
Abstract
Sirtuin 6 (SIRT6) has been reported to play a key role in cognitive function and mood regulation, yet its role in mood disorders is not completely understood. Here, we confirmed that knockdown of hippocampal SIRT6 alleviated depression-like behaviors induced by chronic unpredictable stress (CUS) in mice. Our in vitro data showed that SIRT6 negatively regulated protein kinase B (AKT) signaling by deacetylating histone 3 at Lys9 and Lys56. Knockdown of SIRT6 significantly increased AKT phosphorylation activity, while decreased collapsin response mediator protein 2 (CRMP2) phosphorylation activity. Furthermore, pharmacologic inhibition of SIRT6 by ferulic acid (FA) (40 or 80 mg· kg−1 per day, i.g.) could activate AKT/CRMP2 pathway in vitro, which has been proved to exert an antidepressant-like effect on CUS-induced depressive models. In conclusion, our study suggested that hippocampal SIRT6 contributes to the performance of depression-like behaviors by suppressing AKT/CRMP2 signaling, and FA ameliorates CUS-induced depression-like behaviors in mice as a potential pharmacologic inhibitor of SIRT6.
Similar content being viewed by others


Introduction
Major depression is a highly prevalent psychiatric disorder with causing severe burden on the family and society [1]. Stress plays an important role in the pathophysiology and pathogenesis of major depression disorder (MDD) [2,3,4]. However, the current understanding of the etiology of MDD is incomplete and the mechanisms remain largely unknown. Due to the intricate mechanism of depression, many clinical antidepressants are inefficient.
Sirtuins (SIRTs) are a family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases that regulates many cellular physiological processes, including metabolism, autophagy, differentiation and development [5]. SIRTs have recently been implicated in mood disorders in mice and human [6]. SIRT1 and SIRT2 mRNA were significantly down-regulated in depressive disorders [7, 8]. Inhibition of SIRT1 led to an increase in depression-like behaviors. Conversely, SIRT1 activation prevented depressive phenotypes elicited by chronic stress [9]. However, other evidence has shown the opposite results. SIRT1 overexpression increased stress vulnerability in mice, and brain-specific SIRT1 knockout mice showed less depressive phenotypes [10]. Furthermore, pharmacologic and genetic inhibition of SIRT2 blocked the development of depressive and anxiety behaviors [11, 12]. Nevertheless, overexpression of hippocampal SIRT2 protected against the chronic unpredictable stress (CUS)-induced depression-like behaviors [13]. Recent evidence has suggested the involvement of SIRT6 in the pathophysiology of mood disorders [14, 15], providing a potential mechanism underlying depression.
Studies revealed that up-regulation of SIRT6 activity led to depression-like behaviors in rat models by blocking AKT/GSK-3β signaling [15]. AKT is the key molecule activated downstream of the phosphatidylinositide-3 kinase (PI3K) signaling, and its dysregulation plays important roles in the pathogenesis of many psychiatric diseases [16]. Such as, AKT2-knockout mice displayed increased anxiety behavior [17]. Glycogen synthase kinase 3β (GSK-3β) is a ubiquitous protein kinase having a pivotal role in neurodevelopment and is identified as a downstream target of the PI3K/AKT signaling cascade [18]. Several studies showed that GSK-3β has been implicated in depression and other neuropsychopathies. An increase in GSK-3β activity was prone to develop depression [19]. Conversely, inhibition of GSK-3β activity exhibited antidepressant action in mice [20]. The PI3K/AKT pathway could facilitate GSK-3β phosphorylation to repress GSK-3β activity and thus participated in regulation of depression. However, the function and role of SIRT6/AKT/GSK-3β in depression has not been studied sufficiently, such as how the SIRT6 interacted with AKT, and more in vivo confirmation of its involvement.
Collapsin response mediator protein 2 (CRMP2) is a microtubule-associated protein that is highly expressed in specific regions in the developing and adult nervous systems [21]. Increasing evidence suggests that CRMP2 has been implicated in various neurological and psychiatric disorders, such as schizophrenia [22], ischemia [23, 24] and Alzheimer’s disease [25,26,27]. Remarkably, loss of brain-specific CRMP2 leads to neuronal development deficits and behavioral impairments in mice [22, 28]. In addition, other evidence illustrated that CRMP2 function is robustly regulated by post-translational modification, [21, 29,30,31,32,33] such as phosporylation, glycosylation, proteolysis, and SUMOylation. Especially phosphorylation of CRMP2 has been studied extensively. It has been shown that CRMP2 was phosphorylated by activated GSK-3β at Thr514 [24, 29]. Importantly, excessive CRMP2 phosphorylation results in dendritic development abnormalities and axonal injury [24, 34], whereas dephosphorylated or non-phosphorylated CRMP2 fosters axonal regeneration and microtubule formation [35, 36]. These findings indicated that CRMP2 plays an important role in neuronal development and maturation. However, the precise mechanism by which AKT/GSK-3β/CRMP2 pathway regulates neuronal plasticity and behaviors remains to be determined.
In this study, our in vivo and in vitro data comprehensively explained the function and mechanism of SIRT6 in response to chronic stress and antidepressants.
Materials and methods
Regents and antibodies
Resveratrol (Cat#R5010) were obtained from Sigma. OSS_128167 (Cat#HY-107454) was purchased from MedChemExpress (MCE, shanghai, China). LY294002 (Cat#S1737) was brought from Beyotime Biotechnology (Shanghai, China). Ferulic acid (Cat#IF0050) was purchased from Solarbio Life Sciences (Beijing, China). Anti-acetylated-lysine (Cat#9441), anti-H3K56ac (Cat#4243), anti-H3K9ac (Cat#9649), anti-IgG (Cat#2729 S), anti-SIRT6 (Cat#12486S), anti-phospho-AKT (Ser473) (Cat#4060), anti-AKT (Cat#9272) and anti-CRMP2 (Cat#9393) antibodies were obtained from Cell Signaling Technology. Anti-H3K56ac (Cat#1710259) and anti-H3K4me3 (Cat#2717639) antibodies were purchased from Millipore. Anti-Myc (Cat#TA150121) and anti-GFP (Cat#TA150041) antibodies were provided by Origene. Anti-phospho-CRMP2 (Thr514) (Cat#ab62478) was purchased from Abcam. Anti-GAPDH (Cat#TA-08) and horseradish peroxidase (HRP)-conjugated secondary antibodies (Cat#ZB2301/2305) were brought from ZSGB-Bio (Beijing, China). Secondary fluorescence antibodies (DyLight488, Cat#23220; DyLight594, Cat#23420) were brought from Abbkine (Wuhan, China).
Animals and chronic stress
Male 8-week-old C57BL/6 (20–22 g) mice were purchased from the Vital River Laboratory Animal Technology (Beijing, China). They were housed in a 12-h light/dark cycle standard conditions (temperature, 22 ± 2 °C; humidity, 40%–50%) with free access to food and water. All experiments were conducted following the National Institutes of Health (NIH) guidelines and approved protocols from the Shaanxi Normal University Animal Care Committee.
After 2 weeks, mice were randomly divided into two groups (n = 12 per group): control group and chronic unpredictable stress (CUS) group. We selected eight different mild stressors as described previously [2]. The different types of mild stressor used are detailed in the Table 1. CUS procedure was performed as described in Fig. 1a. Mice were subjected daily for 21 days to two different mild stressors accordingly to a random schedule to avoid any kind of habituation. Mice were subjected to the first stressor for 6 h each day, and then subjected to the second stressor. Control mice were left undisturbed in their home cages, except for behavioral tests.
Behavioral evaluation
For the sucrose preference test (SPT), mice were given a free choice between two bottles, containing either 2% sucrose solution or water for 24 h. After 12 h, the position of the bottles was switched. The consumption of water and sucrose solution was calculated to estimate the total preference for sucrose by weighing the bottles. For the forced swim test (FST), mice were placed individually into a vertical glass cylinder (height 25 cm, diameter 12 cm), which was filled with 12-cm deep water at 23 °C. In each test session, animals were placed in the cylinder for 5 min and videotaped. For the tail suspension test (TST), mice were suspended by its tail with tape (50 cm above the floor), and period of cumulative immobility during 5 min was videotaped by an investigator. The experimenter was blinded to the experimental conditions for analysis of the behavioral tests.
FA treatment
Male C57BL/6 mice were randomly divided into four groups (n = 10 per group): control group, CUS group, and CUS + FA group (40 or 80 mg· kg−1 per day, intragastric (i.g.)). Mice were administered FA for 14 days consecutive after the 15-day CUS as schematized in Fig. 6a. The doses were selected on the basis of previously published reports [37].
Cell culture and transfection
Human HEK293T and mouse neuroblastoma HT-22 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, Cat#SH30022) containing 10% fetal bovine serum (BI, Cat#abs972), 100 U· mL−1 penicillin and 100 mg· mL−1 streptomycin (Hyclone, Cat#SV30010) in a humidified incubator at 37 °C with 5% CO2. Cells were plated into a 6-well and cultured overnight, and then transfection was carried out using Lipo8000 reagent (Beyotime, Cat#C0533).
Primary neurons
Primary hippocampal neurons were prepared from E17.5-E18.5 mouse brains as previously described [38]. After the pregnant mouse was killed in a chamber filled with carbon dioxide (RWD Life science, Shenzhen, China; Cat#R-51031), the embryos were immediately removed, and the brains were placed in ice-cold isotonic buffer (137 mM NaCl, 5 mM KCl, 0.2 mM NaH2PO4, 0.2 mM KH2PO4, 5.5 mM glucose, and 6 mM sucrose, pH 7.4). Following washes, the hippocampuses were extracted and minced, digested in papaya (Solarbio, Beijing, China; Cat#G8430) and DNA enzymes (Solarbio, Beijing, China; Cat#D8070), and resuspended in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. The DMEM was exchanged 24 h later for Neurobasal medium (Gibco, Cat#1967789) supplemented with GlutaMAX (Gibco, Cat#1919082) and B27 supplement (Life Technologies, Cat#12587-010).
Plasmids constructs
Human SIRT6 cDNA was inserted into a pcDNA3.0-Flag or pcDNA4-HisMax vector. SIRT6 H133Y vector was generated by the site-directed mutagenesis kit (YEASEN, Shanghai, China; Cat No. 11003) according to the protocol. Human histone H3B cDNA was cloned into a pCMV-Myc vector. H3K9R and H3K56R vectors were constructed by the MutanBEST kit (Takara; Code No. R401). The wild-type and mutant of SIRT6 and H3B were verified by sequencing (Tsingke, Xi’an, China).
RNA interference and lentiviral purification
Human SIRT6 short hairpin RNA (shRNA) (5′-GCCTCTGACTTGCTGTGTTGT-3′), mouse SIRT6 shRNA (5′-GGTCATTGTCAACCTGCAACC-3′), human AKT1 shRNA (5′-GGACAAGGACGGGCACATTAA-3′) and non-targeting shRNA were synthesized by Tsingke, and then inserted into a pGreenPuro vector. To generate expressing SIRT6 shRNA lentivirues, HEK293T cells were cotransfected by expression vector with packaging vectors psPAX2 and pMD2.G. After incubation for 48 h, cells supernatant was harvested and purified by ultracentrifugation. The titers of LV-shCtrl and LV-shSIRT6 were measured as 5.7 × 108 TU/mL and 6.5 × 108 TU/mL, respectively.
SIRT6 deacetylase activity assay
The deacetylase activity of SIRT6 was measured using a SIRT6 Activity Assay Kit (Abcam, Cat#ab156068) according to manufacturer’s instructions. Briefly, the reaction compound mixture contained SIRT6 assay buffer, 0.1 mM fluoro-substrate peptide, 0.1 mM fluoro-deacetylated peptide, 8 mM NAD, 200 μM FA, and 1.5 µg recombinant SIRT6 setting the final reaction volume of 50 µL. The mixture was incubated for 50 min at room temperature. The fluorescence intensity was measured using a microtiter plate fluorometer at 1–2 min intervals at Ex/Em = 480–500 nm/520–540 nm. All tests were performed in triplicate.
Stereotaxic surgery
Male C57BL/6 mice were randomly divided into three groups (n = 12 per group): the control group, CUS + LV-shCtrl and CUS + LV-shSIRT6. Mice induced by 21-day CUS were anesthetized intraperitoneally with 2% sodium pentobarbital (50 mg· kg−1, (i.p.)) and placed in a stereotaxic apparatus. A total volume of 1 μL of lentivirus expressing a GFP protein was bilaterally injected into the hippocampus (AP, −2.0 mm; ML, ±1.5 mm; DV, −1.5 mm) at 0.1 μL/min [39]. After microinjection was completed, mice recovered on a heating pad upon revival. Mice were allowed to recover for 14 days before behavioral experiments. Successful transduction in the hippocampus was determined by immunostaining with GFP antibody.
Quantitative real-time PCR analysis
Total RNA was extracted using the RNAiso Reagent according to the manufacturer’s instructions (TaKaRa, Cat#9108). The synthesis of cDNA library was reversed using the PrimeScript RT Master Mix (TaKaRa, Cat#RR036). Quantitative real-time PCR analysis was performed using the TB Green qPCR Master Mix (TaKaRa, Cat#RR430). GAPDH was used as an internal control for the normalization. All primers used in this study were shown as follows: SIRT6, 5′-ATGTCCCAAGTGTAAGACGCAGTA-3′ and 5′-CTCATCAGCGAGCATCAGGTC-3′; GAPDH, 5′-CATCTTGGGCTACACTGAGGACC-3′ and 5′-TCATACCAGGAAATGAGCTTGACA-3′.
Western blotting assay
Total proteins of tissue and cells were extracted by RIPA lysis buffer containing protease inhibitor. In all, 30 μg proteins were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Cat#IPVH00010). The membrane was blocked with 5% non-fat milk and incubated with individual primary antibody SIRT6 (1:1000) phospho-AKT/AKT (1:1000) phospho-CRMP2/CRMP2 (1:1000) GAPDH (1:5000) for 1 h at room temperature. Then blots were incubated with the indicated HRP-conjugated secondary antibodies for 1 h at room temperature. All blots were visualized with the SuperSignal West Pico Substrate (Betyotime, Cat#D3308-1).
Immunofluorescence staining
Hippocampal sections were fixed with 4% paraformaldehyde and were permeabilized with 0.5% Triton X-100 in phosphate buffered saline (PBS), and then blocked with 2% bovine serum albumin (BSA, Albumin, Cat#180728) at room temperature. The sections were incubated overnight with primary antibodies of phospho-AKT (1:200) and phospho-CRMP2 (1:100) at 4 °C. Next, the sections were incubated with secondary fluorescence antibodies for 1 h at room temperature, and then stained using 5 μg· mL−1 DAPI (Cell Signaling, Cat#4083 S) for 5 min. Fluorescence images were captured under a ZEISS microscope (Germany).
Immunoprecipitation assay
Cells were extracted by the immunoprecipitation lysis buffer (ThermoFisher; Cat#87787). In total, 500 μg proteins were precleared with protein A/G beads (Santa Cruze Biotechnology; Cat#sc-2003) for 1 h at 4 °C. After incubated with 5 μg anti-SIRT6, anti-AKT, and anti-IgG respectively overnight at 4 °C, protein A/G beads were then added and incubated for 3 h at 4 °C. After cold PBS washing, the immunoprecipitates were suspended and loaded into SDS-PAGE for immunoblotting analysis with the anti-AKT and anti-acetylated-lysine (1:1000) antibody, respectively.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed according to the standard procedures [40]. Briefly, HEK293T cells were washed and crosslinked with 1% formaldehyde for 10 min at room temperature. Then genomic DNA was lysed, sonicated and quantified. In all, 50 μg DNA was precleared for 1 h with protein A/G beads, and incubated with 5 μg anti-SIRT6, anti-H3K56 and anti-H3K56 respectively overnight at 4 °C. Next, immunoprecipitated complexes were bound to protein A/G beads for 3 h at 4 °C and washed successively. Genomic DNA was purified by the EasyPure Genomic DNA kit (Transgen Biotech, Beijing, China; Cat#EE101) according to the protocol. GAPDH was used as negative control [41]. Immunoprecipitated DNA was carried out using specific primers for AKT1 promoter (human AKT1 promoter: forward, 5′-TGCTGGCCTGGTGTATACG-3′; reverse, 5′-CAGAGGGCTGGACTCAAAGAC-3′ and mouse AKT promoter: forward, 5′-ATTTCCATCCTGGGCGATAGC-3′; reverse, 5′-CCAGAAGCCCGACTTGATAGTAAC-3′).
Statistical analysis
The data were expressed as the mean ± SEM. The behavioral data were performed with the investigator blind to the experimental groups. Differences among multiple groups were analyzed with one-way ANOVA followed by Dunnett’s post hoc test. Comparisons between two groups were performed using unpaired Student’s t-test. Values P < 0.05 was considered statistically significant. All data were analyzed by GraphPad Prism 7.0 software.
Results
Chronic stress increases the mRNA and protein level of hippocampal SIRT6
Chronic stress has been reported to produce depression-like behavior in mice [2, 9]. As shown in Fig. 1b, C57BL/6 mice were subjected to a 3-week period of CUS. We then assessed depressive phenotypes using the SPT, FST and TST (Fig. 1c–e). CUS-induced mice showed a deficit in nature reward in the SPT (Fig. 1c) and an increase in the immobility time in the FST and TST compared with control mice (Fig. 1d, e). We investigated whether chronic stress alters the mRNA and protein level of SIRT6 in the hippocampus. Real-time PCR analysis showed that compared with control mice, the mRNA expression of SIRT6 was significantly increased in the whole hippocampus of CUS mice (Fig. 1f). In addition, we separated CA1-CA3 and DG areas from the hippocampus and separately checked the expression of SIRT6 (Fig. 1g). Western blotting confirmed that CUS significantly increased SIRT6 protein level than those of control mice (Fig. 1h).

Chronic stress leads to an increase in mRNA and protein level of hippocampal SIRT6. a Time course and design of CUS. b Experimental timeline is shown for in vivo CUS-induced mice model. c–e SPT, FST, and TST were performed on the 21th day. The data are expressed as mean ± SEM (n = 12 per group). *P < 0.05, **P < 0.01 compared with CON. f Quantitative real-time PCR analysis of SIRT6 mRNA expression in the hippocampus. The data are expressed as mean ± SEM (n = 12 per group). ***P < 0.001 compared with CON. g Western blotting analysis showed that CUS increased the protein level of SIRT6 in hippocampal CA1-CA3 and DG area. h Quantitative analysis of SIRT6 protein level using Image J software. The data are expressed as mean ± SEM (n = 3 per group). ***P < 0.001 compared with CON
Knockdown of hippocampal SIRT6 attenuates depression-like behaviors
To directly testify whether SIRT6 up-regulation is involved in depression-like behaviors, mice were bilaterally microinjected with LV-shSIRT6 or LV-shCtrl into hippocampal CA1-CA3 area after 21-day CUS (Fig. 2a). We examined the efficiency of lentivirus expressing GFP by histological observation (Fig. 2b). Western blotting showed that the protein level of SIRT6 was decreased in CA1-CA3 area in LV-shSIRT6 mice, but no significant difference in DG area (Fig. 2c, d). LV-shSIRT6 mice showed significant preference in the SPT compared with LV-shCtrl mice (Fig. 2e). In the FST and TST, the immobility time of the LV-shSIRT6 mice was markedly decreased (Fig. 2f, g). Our data indicated that knockdown of SIRT6 in hippocampus could alleviate depression-like behaviors induced by CUS, suggesting a direct link between hippocampal SIRT6 up-regulation and depression-like behavior.

Knockdown of hippocampal SIRT6 prevents CUS-induced depression-like behaviors. a Experimental timeline was shown for in vivo lentiviral-mediated SIRT6 knockdown studies. After 21 days of chronic stress, mice were microinjected for 14 days with LV-shCtrl or LV-shSIRT6 into the hippocampus. b Corresponding images of lentiviral infection in the hippocampus. Scale bar, 200 μm. c, d Representative blots of SIRT6 detected by Western blotting with tissues from the hippocampal CA1-CA3 and DG area. The data are expressed as mean ± SEM (n = 3 per group). ***P < 0.001 compared with LV-Ctrl. n.s. no significant difference. e–g Knockdown of hippocampal SIRT6 ameliorated CUS-induced depression-like behaviors, including SPT (F2, 33 = 8.902, P = 0.0008), FST (F2, 33 = 11.69, P = 0.0001), and TST (F2, 33 = 7.938, P = 0.0015). The data are expressed as mean ± SEM (n = 12 per group). ##P < 0.01, ###P < 0.001 compared with CON; **P < 0.01, ***P < 0.001 compared with LV-shCtrl+CUS
AKT is a potential downstream target of SIRT6
Several studies have revealed an involvement of SIRT6 in the regulation of AKT/GSK-3β signaling [15, 41], proteins known to be associated with antidepressant action in rodent models. To elucidate the mechanism by which SIRT6 regulates AKT signaling, we firstly checked the interaction of SIRT6 and AKT using immunoprecipition assay. However, immunoprecipitation assay showed that SIRT6 could not coprecipitate with AKT (Fig. 3a). To exclude off-target effect, SIRT6 shRNA was used to assess the effect of AKT acetylation. Consistently, SIRT6 shRNA failed to affect AKT acetylation level directly (Fig. 3b). Considering that SIRT6 is a nuclear histone deacetylase, and directly regulate various proteins expression by deacetylating H3K9 and H3K56 [42], we performed ChIP assay to examine whether SIRT6 regulates AKT signaling at the level of chromatin. As we expected, after transfection of His-SIRT6, we confirmed that SIRT6 could bind to the promoter of AKT1 (Fig. 3c). Furthermore, we found that overexpression of SIRT6 led to a decrease of H3K9 and H3K56 acetylation in the promoter of down-regulated AKT1 signaling (Fig. 3d). In addition, immunoprecipitation assay confirmed that acetylation of wild-type histones was reduced by SIRT6, while acetylation of H3K9R and H3K56R mutants was not decreased (Fig. 3e). These data demonstrated that SIRT6 directly suppresses AKT signaling by deacetylating H3K9 and H3K56 (Fig. 3f, g).

SIRT6 negatively regulates AKT signaling. a HEK293T cells were transfected with Flag or Flag-SIRT6 for 48 h, and then immunoprecipitated with anti-SIRT6. IgG was used as a negative control. b HEK293T cells were transfected with shCtrl or shSIRT6 for 48 h, cells lysates was immunopreciptated with anti-AKT, acetylated AKT was performed using anti-acetylated-lysine. c ChIP analysis using anti-SIRT6 to detect SIRT6 at the promoter of AKT in SIRT6 overexpression HEK293T cells. The data are expressed as mean ± SEM (n = 3). ***P < 0.001 compared with His. d ChIP analysis using anti-H3K9ac or anti-H3K56ac to detect histone at the promoter of AKT in SIRT6 overexpression HT-22 cells. The data are expressed as mean ± SEM (n = 4). ***P < 0.001 compared with Myc-H3 + SIRT6. e Immunoprecipitation analysis using anti-Myc to detect acetylated-histone in SIRT6 overexpression HEK293T cells. f, g SIRT6 condensed chromatin and inhibited AKT signaling by deacetylating H3K9 and H3K56. Conversely, suppression of SIRT6 relaxed chromatin and promoted AKT signaling by increasing H3K9 and H3K56 acetylation
Genetic suppression of SIRT6 triggers AKT/CRMP2 signaling
CRMP2 is a potential downstream target of AKT/GSK-3β signaling [29, 43], and we found that CRMP2 was also involved in the downstream signaling pathway of SIRT6. LV-shSIRT6 significantly increased AKT phosphorylation activity in HEK293T cells, whereas reduced CRMP2 phosphorylation activity (Fig. 4a). Conversely, LV-shAKT1 significantly reversed the phosphorylation activity of CRMP2 (Fig. 4b). To further characterize the potential role of SIRT6 in regulating AKT/CRMP2 signaling, HEK293T cells were transfected with SIRT6 H133Y encoding a catalytically inactive mutant of SIRT6 [42]. As shown in Fig. 4c, overexpression of SIRT6 H133Y could remarkably elevate phosphorylation activity of AKT and supress phosphorylation activity of CRMP2. In order to consolidate the in vitro SIRT6/AKT/CRMP2 pathway, primary hippocampal neurons were infected for 48 h with LV-shSIRT6 and LV-shCtrl, respectively. Knockdown of SIRT6 could also activate AKT/CRMP2 pathway in primary hippocampal neurons (Fig. 4d). Taken together, our results confirmed that SIRT6 potently blocks AKT/CRMP2 signaling.

Suppression of SIRT6 triggers AKT/CRMP2 signaling. a HEK293T cells were infected with LV-shCtrl or LV-shSIRT6 for 48 h, followed by Western blotting. **P < 0.01 compared with LV-shCtrl. b HEK293T cells were transfected with shCtrl or shAKT1 for 48 h, followed by Western blotting. *P < 0.05 ***P < 0.001 compared with LV-shCtrl. c HEK293T cells were transfected with Flag or Flag-SIRT6 H133Y plasmid for 48 h, followed by Western blotting. *P < 0.05, ***P < 0.001 compared with His. d Primary hippocampal neurons were infected by LV-shSIRT6 or LV-shCtrl. After 48 h, SIRT6, phospho-AKT and phospho-CRMP2 expression were determined by Western blotting. **P < 0.01 compared with LV-shCtrl. All data are expressed as mean ± SEM (n = 3)
Pharmacologic inhibition of SIRT6 activates AKT/CRMP2 signaling
We further investigated the effect of pharmacologic inhibition of SIRT6 on AKT/CRMP2 signaling. SIRT6 inhibitor OSS_128167 effectively promoted AKT phosphorylation and inhibited CRMP2 phosphorylation in primary neurons (Fig. 5a). Simultaneously, the phosphorylation activity of CRMP2 was significantly reversed by PI3K inhibitor LY294002 (Fig. 5a). Ferulic acid (FA) has been proved to exert an antidepressant-like effect on chronic stress-induced mouse model [44, 45]. However, the mechanism of its antidepressant effect is not clear. The relationship between FA and SIRT6 has not been investigated. Occasionally we found that it seems that FA has the same effect as OSS_128167. FA significantly increased AKT phosphorylation and decreased CRMP2 phosphorylation as effective as OSS_128167 (Fig. 5b). Then we strictly examined the effect of FA on the regulation of SIRT6 in primary neurons in a concentration-dependent manner. SIRT6 expression was significantly decreased by FA treatment at the concentration of 50–400 μM (Fig. 5c). Simultaneously, FA activated AKT/CRMP2 signaling pathway (Fig. 5c). Our data further showed that activation of SIRT6 by resveratrol treatment on primary neurons was attenuated by FA (Fig. 5d), and the inhibition of AKT/CRMP2 signaling by resveratrol was also buffered by FA (Fig. 5d). Using HT-22 cells, a commonly used hippocampus-like cell model system, we can see that AKT/CRMP2 signaling activated by FA was largely abrogated by overexpression of SIRT6 (Fig. 5e). To seek direct evidence for pharmacologic inhibition of SIRT6 by FA, we tested whether FA has an important effect on SIRT6 deacetylase activity and its substrates. As shown in Supplementary Fig. S1A, SIRT6 deacetylase activity was down-regulated by FA in dose-dependent manner. In addition, we checked the expression of SIRT6 enzyme substrates using Western blotting analysis. FA significantly increased the expression of H3K56ac and H3K9ac by inhibiting SIRT6 deacetylase activity (Supplementary Fig. S1B, C). These results indicated that FA is a potential pharmacologic inhibitor of SIRT6, and AKT/CRMP2 signaling activated by FA can be attributed to its ability to block SIRT6 activity.

Pharmacological inhibition of SIRT6 activates AKT/CRMP2 in primary neurons. a Primary hippocampal neurons were treated for 6 h with respectively 100 μM OSS_128167 and 50 μM LY294002, followed by Western blotting. *P < 0.05, **P < 0.01 compared with negative control (NC). b Primary neurons were pretreated with OSS_128167 and 200 μM FA, followed by Western blotting. *P < 0.05, **P < 0.01, ***P < 0.001 compared with NC. c Primary neurons were pretreated for 24 h with different concentrations of FA, followed by Western blotting. **P < 0.01, ***P < 0.001 compared with 0 μM. d Primary neurons were incubated for 24 h with FA and 100 μM resveratrol, followed by Western blotting. *P < 0.05, **P < 0.01, ***P < 0.001 compared with resveratrol (Res). e HT-22 cells were transfected with His-SIRT6, and then incubated with FA, followed by Western blotting. *P < 0.05 compared with FA + His. About all data are expressed as mean ± SEM (n = 3)
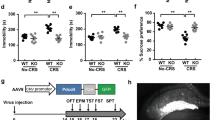
Pharmacologic inhibition of SIRT6 alleviates CUS-induced depression-like behaviors in mice
Mice underwent a 21-day chronic stress. From day 15, mice were given FA (40 or 80 mg· kg−1) for 14 days continuously (Fig. 6a). Analysis of variance revealed that administration of FA treatment could reverse the depression-like behaviors induced by CUS (Fig. 6b–d). We subsequently detected the expressions of proteins in the hippocampus. Compared with the CUS mice, mice with FA treatment exhibited reduced protein level of SIRT6 (Fig. 6e, f). In addition, mice with FA treatment exhibited a remarkable up-regulation of phosphorylated AKT and downregulation of phosphorylated CRMP2 compared with CUS mice (Fig. 6e, f). Consistently, immunostaining analysis showed that FA obviously increased AKT phosphorylation expression, whereas decreased CRMP2 phosphorylation expression in the hippocampus (Fig. 6g). These data indicated that FA treatment protects against CUS-induced depression-like behaviors by regulating SIRT6/AKT/CRMP2 signaling pathway.

FA attenuates CUS-induced depression-like behaviors in mice as a pharmacological inhibitor of SIRT6. a Experimental procedure for the CUS-induced mouse model. Mice underwent a 21-day chronic stress. From day 15, mice were administered FA (40 or 80 mg· kg−1) for 14 days. b–d FA treatment completely prevented CUS-induced depression-like behaviors, including SPT (F3, 35 = 8.167, P = 0.0003), FST (F3, 36 = 9.859, P < 0.0001), and TST (F3, 36 = 9.907, P = 0.0001). The data are expressed as mean ± SEM. (n = 10 per group). ###P < 0.001 compared with CON group; *P < 0.05, ***P < 0.001 compared with CUS group. e, f Western blotting analysis of SIRT6, phospho-AKT and phospho-CRMP2 protein level in the hippocampus. The data are expressed as mean ± SEM (n = 3 per group). #P < 0.05, ##P < 0.01 compared with CON group; *P < 0.05, **P < 0.01 compared with CUS group. g Immunostaining images showed that pharmacological inhibition of SIRT6 by FA treatment increased the expression of phospho-AKT and decreased the expression of phospho-CRMP2 in the hippocampus. Scale bar, 100 μm
Discussion
In mammals, SIRT6 is known to be a longevity protein that regulates cellular and individual growth, development, and lifespan. Overexpression of SIRT6 could improve health and extend lifespan in mice [46], whereas ablation of SIRT6 resulted in elevated reactive oxygen species (ROS) levels and increased vulnerability to oxidative injury [42]. In addition, loss of SIRT6 resulted in neuronal development defects [47]. However, the pricise mechanisms by which SIRT6 mediates depression-like behavior remain to be investigated. In this study, our results showed that hippocampal SIRT6 expression was significantly up-regulated in CUS-induced mouse model, consistent with previous studies [15]. And we further confirmed the involvement of hippocampal SIRT6 in CUS-induced depressive model using lentivirus-mediated knockdown in vivo. Knockdown of hippocampal SIRT6 could strongly prevent CUS-induced depression-like phenotypes. However, another animal study indicated that chronic stress exposure leads to the reduction of prefrontal SIRT6 activity in mice, and a SIRT6 activator has an antidepressant-like effect [14]. Thus, SIRT6-mediated control of depression may rely on the cell type, genetic background, and brain region.
It is confirmed that the PI3K signaling participates in the regulation of many neuronal processes, including neuronal differentiation, dendritic formation, and synaptic plasticity [43]. Increasing evidence showed that the PI3K/AKT signaling has been discussed to mediate antidepressant effects. Pharmacologic activation of PI3K/AKT pathway extremely mitigated depression-like impairments induced by stress [48, 49]. Previous studies indicated that overexpression of SIRT6 in HEK293T cells dephosphorylated AKT and inhibited AKT signaling [50]; and up-regulation of hippocampal SIRT6 resulted in depressive phenotypes by blocking AKT/GSK-3β signaling [15]. Consistently, our results indicated that pharmacological inhibition of SIRT6 strongly and effectively defeated depressive behaviors through stimulating AKT signaling. Moreover, our data showed that SIRT6 not only strongly blocked AKT phosphorylation activity, but also slightly decreased total AKT expression. And our further study revealed that SIRT6 condensed chromatin and inhibited AKT signaling through its ability to deacetylating H3K9 and H3K56. In line with our data, SIRT6 completely blocked IGF/AKT signaling in the development of cardiac hypertrophy [41]. In addition, given that SIRT6 is the pivotal deacetylase, it is possible whether SIRT6 is involved in directly deacetylating AKT. However, our present study showed that SIRT6 directly failed to affect AKT acetylation activity. To date, a few studies indicated that SIRT1 can be able to directly deacetylate AKT and alter its phosphorylation activity [51], but not SIRT6.
Besides, CRMP2 has been well characterized as a target of neuronal plasticity and behavioral modulation [22], which belongs to a downstream molecule of AKT/GSK-3β signaling. Increasing evidence suggests that CRMP2 is a multifunctional molecule that regulates various aspects of neuronal development, including axon guidance, dendritic morphogenesis and synaptic plasticity [29]. CRMP2 overexpression effectively protects against acute axonal degeneration [29]. CRMP2 phosphorylation, an inactive form, led to the impairment of neuronal plasticity and neural function [21]. Similarly, we found that an increase of CRMP2 phosphorylation induced by CUS was correlated with depression-like behavior. Furthermore, our data showed that AKT1 shRNA and LY294002 resulted in an increase of CRMP2 phosphorylation activity. Combined with previous reports [43, 52], CRMP2 activity was regulated by SIRT6/AKT signaling pathway.
We reported that FA is a small molecule that specifically inhibits SIRT6 expression dose-dependently. FA is an effective component extracted from Angelica sinensis (danggui), which has shown multiple beneficial biological activities [53]. The antidepressant-like activity of FA has been reported in a few animal models [54]. However, the detailed mechanism of FA on depression is still unknown. The combination of our in vivo and in vitro data can comprehensively explain the function and mechanism of FA in response to chronic stress. Our findings with pharmacological inhibition of SIRT6 indicated that FA strongly and effectively activated AKT/CRMP2 signaling pathway, and ameliorated CUS-induced depression-like behaviors and played a neuroprotective effect by SIRT6/AKT/CRMP2 pathway (Fig. 7).

The regulation role of SIRT6/AKT/CRMP2 in depression-like behavior. Schematic view of SIRT6/AKT/CRMP2 signaling pathway involvement in depression-like behaviors in mice
Accumulating evidence shows that chronic stress activates the inflammatory and oxidative stress system, lead to a high risk of vulnerability to depression. Pharmacological inhibition of PPARγ/NF-κB/NLRP3 inflammasome axis can modulate the inflammatory response and further have an antidepressant effect [55]. Pharmacological treatment by increasing antioxidant pool relieves stress-induced behavioral and cognitive impairments in vivo experiment [56]. In addition, recent evidence shows that FA has anti-inflammatory, anti-oxidative, and anti-apoptotic properties [53]. However, whether FA can prevent the inflammasome axis and oxidative stress to fight against depression remains to be further investigated.
In conclusion, our in vivo studies confirmed that down-regulation of hippocampal SIRT6 attenuates CUS-induced depressive phenotypes in mice. The beneficial effect of suppression of SIRT6 is related to AKT/CRMP2 signaling pathway. In addition, our study showed that hippocampal SIRT6/AKT/CRMP2 signaling pathway was involved in the mechanism of antidepressant effect of FA.
References
Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902.
Nasca C, Bigio B, Zelli D, Nicoletti F, McEwen BS. Mind the gap: glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol Psychiatry. 2015;20:755–63.
Duman RS. Neurobiology of stress, depression, and rapid acting antidepressants: remodeling synaptic connections. Depress Anxiety. 2014;31:291–6.
Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338:68–72.
Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014;25:138–45.
Alageel A, Tomasi J, Tersigni C, Brietzke E, Zuckerman H, Subramaniapillai M, et al. Evidence supporting a mechanistic role of sirtuins in mood and metabolic disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2018;86:95–101.
Lo Iacono L, Visco-Comandini F, Valzania A, Viscomi MT, Coviello M, Giampà A, et al. Adversity in childhood and depression: linked through SIRT1. Transl Psychiatry. 2015;5:e629.
Abe N, Uchida S, Otsuki K, Hobara T, Yamagata H, Higuchi F, et al. Altered sirtuin deacetylase gene expression in patients with a mood disorder. J Psychiatr Res. 2011;45:1106–12.
Abe-Higuchi N, Uchida S, Yamagata H, Higuchi F, Hobara T, Hara K, et al. Hippocampal sirtuin 1 signaling mediates depression-like behavior. Biol Psychiatry. 2016;80:815–26.
Libert S, Pointer K, Bell EL, Das A, Cohen DE, Asara JM, et al. SIRT1 activates MAO-A in the brain to mediate anxiety and exploratory drive. Cell. 2011;147:1459–72.
Zhang Z, Zhang P, Qi GJ, Jiao FJ, Wang QZ, Yan JG, et al. CDK5-mediated phosphorylation of Sirt2 contributes to depression-like behavior induced by social defeat stress. Biochim Biophys Acta Mol Basis Dis. 2018;1864:533–41.
Muñoz-Cobo I, Belloch FB, Díaz-Perdigón T, Puerta E, Tordera RM. SIRT2 inhibition reverses anhedonia in the Vglut1+/− depression model. Behav Brain Res. 2017;335:128–31.
Liu R, Dang W, Du Y, Zhou Q, Jiao K, Liu Z. SIRT2 is involved in the modulation of depressive behaviors. Sci Rep. 2015;5:8415.
Li W, Zhu Y, Liu X, Hou J, Fang J, Shen JX, et al. Phencynonate mediates antidepressant response by activating sirtuin 6-SOD2/Prdx6 pathway. Biochem Biophys Res Commun. 2018;505:898–904.
Mao Q, Gong X, Zhou C, Tu Z, Zhao L, Wang L, et al. Up-regulation of SIRT6 in the hippocampus induced rats with depression-like behavior via the block Akt/GSK3b signaling pathway. Behav Brain Res. 2017;323:38–46.
Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–27.
Leibrock C, Ackermann TF, Hierlmeier M, Lang F, Borgwardt S, Lang UE. Akt2 deficiency is associated with anxiety and depressive behavior in mice. Cell Physiol Biochem. 2013;32:766–77.
Papazoglou IK, Jean A, Gertler A, Taouis M, Vacher CM. Hippocampal GSK3β as a molecular link between obesity and depression. Mol Neurobiol. 2015;52:363–74.
Ackermann TF, Kempe DS, Lang F, Lang UE. Hyperactivity and enhanced curiosity of mice expressing PKB/SGK-resistant glycogen synthase kinase-3 (GSK-3). Cell Physiol Biochem. 2010;25:775–86.
Qiao J, Rong L, Wang Z, Zhang M. Involvement of Akt/GSK3β/CREB signaling pathway on chronic omethoate induced depression-like behavior and improvement effects of combined lithium chloride and astaxanthin treatment. Neurosci Lett. 2017;649:55–61.
Quach TT, Honnorat J, Kolattukudy PE, Khanna R, Duchemin AM. CRMPs: critical molecules for neurite morphogenesis and neuropsychiatric diseases. Mol Psychiatry. 2015;20:1037–45.
Zhang H, Kang E, Wang Y, Yang C, Yu H, Wang Q, et al. Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nat Commun. 2018;9:16229.
Nada SE, Tulsulkar J, Raghavan A, Hensley K, Shah ZA. A derivative of the CRMP2 binding compound lanthionine ketimine provides neuroprotection in a mouse model of cerebral ischemia. Neurochem Int. 2012;61:1357–63.
Wilson SM, Ki Yeon S, Yang XF, Park KD, Khanna R. Differential regulation of collapsin response mediator protein 2 (CRMP2) phosphorylation by GSK3β and CDK5 following traumatic brain injury. Front Cell Neurosci. 2014;8:135.
Yang Z, Kuboyama T, Tohda C. A systematic strategy for discovering a therapeutic drug for Alzheimer’s disease and its target molecule. Front Pharmacol. 2017;8:340.
Ip JP, Fu AK, Ip NY. CRMP2: functional roles in neural development and therapeutic potential in neurological diseases. Neuroscientist. 2014;20:589–98.
Hensley K, Kursula P. Collapsin response mediator protein-2 (CRMP2) is a plausible etiological factor and potential therapeutic target in Alzheimer’s disease: comparison and contrast with microtubule-associated protein Tau. J Alzheimers Dis. 2016;53:1–14.
Nakamura H, Yamashita N, Kimura A, Kimura Y, Hirano H, Makihara H, et al. Comprehensive behavioral study and proteomic analyses of CRMP2-deficient mice. Genes Cells. 2016;21:1059–79.
Zhang JN, Koch JC. Collapsin response mediator protein-2 plays a major protective role in acute axonal degeneration. Neural Regen Res. 2017;12:692–5.
Saitoh F, Hagiwara H, Wakatsuki S, Araki T. Carboxymethylation of CRMP2 is associated with decreased Schwann cell myelination efficiency. Neurosci Res. 2018;18:30492–94.
Zhang JN, Michel U, Lenz C, Friedel CC, Köster S, d’Hedouville Z, et al. Calpain-mediated cleavage of collapsin response mediator protein-2 drives acute axonal degeneration. Sci Rep. 2016;6:37050.
Dustrude ET, Wilson SM, Ju W, Xiao Y, Khanna R. CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J Biol Chem. 2013;288:24316–31.
Moutal A, Dustrude ET, Largent-Milnes TM, Vanderah TW, Khanna M, Khanna R. Blocking CRMP2 SUMOylation reverses neuropathic pain. Mol Psychiatry. 2018;23:2119–21.
Liu Y, Lin D, Liu C, Zhao Y, Shen Z, Zhang K, et al. Cyclin-dependent kinase 5/Collapsin response mediator protein 2 pathway may mediate sevoflurane-induced dendritic development abnormalities in rat cortical neurons. Neurosci Lett. 2017;651:21–9.
Nagai J, Owada K, Kitamura Y, Goshima Y, Ohshima T. Inhibition of CRMP2 phosphorylation repairs CNS by regulating neurotrophic and inhibitory responses. Exp Neurol. 2016;277:283–95.
Cole AR, Soutar MP, Rembutsu M, van Aalten L, Hastie CJ, McLauchlan H, et al. Relative resistance of Cdk5-phosphorylated CRMP2 to dephosphorylation. J Biol Chem. 2008;283:18227–37.
Zheng X, Cheng Y, Chen Y, Yue Y, Li Y, Xia S, et al. Ferulic acid improves depressive-like behavior in prenatally-stressed offspring rats via anti-inflammatory activity and HPA axis. Int J Mol Sci. 2019;20:E493.
Zheng D, Sabbagh JJ, Blair LJ, Darling AL, Wen X, Dickey CA. MicroRNA-511 binds to FKBP5 mRNA, which encodes a chaperone protein, and regulates neuronal differentiation. J Biol Chem. 2016;291:17897–906.
Higuchi F, Uchida S, Yamagata H, Abe-Higuchi N, Hobara T, Hara K, et al. Hippocampal microRNA-124 enhances chronic stress resilience in mice. J Neurosci. 2016;36:7253–67.
Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J. 2006;400:179–88.
Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med. 2012;18:1643–50.
Pan H, Guan D, Liu X, Li J, Wang L, Wu J, et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016;26:190–205.
Wu Z, Wang G, Wei Y, Xiao L, Wang H. PI3K/AKT/GSK3β/CRMP-2-mediated neuroplasticity in depression induced by stress. Neuroreport. 2018;29:1256–63.
Liu YM, Hu CY, Shen JD, Wu SH, Li YC, Yi LT. Elevation of synaptic protein is associated with the antidepressant-like effects of ferulic acid in a chronic model of depression. Physiol Behav. 2017;169:184–8.
Zeni ALB, Camargo A, Dalmagro AP. Ferulic acid reverses depression-like behavior and oxidative stress induced by chronic corticosterone treatment in mice. Steroids. 2017;125:131–6.
Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–21.
Zhang W, Wan H, Feng G, Qu J, Wang J, Jing Y, et al. SIRT6 deficiency results in developmental retardation in cynomolgus monkeys. Nature. 2018;560:661–5.
Ludka FK, Constantino LC, Dal-Cim T, Binder LB, Zomkowski A, Rodrigues AL, et al. Involvement of PI3K/Akt/GSK-3b and mTOR in the antidepressant-like effect of atorvastatin in mice. J Psychiatr Res. 2016;82:50–57.
Cunha MP, Budni J, Ludka FK, Pazini FL, Rosa JM, Oliveira Á, et al. Involvement of PI3K/Akt signaling pathway and its downstream intracellular targets in the antidepressant-like effect of creatine. Mol Neurobiol. 2016;53:2954–68.
Shao J, Yang X, Liu T, Zhang T, Xie QR, Xia W. Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell. 2016;7:281–90.
Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins its implication in cardiac hypertrophy and aging. Circ Res. 2014;114:368–78.
Wakatsuki S, Saitoh F, Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Biol. 2011;13:1415–23.
Kumar N, Pruthi V. Potential applications of ferulic acid from natural sources. Biotechnol Rep. 2014;4:86–93.
Chen J, Lin D, Zhang C, Li G, Zhang N, Ruan L, et al. Antidepressant-like effects of ferulic acid: involvement of serotonergic and norepinergic systems. Metab Brain Dis. 2015;30:129–36.
Song MT, Ruan J, Zhang RY, Deng J, Ma ZQ, Ma SP. Astragaloside IV ameliorates neuroinflammationinduced depressive-like behaviors in mice via the PPARγ/NF-κB/NLRP3 inflammasome axis. Acta Pharmacol Sin. 2018;39:1559–70.
Solanki N, Salvi A, Patki G, Salim S. Modulating oxidative stress relieves stress-induced behavioral and cognitive impairments in rats. Int J Neuropsychopharmacol. 2017;20:550–61.
Acknowledgements
We thank Prof. Yi Yang, Ningxia Medical University, for providing antibodies (anti-AKT and anti-phospho-AKT). In addition, we thank Prof. Li-jun Zhao, Shaanxi Normal University, for gifting HT-22 cells. This study was also supported by Fundamental Research Fund for the Central Universities (GK202003054) and Natural Science Basic Research Plan in Shaanxi Province of China (No. 2019JM-285).
Author information
Authors and Affiliations
Contributions
HQ and WL conceived and designed the study, and wrote the manuscript. WL and XL performed the experiments and analyzed the data. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, W., Liu, X. & Qiao, H. Downregulation of hippocampal SIRT6 activates AKT/CRMP2 signaling and ameliorates chronic stress-induced depression-like behavior in mice. Acta Pharmacol Sin 41, 1557–1567 (2020). https://doi.org/10.1038/s41401-020-0387-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0387-5
Keywords
This article is cited by
-
Areca Thirteen Pill Improves Depression in Rat by Modulation of the Chemokine/Chemokine Receptor Axis
Molecular Neurobiology (2023)
-
Roles of SIRT6 in kidney disease: a novel therapeutic target
Cellular and Molecular Life Sciences (2022)
