkai Y, Unoki T, Shim I, Ishii I, Kumagai Y. The capture of cadmium by reactive polysulfides attenuates cadmium-induced adaptive responses and hepatotoxicity. Chem Res Toxicol. 2017;30:2209–17.
Park JD, Liu Y, Klaassen CD. Protective effect of metallothionein against the toxicity of cadmium and other metals. Toxicology. 2001;163:93–100.
Selvaratnam J, Guan H, Koropatnick J, Yang K. Metallothionein-I- and -II-deficient mice display increased susceptibility to cadmium-induced fetal growth restriction. Am J Physiol Endocrinol Metab. 2013;305:E727–35.
Klaassen CD, Liu J, Choudhuri S. Metallothionein: an intracellular protein to protect against cadmium toxicity. Rev Pharmacol Toxicol. 1999;39:267–94.
Liu Y, Liu J, Iszard MB, Andrews GK, Palmiter RD, Klaassen CD. Transgenic mice that overexpress metallothionein-I are protected from cadmium lethality and hepatotoxicity. Toxicol Appl Pharmacol. 1995;135:222–8.
Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–20.
Liu J, Liu Y, Michalska AE, Choo KH, Klaassen CD. Metallothionein plays less of a protective role in cadmium-metallothionein-induced nephrotoxicity than in cadmium chloride-induced hepatotoxicity. J Pharmacol Exp Ther. 1996;276:1216–23.
Leber AP, Miya TS. A mechanism for cadmium- and zinc-induced tolerance to cadmium toxicity: involvement of metallothionein. Toxicol Appl Pharmacol. 1976;37:403–14.
Kennette W, Collins OM, Zalups RK, Koropatnick J. Basal and zinc-induced metallothionein in resistance to cadmium, cisplatin, zinc, and tertbutyl hydroperoxide: studies using MT knockout and antisense-downregulated MT in mammalian cells. Toxicol Sci. 2005;88:602–13.
Urani C, Melchioretto P, Canevali C, Crosta GF. Cytotoxicity and induction of protective mechanisms in HepG2 cells exposed to cadmium. Toxicol In Vitro. 2005;19:887–92.
Mishima A, Yamamoto C, Fujiwara Y, Kaji T. Tolerance to cadmium cytotoxicity is induced by zinc through non-metallothionein mechanisms as well as metallothionein induction in cultured cells. Toxicology. 1997;118:85–92.
Souza V, Escobar Mdel C, Bucio L, Hernandez E, Gutierrez-Ruiz MC. Zinc pretreatment prevents hepatic stellate cells from cadmium-produced oxidative damage. Cell Biol Toxicol. 2004;20:241–51.
Yu HT, Zhen J, Xu JX, Cai L, Leng JY, Ji HL, et al. Zinc protects against cadmium-induced toxicity in neonatal murine engineered cardiac tissues via metallothionein-dependent and independent mechanisms. Acta Pharmacol Sin. 2019;40. https://doi.org/10.1038/s41401-019-0320-y. in press.
Kukongviriyapan U, Apaijit K, Kukongviriyapan V. Oxidative stress and cardiovascular dysfunction associated with cadmium exposure: beneficial effects of curcumin and tetrahydrocurcumin. Tohoku J Exp Med. 2016;239:25–38.
Mohajeri M, Rezaee M, Sahebkar A. Cadmium-induced toxicity is rescued by curcumin: a review. Biofactors. 2017;43:645–61.
Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117.
Wang Y, Fang J, Leonard SS, Rao KMK. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med. 2004;36:1434–43.
Ren L, Qi K, Zhang L, Bai Z, Ren C, Xu X, et al. Glutathione might attenuate cadmium-induced liver oxidative stress and hepatic stellate cell activation. Biol Trace Elem Res. 2019;191:443–52.
Corticeiro S, Freitas R, Figueira E. The role of GSTs in the tolerance of Rhizobium leguminosarum to cadmium. Biometals. 2013;26:879–86.
Maret W. The redox biology of redox-inert zinc ions. Free Radic Biol Med. 2019;134:311–26.
Marreiro DDN, Cruz KJC, Morais JBS, Beserra JB, Severo JS, de Oliveira ARS. Zinc and oxidative stress: current mechanisms. Antioxidants. 2017;6:24.
Brzoska MM, Rogalska J. Protective effect of zinc supplementation against cadmium-induced oxidative stress and the RANK/RANKL/OPG system imbalance in the bone tissue of rats. Toxicol Appl Pharmacol. 2013;272:208–20.
Zhang D, Li Y, Zhang T, Liu J, Jahejo AR, Yang L, et al. Protective effects of zinc and N-acetyl-L-cysteine supplementation against cadmium induced erythrocyte cytotoxicity in Arbor Acres broiler chickens (Gallus gallus domesticus). Ecotoxicol Env Saf. 2018;163:331–9.
Pan J, Huang X, Li Y, Li M, Yao N, Zhou Z, et al. Zinc protects against cadmium-induced toxicity by regulating oxidative stress, ions homeostasis and protein synthesis. Chemosphere. 2017;188:265–73.
Wang CC, Si LF, Guo SN, Zheng JL. Negative effects of acute cadmium on stress defense, immunity, and metal homeostasis in liver of zebrafish: the protective role of environmental zinc dpre-exposure. Chemosphere. 2019;222:91–7.
Rahman MM, Ukiana J, Uson-Lopez R, Sikder MT, Saito T, Kurasaki M. Cytotoxic effects of cadmium and zinc co-exposure in PC12 cells and the underlying mechanism. Chem Biol Interact. 2017;269:41–9.
Tang W, Sadovic S, Shaikh ZA. Nephrotoxicity of cadmium-metallothionein: protection by zinc and role of glutathione. Toxicol Appl Pharmacol. 1998;151:276–82.
Fujishiro H, Kubota K, Inoue D, Inoue A, Yanagiya T, Enomoto S, et al. Cross-resistance of cadmium-resistant cells to manganese is associated with reduced accumulation of both cadmium and manganese. Toxicology. 2011;280:118–25.
Acknowledgements
All cited work from the authors was supported in part by the Kosair Charities Pediatric Heart Research Fund to B.B.K., the US-China Pediatric Research Exchange Training Program to L.C. and B.B.K., the First Hospital of Jilin University Youth Fund (JDYY92018031) to J.Z., the Scientific and Technological Development Program of Jilin Province (20190701067GH) to J.Y.L., the Jilin Province Department of Finance Fund (2018SCZWSZX-005) to J.Y.L., and the National Natural Science Foundation of China (81974027) to H.L.J. All personnel expenses and partial research-related expenses for J.Z. and H.T.Y. were provided by Jilin University through a collaborative research agreement between the University of Louisville and Jilin University, Changchun, China.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Yu, Ht., Zhen, J., Leng, Jy. et al. Zinc as a countermeasure for cadmium toxicity. Acta Pharmacol Sin 42, 340–346 (2021). https://doi.org/10.1038/s41401-020-0396-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0396-4
Keywords
This article is cited by
-
Ameliorative Effects of Zn and Se Supplementation on Heavy Metal Mixture Burden via Increased Renal Metal Excretion and Restoration of Redoxo-Inflammatory Alterations
Biological Trace Element Research (2024)
-
BDNF and KISS-1 Levels in Maternal Serum, Umbilical Cord, and Placenta: The Potential Role of Maternal Levels as Effect Biomarker
Exposure and Health (2024)
-
A State-of-the-Science Review on Metal Biomarkers
Current Environmental Health Reports (2023)
-
Occurrence of 8 trace elements in Rhizoma Cibotii from China and exposure assessment
Environmental Science and Pollution Research (2023)
-
Zinc and selenium attenuate quaternary heavy metal mixture-induced testicular damage via amplification of the antioxidant system, reduction in metal accumulation, inflammatory and apoptotic biomarkers
Toxicological Research (2023)
