
Abstract
Autoimmune diseases are chronic immune diseases characterized by dysregulation of immune system, which ultimately results in a disruption in self-antigen tolerance. Cumulative data show that nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) play essential roles in various autoimmune diseases, such as inflammatory bowel disease (IBD), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), psoriasis, multiple sclerosis (MS), etc. NLR proteins, consisting of a C-terminal leucine-rich repeat (LRR), a central nucleotide-binding domain, and an N-terminal effector domain, form a group of pattern recognition receptors (PRRs) that mediate the immune response by specifically recognizing cellular pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) and triggering numerous signaling pathways, including RIP2 kinase, caspase-1, nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK) and so on. Based on their N-terminal domain, NLRs are divided into five subfamilies: NLRA, NLRB, NLRC, NLRP, and NLRX1. In this review, we briefly describe the structures and signaling pathways of NLRs, summarize the recent progress on NLR signaling in the occurrence and development of autoimmune diseases, as well as highlight numerous natural products and synthetic compounds targeting NLRs for the treatment of autoimmune diseases.
Similar content being viewed by others

Introduction
The immune system is mainly composed of the innate immune system and adaptive immune system. The innate immune system, which plays a pivotal role in the first line of host defense against infection, activates the host immune response by recognizing pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) through the pattern recognition receptors (PRRs), while the adaptive immune system mainly recognizes protein antigens through lymphocyte receptors [1, 2]. Interestingly, some PRRs are also expressed and function in adaptive immune cells, such as regulating antigen presentation [3]. Currently, PRRs involved in the innate immune response are mainly divided into two categories: transmembrane proteins, such as Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic proteins, including retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), and NOD-like receptors (NLRs) [4, 5]. Activation of these receptors induces the activation of different transcription factors and the synthesis of different cytokines, interferons, and chemokines, which exert differential effects depending on their properties [6, 7].
The most well-characterized PRRs are TLRs and NLRs. NLRs are a family of evolutionarily conserved innate immune receptors that were initially shown to respond to intracellular pathogens (bacterial wall components, toxins, uric acids, damaged membrane, and others) and endogenous byproducts of tissue injury. Recently, certain observations have shown that NLRs also play important roles in distinct biological processes such as the regulation of antigen presentation [8], inflammatory reactions [9], embryo development [10], and cell death [11].
Autoimmune diseases are chronic immune diseases characterized by the dysregulation of the immune system, which ultimately results in the dysregulation of tolerance to self-antigens. The overall prevalence of autoimmune diseases is ~3%–5% in the general population. Although the precise mechanisms of autoimmune diseases remain unknown, many factors are believed to contribute to their pathogenesis, including genetic susceptibility, immunologic, and environmental factors [12, 13]. Cumulative evidence has shown that aberrant activation of innate immune signaling is involved in the occurrence and development of autoimmune diseases, and NLRs play essential roles in various autoimmune diseases, including inflammatory bowel disease (IBD) [9], rheumatoid arthritis (RA) [14], systemic lupus erythematosus (SLE) [15], psoriasis [16], multiple sclerosis (MS) [17], type 1 diabetes (T1D) [18], autoimmune thyroiditis (AIT) [19], and autoimmune hepatitis (AIH) [20]. In this review, we summarize recent research advances on NLR signaling and discuss the potential roles of NLRs in the pathogenesis of chronic and idiopathic inflammatory disorders, which may provide novel targets for the prevention and/or treatment of autoimmune diseases.
The NLR family and canonical signaling mechanisms
The structure of NLRs
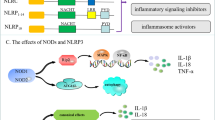
NLRs, which are PRRs, are distributed in the cytosol and play important roles in activating host responses against pathogen infection and cellular stress as intracellular sensors of PAMPs and DAMPs. All NLR proteins contain a C-terminal leucine-rich repeat (LRR), except NLRP10, a centrally located nucleotide-binding domain known as the NACHT domain (named for neuronal apoptosis inhibitor protein (NAIP), major histocompatibility complex (MHC) class II transcription activator (CIITA), incompatibility locus protein from Podospora anserina (HET-E) and telomerase-associated protein 1 (TP1)) that facilitates self-oligomerization and adenosine triphosphate (ATP)-dependent NLR activation, and an N-terminal effector domain (Fig. 1). The LRR domain is similar to the extracellular domain of TLRs, which is responsible for recognizing molecular patterns, including the corresponding components of PAMPs/DAMPs. The characteristic NACHT domain is involved in ATPase activity and control oligomerization. The N-terminal effector domain, which is the most distinguished component of NLRs, binds with various adaptor molecules and downstream effectors to mediate signal transduction and can be divided into five subfamilies according to unique functional characteristics: NLRA, NLRB, NLRC, NLRP, and NLRX1 (Fig. 1).

Human NLRs are divided into five subfamilies: NLRA, NLRB, NLRC, NLRP, and NLRX. All NLRs except NLRP10 contain an N-terminal effector domain, a central NACHT domain and a C-terminal LRR domain. CARD caspase recruitment domain, AD acidic transactivation domain, NACHT, NAIP (neuronal apoptosis inhibitor protein), CIITA (MHC class 2 transcription activator), HET-E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase-associated protein); BIR baculoviral inhibitory repeat-like domain, PYD pyrin domain, FIIND function to find domain, X unidentified, MTS mitochondria-localization sequence, LRR leucine-rich repeat
NLRA
The NLRA subfamily has an N-terminal acidic activation domain and composed of a single member, CIITA, the activation of which is dynamically regulated by a series of posttranslational modifications, such as acetylation, phosphorylation and ubiquitination [21]. The cell lineage-specific mechanisms that control CIITA transcription are responsible for positively regulating the expression of MHCII in different populations of antigen-presenting cells (APCs) [22] (Fig. 2).

Classic NLR signaling pathways are mainly composed of four parts: signal transduction, autophagy, transcriptional activation, and inflammasome activation. NOD1 and NOD2 recognize the bacterial peptidoglycans iE-DAP and MDP, respectively. Then, the NOD1/2 multimers are activated by homophilic CARD-CARD interactions to recruit the serine/threonine kinase RIP2 and form NOD signalosomes, which activate the NF-κB and MAPK signaling pathways. Autophagosomes deliver pathogens to lysosomes for degradation via the essential autophagic adapter proteins ATG5, ATG12 and ATG16L1, and ATG16L1 negatively regulates NOD/RIP2 signaling. NLRP2 and NLRP4 also negatively regulate activation of the NF-κB pathway by modifying TRAF6. IFN-γ stimulates CIITA and NLRC5 activation. Self-assembled CIITA and NLRC5 subsequently activate the transcription of MHCII and MHCI, respectively. Activated NLRP1, NLRP3, NLRC4, and AIM2 inflammasomes recruit ASCs, followed by the activation of caspase-1, as well as the secretion of IL-1β and IL-18. In addition, the activation of caspase-1 also triggers pyroptosis
NLRB
NLRB has an N-terminal baculoviral inhibition of apoptosis repeat (BIR) domain and is likewise composed of only one member, the NAIP, which is largely recognized for its role in host defense and cell survival. NAIP inhibits the activities of caspase-3, caspase-7, and caspase-9, as well as the autocleavage of pro-caspase-9 and the cleavage of pro-caspase-3 by caspase-9, exerting anti-apoptosis effects via multiple signals [23].
NLRC
NLRC, the second-largest subfamily of NLRs, harbors an N-terminal caspase activation and recruitment domain (CARD) and consists of five members (NLRC1-5). The members of this family can interact with other CARD-containing adaptor proteins. NOD1 (NLRC1) and NOD2 (NLRC2) are considered essential NLRs and are the two primary members of the NLRC subfamily [24]. These proteins can act on the downstream serine/threonine kinase RIP2, resulting in the activation of nuclear factor kappa B (NF-κB). Conversely, NLRP2 and NLRP4 negatively regulate the NF-κB signaling pathway by inhibiting the expression of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) [25] (Fig. 2).
NLRP
The NLRP subfamily is the largest subfamily of NLRs and is distinguished by the presence of a pyrin domain (PYD), which is a conserved sequence motif found in over 20 human proteins. Instead of being involved in the transcriptional activation of inflammatory mediators, NLRPs participate in pyroptosis and are components of the inflammasomes that regulate caspase-1 activation. NLRPs consist of 14 members (NLRP1-14) and can sense both pathogen-associated and sterile activators. Pathogen-associated activators include different PAMPs derived from bacteria (peptidoglycan (PGN), RNA, DNA, anthrax lethal toxin (anthrax LT), flagellin, and muramyl dipeptide (MDP)), viruses (protein and RNA) and fungi (zymosan, mannan, and hyphae) [26]. In addition, sterile factors that activate inflammasomes include autogenous DAMPs (monosodium urate (MSU)/calcium pyrophosphate dihydrate (CPPD) crystals, hyaluronan, amyloid β, and cholesterol crystals) and environmental factors (alloy particles, silica, asbestos, alum, ultraviolet radiation, and skin irritants) [27].
NLRX
NLRX1, the only described member of the NLRX subfamily, is characterized by an N-terminal mitochondrial-addressing sequence that allows targeting to the mitochondrial matrix (Fig. 1). NLRX1 is present in the mitochondrial matrix and widely exerts a negative effect on antiviral immune responses in an inflammasome-independent manner, such as by negatively regulating the mitochondrial antiviral signaling protein (MAVS)-mediated signaling pathway during hepatitis C virus infection and typically regulating the NF-κB signaling pathway by inhibiting the binding of TRAF6 to the inhibitor of NF-κB kinase (IKK) [28].
Canonical signaling mechanisms of NLRs
The signaling mechanisms of the NLR family are relatively conserved. When a PAMP is recognized by the LRR domain of an NLR, conformational changes in the NLR occur, which causes ADP/GDP and ATP/GTP to exchange and assemble to form a polymer scaffold, thus triggering downstream signal transduction.
NOD1 and NOD2 signaling
NOD1 and NOD2 have been shown to be critical receptors involved in the cytoplasmic recognition of minimal peptidoglycan motifs from bacterial pathogens. Specifically, NOD1 recognizes γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP), which is primarily found in gram-negative bacteria, whereas NOD2 is activated by MDP, a common peptidoglycan motif in both gram-positive and gram-negative bacteria [29, 30]. Upon specific binding to the ligand, the NACHT domain oligomerizes and initiates the recruitment of interacting proteins, leading to the activation of homotypic CARD and the formation of a signalosome including CARD and the CARD-containing kinase RIPK2 (also called RIP2/RICK) [31, 32]. Afterward, RIPK2 induces the activation of the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways through IKK and transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), thus promoting the transcription of proinflammatory genes [33]. In addition, NOD2 induces autophagy to remove pathogens by recruiting Atg5, Atg12 and Atg16L1 to the plasma membrane at the site of bacterial entry [34] (Fig. 2).
Signaling involving inflammasome assembly
The inflammasome is an oligomeric protein complex that consists of three parts: an NLR molecule (recognition of DAMP/PAMP), an effector molecule (cleavage of pro-interleukin (IL)-Iβ or pro-IL-18) and coupling molecules. The structures of all inflammasomes are roughly the same, and the only difference lies in the diversity of NLR molecules [35]. Among the NLR family, several members participate in the formation of inflammasomes, including NLRC4, NLRP1, NLRP3, NLRP2, NLRP6, NLRP7, and NLRP12. The roles of the remaining identified NLRs in inflammasome formation have not yet been reported [26]. These NLRs are essential activators that play crucial roles in regulating inflammasome activity upon detecting infection or cell damage.
The NLRP3 inflammasome has been one of the most widely studied inflammasomes in recent years. In response to PAMPs/DAMPs, dimerized NLRP3 molecules polymerize the two PYD effector domains and activate apoptosis-associated speck-like protein containing a CARD (ASC) through a homophilic CARD–CARD interaction, which links activated inflammasome sensors to the effector molecule pro-caspase-1 and promotes the autocatalytic activation of caspase-1 via the recruitment of pro-caspase-1 [36]. Thus, the duplex of NLRP3, ASC (PYD + CARD) and the effector complex (CARD + caspase-1) collectively form a structure called the inflammasome [37]. The main function of caspase-1 is to cleave two inactive cytokine precursors, pro-IL-1β and pro-IL-18, into active IL-1β and IL-18, respectively [38]. In some cases, caspase-1 can also cleave gasdermin D at Asp276 and Asp275 to generate an N-terminal cleavage product that triggers a particular type of inflammatory death known as pyroptosis [39]. In addition, it has been reported that some activators, such as lipopolysaccharide (LPS) and ATP, can induce mitochondrial dysfunction and the release of mitochondrial reactive oxygen species (mtROS) and oxidized mitochondrial DNA (mtDNA), activating the NLRP3 inflammasome [40]. Most NLRP3 stimuli induce K+ efflux, which is necessary and sufficient for NLRP3 activation [41]. However, activation of the NLRP3 inflammasome needs to be tightly controlled. NEK7, a serine/threonine kinase required for mitotic spindle formation, is indispensable for NLRP3 inflammasome activation [42]. A recent study has shown that the centrosomal Spata2/CYLD/polo-like kinase 4 (PLK4) signaling axis suppresses NLRP3 inflammasome activation by PLK4-mediated NEK7 phosphorylation, thus inhibiting inflammasome overactivation. In addition, the association between NLRP3 and ASC is also attenuated by silencing β-catenin, the master regulator of the canonical Wnt/β-catenin signaling pathway [43]. Similar to NLRP3, both NLRP1 and NLRC4 have also been shown to assemble inflammasomes via similar mechanisms [44, 45] (Fig. 2).
NLRs in autoimmune diseases
Autoimmune diseases are chronic immune diseases characterized by inappropriate immune responses to the body’s own cells, tissues, or organs. Although numerous progressive treatment strategies can ameliorate the progression of these diseases, autoimmune diseases are still incurable, and the long-term prognoses are unsatisfactory. Therefore, it is urgent to explore novel, individualized therapeutic targets. Accumulating evidence has shown that NLRs are involved in the pathogenesis and development of multiple autoimmune diseases (Fig. 3, Table 1).

NLRs are involved in the development of multiple autoimmune diseases, including IBD, RA, SLE, psoriasis, MS, and T1D. Abnormal expression of NLRs and excessive inflammasome activation drive the pathogenesis of autoimmune diseases
IBD
IBD is a chronic inflammatory disorder of the gastrointestinal tract that is characterized by complicated and relapsing inflammation and includes ulcerative colitis (UC) and Crohn’s disease (CD). Host genes, the environment, and other factors lead to an imbalance in the mucosal immune response to the commensal intestinal microbiota, and innate immunity plays a direct role in the pathogenesis of this disease.
NOD1 and NOD2 are expressed throughout the intestinal epithelium and in various intestinal immune cells, such as T cells, B cells, and monocytes, and can induce the NF-κB, MAPK, and interferon (IFN) signaling pathways [46]. NOD1/2 signaling in the context of maintaining intestinal homeostasis under different conditions is complicated. On the one hand, intestinal intraepithelial lymphocytes (IELs) are mainly distributed in the intestinal epithelial cell layer and play a protective role in IBD. Jiang et al. reported that IELs were reduced significantly in Nod2−/− mice, and the recognition of gut microbiota by NOD2 was important in maintaining the homeostasis of IELs [47]. Furthermore, Nod1−/−;Nod2−/− mice had increased paracellular permeability, decreased E-cadherin expression and increased susceptibility to dextran sulfate sodium (DSS) based on the effects of commensal and probiotic bacteria [48]. On the other hand, NOD2 signaling promoted hyperresponsive macrophages and colitis in IL-10-deficient mice, and the loss of Nod2 in IL-10−/− mice resulted in significant amelioration of chronic colitis [49]. In addition, Nod2 deletion in SAMP mice, a murine model of spontaneous ileitis, decreased the severity of chronic ileitis by reducing Th2 cytokine production and Th2 transcription factor activation [50].
Members of the NLRP subfamily are also closely related to the course of IBD. Among them, NLRP3 has regularly been a research focus. In 2017, Lazaridis et al. reported for the first time that the NLRP3 inflammasome is more highly activated in CD patients than in healthy controls [51]. In 2018, Ranson et al. demonstrated that the expression of NLRP3 was upregulated in active UC and CD [52]. Activation of the NLRP3 inflammasome is a key step in the initiation of IBD, leading to tissue damage and clinical manifestations. Recently, a study by Chen et al. showed that NEK7, an important component of the NLRP3 inflammasome in macrophages, interacted with NLRP3 to affect IBD via pyroptosis [53]. However, NLRP3 also exhibits protective effects in mucosal immunity. When the integrity of the intestinal epithelium is impaired, activation of the NLRP3 inflammasome can promote the repair and regeneration of the intestinal mucosa. Nlrp3−/− mice were found to be more susceptible to DSS- and 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced experimental colitis [54]. More interestingly, a study unexpectedly revealed that hyperactive NLRP3 inflammasomes remodeled the gut microbiota and neutralized intestinal inflammation by inducing the production of regulatory T cells (Tregs) in Nlrp3R258W mutant mice [55]. In addition, Nlrp1b−/− mice exhibited significant increases in morbidity, inflammation, and colitis-associated tumorigenesis [56]. In contrast, a recent study by Tye et al. reported that the NLRP1 inflammasome was a key negative regulator of butyrate-producing protective commensal bacteria and exacerbated DSS-induced colitis by limiting the production of Clostridiales in the gastrointestinal tract [57]. The NLRP6 inflammasome also plays a role in maintaining homeostasis between the microflora and the host in the digestive tract [58]. Overall, it is critical to clarify the exact mechanisms of inflammasomes for the effective treatment of IBD.
RA
RA is a chronic inflammatory arthropathy that is characterized by synovitis and irreversible joint injury. The exact cause of RA is not clear yet. As the master regulator of MHCII gene expression, the CIITA locus is a strong autoimmune risk factor that regulates the expression of two major risk loci, DRB1 and DQB1. Notably, Ronninger et al. showed that a single-nucleotide polymorphism (SNP) in the CIITA promoter, rs3087456 (-168 A/G), is involved in RA in Scandinavian populations [59]. NLRC subfamily members NOD1 and NOD2 have also been correlated with RA. NOD1 can promote the production of inflammatory mediators in the synovial tissue of RA patients [60]. Franca et al. observed that NOD2/RIP2 signaling was upregulated in peripheral blood mononuclear cells (PBMCs) isolated from RA patients after stimulation with MDP [61]. Similarly, proinflammatory cytokine and NF-κB levels in fibroblast-like synoviocytes (FLSs) in RA can be reduced by downregulating NOD2 expression [62]. In addition, transient transfection with siRNA against NLRC5 can significantly reduce the level of proinflammatory cytokines (TNF-α and IL-6) and inhibit the proliferation of FLSs in the synovial tissues (STs) of rats with adjuvant-induced arthritis by suppressing activation of the NF-κB signaling pathway; thus, NLRC5 may be a candidate therapeutic target for RA treatment [63].
Recently, increasing evidence has suggested that the NLRP3 inflammasome participates in the pathogenesis of RA. Jenko et al. demonstrated that the interaction between NLRP3 rs35829419 and CARD8 rs2043211 facilitated RA susceptibility and that the latter promoted early-stage and long-term disease activity [64]. A study showed that arthritic symptoms and cartilage erosion in collagen-induced arthritis (CIA) mice could be effectively attenuated by treatment with a selective NLRP3 inhibitor, suggesting that NLRP3 is associated with the susceptibility and disease activity of RA [14]. Knockdown of NLRP3 can inhibit Th17 cell differentiation and alleviate RA, implied that the NLRP3 inflammasome contributes to the pathogenesis of this disease by promoting Th17 cell differentiation [65]. A new study in 2020 proposed that allosteric enhancement of calcium-sensing receptor signaling led to the exacerbation of RA by driving NLRP3 inflammasome activation [66]. Other members of the NLRP subfamily also participate in the pathological process of arthritis. The NLRP1 inflammasome has been reported to play a protective role in RA inflammation by reducing the activation of caspase-1 [67]. Moreover, further analysis has revealed that NLRP2 rs703468 and rs2217659, which are located on chromosome 19q13.42, are prominently associated with RA [68]. NLRP6, which mediates the interaction between TAK1-binding protein 2/3 and tripartite motif 38 in RA-FLSs in response to TNF-α, may also be a potential therapeutic target for RA [69]. NLRP12 has emerged as a negative regulator of inflammation. Prado et al. demonstrated that NLRP12 controlled the severity of RA by acting as a checkpoint inhibitor of Th17 cell differentiation [70].
SLE
SLE is a chronic autoimmune disease characterized by the abnormal accumulation of autoreactive T lymphocytes and the production of autoantibodies against self-antigens that eventually lead to a wide spectrum of clinical manifestations involving multiple systems and organs.
NOD2 is expressed in many types of immune cells, including dendritic cells, macrophages, T lymphocytes, and B lymphocytes [71,72,73]. Compared to those of IBD and RA, studies exploring the expression and function of NOD2 in SLE are very limited. Yu et al. reported that NOD2 expression in monocytes and plasmacytoid dendritic cells in SLE patients was significantly increased compared to that in healthy controls. Bacterial exposure increased the expression of NOD2 in monocytes, which resulted in the production of proinflammatory cytokines by PBMCs and exacerbated SLE conditions [72]. However, the detailed cellular regulatory mechanisms of NOD2 activation and the differential functions of NOD2 in other immune cell types in SLE need to be clarified.
Recent evidence suggests the critical role of inflammasomes in the predisposition of an individual to SLE. Pontillo et al. analyzed 14 SNPs in 7 inflammasome genes (NLRP1, NLRP3, NLRC4, AIM2, CARD8, CASP1, and IL1B) and first demonstrated that the NLRP1 rs2670660 SNP and the NLRP1 rs12150220-rs2670660 A-G haplotype increased the risk of SLE, especially the development of nephritis, rash and arthritis [74]. A study by Kahlenberg et al. reported that NLRP3 was an important intracellular sensor in SLE. Neutrophil extracellular traps (NETs) and cathelicidin LL-37-mediated NLRP3 inflammasome activation was enhanced in lupus macrophages, and induced inflammatory responses by stimulating IL-1β and IL-18 secretion [75]. Moreover, the binding of self double-stranded DNA (dsDNA) and anti-dsDNA antibodies also activated the NLRP3 inflammasome by inducing ROS synthesis and K+ efflux, which induced IL-1β production and the Th17 cell response [76]. In addition, NLRP3 is activated in podocytes, contributing to the pathogenesis of podocyte injury and the development of proteinuria in patients with lupus nephritis (LN) [77]. In lupus-prone NZM2328 and MRL/lpr mice, IgG induced both RIP3-dependent necroptosis and NLRP3 inflammasome pathway activation in a podocyte cell line during LN [15]. Conversely, Yang et al. surprisingly showed that the expression of NLRP3/NLRP1 inflammasomes was significantly downregulated in PBMCs from patients with SLE [78]. These conflicting results may be due to the direct inhibitory effect of NLRP3/NLRP1 inflammasomes by the overactivation of T cells, as well as the increased serum levels of IFN-I in SLE patients.
Psoriasis
Psoriasis is one of the most common skin inflammatory diseases, with a global incidence of ~1–3%. This disease is characterized by hyperproliferation and abnormal differentiation of epidermal keratinocytes, accompanied by the infiltration of immune cells in the skin.
Since epidermal thickening is one of the most important features of psoriasis, Tervaniemi et al. focused on the psoriatic epidermis and assayed the transcriptomes of split-thickness skin grafts utilizing RNA 5′-end sequencing. The researchers highlighted the pivotal role of NLR signaling pathways and demonstrated that the NLR signaling genes NOD2, PYCARD, CARD6, and IFI16 were upregulated in the psoriatic epidermis [16]. Likewise, integrative methylome and transcriptome analysis also revealed that NLR signaling was a key biological pathway for psoriasis in the Chinese Han population [79].
Mutations of the CARD14 gene, which is one of the members of the CARD family, have been associated with several psoriasis clinical phenotypes, including psoriasis vulgaris, pustular psoriasis and pityriasis rubra pilaris. In 2012, Jordan et al. established that psoriasis susceptibility locus 2 (PSORS2) was due to gain-of-function mutations in the CARD14 gene, where the c.349 G > A and c.413 A > C substitutions led to enhanced NF-κB activation and upregulated psoriasis-associated genes in keratinocytes [80]. Further research identified 15 additional rare missense variants within CARD14 in psoriasis cases compared to healthy controls [81]. Recent studies reported that gain-of-function mutations in CARD14 resulted in spontaneous psoriasis-like skin inflammation through enhanced keratinocyte responses to IL-17A. Hyperactivation of CARD14 alone was sufficient to drive IL-23/IL-17-mediated psoriasis in vivo [82]. In addition, it is well recognized that inflammasomes also participate in the pathogenesis of psoriasis. Carlström et al. demonstrated that certain polymorphisms in NLRP3 rs10733113 and CARD8 rs2043211 are associated with psoriasis susceptibility [83].
MS
MS is the most common demyelinating disease of the central nervous system (CNS), especially the optic nerve, spinal cord, and brainstem, and is characterized by multiple lesions, remission, and recurrence of the disease.
Accumulating evidence supports the correlations between NOD1/2-mediated pathways and the development of experimental autoimmune encephalomyelitis (EAE), which is an ideal animal model to explore the pathogenesis of MS. APCs invade the CNS upon activation by cytosolic PGNs through NOD1 and NOD2, which mediate the recruitment and phosphorylation of RIP2, increasing the activity of neuronal nitric oxide synthase (NOS) and the accumulation of nitric oxide (NO) in oligodendrocyte precursor cells and causing the neuroinflammation that drives EAE pathogenesis [84, 85]. In addition, CIITA polymorphism -168A/G (rs3087456) not only participates in RA but also influences the severity of MS in combination with CIITA + 1614G/C (rs4774), a missense variant connected to SLE susceptibility [86, 87].
Indeed, increasing evidence suggests that the activation of inflammasomes also correlates with the severity of MS. Maver et al. observed that a potentially pathogenic homozygous missense variant in NLRP1 (Gly587Ser) promoted the production of proinflammatory cytokines, as well as the global activation of NLRP1-mediated signaling pathways in MS [88]. Gris et al. also demonstrated that the NLRP3 inflammasome played a critical role during EAE development by mediating Th1 and Th17 responses. Nlrp3−/− mice exhibited improved spinal cord histology and reduced disease severity through the reduced production of IFN-γ and IL-17 [89]. In 2020, Malhotra et al. noted that the NLRP3 inflammasome was a prognostic factor and therapeutic target in patients with primary progressive MS [90]. Inhibition of the NLRP3 inflammasome prevents cognitive deficits by suppressing the transformation of astrocytes to the neurotoxic A1 phenotype [91].
Interestingly, several studies have suggested that NLRs have dual roles in MS onset and progression [92]. The gain-of-function of NLRP3 in CD4+ T cells can ameliorate EAE by promoting Th2 and Treg differentiation [93]. In a recent study, NLRP12 was reported to decrease IFN-γ and IL-2 production by suppressing T-cell proliferation and the Th1 response while promoting the Th2 response in T-cell-mediated EAE, suggesting that NLRP12 exerts an anti-inflammatory effect on MS by regulating the Th1/Th2 balance [94]. In addition, NLRC3 exerts a negative effect by regulating the p38 signaling pathway in APCs; in particular, vaccination with NLRC3-overexpressing dendritic cells ameliorated the pathogenesis of EAE [95]. NLRX1 also plays a crucial role in controlling the early stages of CNS inflammation by intrinsically inhibiting the autoreactive T-cell response and protecting against the activation of innate immune cells in the CNS [96].
Other autoimmune diseases
NLRs also play an important role in other relatively rare autoimmune diseases. T1D, formerly known as insulin-dependent diabetes, is an autoimmune disease characterized by insufficient insulin levels in the body, which destroys pancreatic β cells. It has been shown that NOD2 participates in T1D by inducing Th1 and Th17 cells in the pancreatic lymph nodes through a mechanism that is potentially mediated by the gut microbiota. Nonobese diabetic mice that are deficient in Nod2 are protected from T1D development [18, 97]. Two SNPs, rs3087456 and rs11074932, in CIITA have also been reported to be associated with susceptibility to T1D [98]. Analogously, NLRP3 ablation reduces pancreatic β cell impairment by reducing the expression of the chemokines CCL5 and CXCL10, demonstrating that NLRP3 deficiency contributes to the improvements in T1D [99]. In addition, inflammasomes also correlate with the pathogenesis of AIT, a representative organ-specific autoimmune disorder accompanied by mild albuminuria and nephrotic syndrome, and AIT patients exhibit prominently increased expression of AIM2, ASC, NLRC4, NLRP1, NLRP3, caspase-1, pro-IL-1β, and pro-IL-18 [19]. AIH is a chronic progressive liver inflammatory disease characterized by elevated serum transaminase, high γ-globulinemia and positive autoantibodies and can rapidly deteriorate into liver cirrhosis and liver failure. Luan et al. found that NLRP3-dependent IL-1β promoted the pathogenesis of AIH; in particular, NLRP3−/− and caspase-1−/− mice were protected from hepatitis and exhibited decreased levels of serum aminotransferase/aspartate transaminase, suggesting potential therapeutic strategies for AIH [20]. Vitiligo is a common acquired pigmented dermatosis that is characterized by localized or generalized complete depigmentation of the skin and mucosa. Increased expression of NLRP1 and IL‐1β has been observed in the perilesional skin of patients with active vitiligo [100]. Li et al. also showed that NLRP3 inflammasome activation induced by oxidative stress in keratinocytes promoted the cutaneous T-cell response [101]. In addition, NLRP3-dependent endothelial cell pyroptosis plays a significant role in coronary endothelial damage in Kawasaki disease (KD), a kind of acute febrile and eruptive pediatric disease with systemic vasculitis [102]. Celiac disease is a chronic autoimmune disorder induced by intolerance to gluten peptides that develops in genetically susceptible individuals. The p31–43 peptide from α-gliadin participates in the pathogenesis of celiac disease at an early stage by inducing NLRP3-dependent mucosal damage [103]. Moreover, over 25 defense-related genes, including CIITA, are significantly upregulated in intestinal epithelial cells in patients with active celiac disease [104].
NLRs in autoimmune diseases treatment
NLRCs in autoimmune disease therapy
The NLRC family is the second-largest subfamily of NLRs, of which NOD1 and NOD2 are two primary members and can directly activate multiple inflammatory pathways that result in cytokine production by binding to the CARD domain of RIP2 kinase. Theoretically, antagonists of NOD1 or NOD2 could have applications in several acute and chronic autoimmune diseases. However, since there is no way to measure a compound’s direct binding capacity to either NOD1 or NOD2, several groups have recently explored the effect of inhibiting the common NOD1 and NOD2 signaling kinase RIP2, a multidomain, dual specificity and tractable downstream kinase that is ubiquitously expressed (Table 2).
Early kinase inhibitors such as p38 inhibitors also effectively inhibit the activity of RIP2 kinase. Hollenbach et al. reported that SB203580 ameliorated DSS-induced experimental colitis by inhibiting the p38/MAPK and NOD2/RIP2/NF-κB signaling pathways [105]. GSK583, a selective 4-aminoquinoline-based next-generation RIP2 inhibitor, effectively blocked the downstream signaling of NOD2 and exhibited a robust ability to modulate inflammatory responses in the intestinal mucosa of IBD patients [106]. In addition, a recent study indicated that 2E7, a monoclonal antibody against bacterial PGN subunits, suppressed the development of autoimmune arthritis and EAE by blocking NOD2-mediated pathways [107]. Conversely, MIS416, a novel and myeloid-directed microparticle derived from Propionibacterium acnes, downregulated inflammatory T-cell activity via negative feedback pathways induced by the activation of TLR9 and NOD2, thus reducing EAE disease severity [108]. A phase 1/2 clinical trial of MIS416 was completed in 2012, but it was disappointing that the development of MIS416 was terminated in June 2017 because it was not beneficial as a treatment for secondary progressive multiple sclerosis (SPMS).
NLRC5 has also been proven to be a candidate therapeutic target for autoimmune disease treatment. In RA, NLRC5 is overexpressed in synovial tissues and cells, accompanied by the overexpression of inflammatory cytokines and the hyperproliferation of FLSs [63]. It has been reported that the overexpression of long noncoding RNA Fer-1-like protein 4 (FER1L4), a tumor suppressor in various cancers, can decrease the expression of NLRC5 and inflammatory cytokines in STs and FLSs, suggesting that FER1L4 regulates RA by targeting NLRC5. Coincidently, another long noncoding RNA, maternally expressed gene 3 (MEG3), had an analogous pathway. Treatment with the methylation inhibitor 5-aza-2-deoxycytidine (5-azadC) inhibited hypermethylation of the FER1L4 and MEG3 promoters, as well as the expression of NLRC5 in CFA-induced synovial tissues and cells [109, 110].
NLRPs in autoimmune diseases therapy
Based on studies focusing on the effects of NLRPs on autoimmune diseases, many approaches have been developed to target the regulation of NLRPs, of which NLRP3 is the best-studied and most well-characterized member of the NLRP subfamily (Table 2).
NLRPs in IBD therapy
Although the precise pathogenic mechanism of IBD is unknown, recent studies have revealed that uncontrolled activation of the NLRP3 inflammasome plays a major role in the pathogenesis of IBD, and the secretion of mature IL-1β and IL-18 is associated with exacerbated colitis. To date, many NLRP3 inhibitors have been implemented as pharmacological agents in the treatment of IBD.
It has been reported that many flavonoids have significant immunomodulatory effects. Wogonoside is a glucuronide metabolite of the bioactive flavonoid wogonin that can significantly ameliorate DSS-induced colitis. The underlying mechanisms of the protective effect are attributed to the inhibition of NF-κB activation and NLRP3 inflammasome formation by suppressing caspase-1 activity in the colon [111]. Natural products with similar mechanisms include alpinetin [112], oroxylin A [113], and naringin [114].
mtROS and mtDNA derived from damaged mitochondria play pivotal roles in the initiation and regulation of NLRP3 inflammasome activation. Asiatic acid is a natural triterpenoid compound extracted from the Chinese herb Centella asiatica. Guo et al. reported that asiatic acid suppressed NLRP3 inflammasome activation by inhibiting mtROS generation and preventing mitochondrial membrane potential collapse, which ameliorated DSS-induced experimental colitis in mice [115]. Dimethyl fumarate is a first-line drug for patients with relapsing MS that can dose-dependently achieve significant amelioration of colonic pathological damage in DSS-induced mice. The underlying mechanisms involve inhibiting the NLRP3 inflammasome by activating Nrf2, which decreases mtROS generation and mtDNA release [116].
In addition, microRNAs (miRNAs) are emerging as important regulators in the maintenance of intestinal homeostasis. Neudecker et al. discovered that miR-223 restrained pathological intestinal inflammation by controlling myeloid-specific NLRP3 inflammasome activity. DSS-induced colitis is exacerbated in miR-223-/y mice compared to wild-type mice, and nanoparticle delivery of a miR-223 mimetic attenuates intestinal inflammation [117]. MCC950, a potent, highly specific and well-characterized small molecule inhibitor, effectively inhibits both canonical and noncanonical activation of the NLRP3 inflammasome. MCC950 treatment significantly ameliorates the disease activity index of the Winnie spontaneous chronic colitis mouse model, which mimics UC, accompanied by decreased release of proinflammatory cytokines, chemokines, and NO in colonic explants [118].
Wang et al. indicated that in addition to targeting NLRP3, secoisolariciresinol diglucoside (SDG) ameliorated DSS-induced colitis by inhibiting NLRP1 inflammasome activation and partly disrupting NF-κB activation [119]. More interestingly, apigenin, a widely distributed dietary flavone, modulates the composition of the gut microbiota through NLRP6 signaling to protect mice from DSS-induced acute colitis [120].
NLRPs in RA therapy
Traditional Chinese medicine has been widely used to treat arthritis for thousands of years. Taraxasterol, one of the bioactive components of Taraxacum officinale, exerts protective effects on IL-1β-induced human RA-FLSs and CIA mice by modulating the NF-κB and NLRP3 inflammasome signaling pathways [121]. Similarly, the Curculigo orchioides extract curculigoside A is a considerable therapeutic compound for alleviating RA progression by maintaining the oxidant/antioxidant balance and downregulating the expression of NF-κB and NLRP3 [122]. Other synthetic inhibitors also play potential preventive or therapeutic roles in RA. Methotrexate, which is usually used as an initial cancer therapy that inhibits dihydrofolate reductase, exhibits satisfactory effects on RA by attenuating the activation of NF-κB and NLRP3/caspase-1 proinflammatory pathways, as well as regulating related metabolic networks [123].
NLRPs in SLE therapy
Blocking NLRP3 activity has yielded fruitful results in SLE. Activation of the NLRP3 inflammasome requires 2 signals: a priming signal through PRRs that activates the NF-κB or AP-1 pathways to upregulate the expression of NLRP3 and other inflammasome components and an activating signal that promotes NLRP3 oligomerization to assemble the inflammasome complex [124]. Icariin significantly attenuates renal disease in MRL/lpr mice by inhibiting NF-κB activation and TNF-α production, leading to reductions in the formation of the NLRP3 inflammasome and the production of IL-1β [125]. Similarly, sophocarpine is a quinolizidine alkaloid that is widely used in traditional Chinese medicine and suppresses NLRP3 inflammasome activation by inhibiting the NF-κB pathway, thereby attenuating murine LN [126]. Other NLRP3 signaling inhibitors, such as piperine [127], curcumin [128], and procyanidin B2 [129], have also been shown to be effective in the treatment of SLE.
Some botanical and synthetic inhibitors that target oxidative stress, including ROS production and mitochondrial alterations, may also have potential preventive or therapeutic effects on SLE. Bonomini et al. demonstrated that melatonin supplementation protected against renal injury by enhancing the Nrf2 antioxidant signaling pathway and decreasing renal NLRP3 inflammasome activation in chronic pristane-induced LN [130]. Additionally, baicalein is a flavonoid derived from the roots of the traditional Chinese herbal medicine Scutellaria baicalensis Georgi that reduces proteinuria and improves renal function through NLRP3 inflammasome inhibition by inducing Nrf2/HO-1 signaling [131].
Autophagy is an essential cellular mechanism by which cellular material including damaged organelles, aggregated and misfolded proteins is delivered to lysosomes for degradation, thus negatively regulates the NLRP3 inflammasome activation and the innate immune response [132]. Therefore, targeting autophagy, which affects the activation of NLRP3, can also be used for SLE treatment. Lin et al. reported that a major absorbable intestinal bacterial metabolite of ginsenosides, M1, exerted therapeutic effects on a mouse model of accelerated and severe lupus nephritis (ASLN) by activating the sirtuin3/autophagy axis and inhibiting NLRP3 inflammasome activation [133]. Additionally, miRNAs that regulate target gene expression are still a research hotspot at present. Let-7f-5p, a novel inflammation-related miRNA, directly ameliorates inflammation by repressing NLRP3 expression in bone marrow-derived mesenchymal stem cells from SLE patients [134].
NLRPs in psoriasis therapy
Irrera et al. reported that psoriasis-like lesions and epithelial thickness were reduced in the skin of NLRP3−/− mice compared with the skin of wild-type mice challenged with imiquimod (IMQ). BAY 11-7082 significantly ameliorated psoriasis-like dermatitis by inhibiting both the NF-κB and NLRP3 inflammasome pathways [135]. Deng et al. discovered that cycloastragenol, an active triterpenoid saponin component, selectively modulated the function of macrophages by suppressing NLRP3 inflammasome complex assembly and NLRP3 inflammasome-mediated pyroptosis, thus improving IMQ-induced psoriasiform dermatitis [136]. Datura Metel L. has been confirmed to have a marked therapeutic effect on psoriasis, decreasing the mRNA expression levels of IKKα, NF-κB, ASC, NLRP3, and caspase-1, as well as the production of the proinflammatory cytokines IL-1β, IL-6, TNF-α, monocyte chemotactic protein 1 (MCP-1) and IFN-γ [137]. Apart from conventional therapy, some novel treatment strategies have been developed. Since miRNA155 expression levels are markedly elevated during the pathology of psoriasis and may be dependent on NLRP3 inflammasome activation, Luo et al. proposed silencing miRNA155 to treat the disease [138].
NLRPs in MS therapy
The star molecule MCC950 is a potent and selective NLRP3 inflammasome inhibitor that ameliorates the severity of EAE by blocking activation of the NLRP3 inflammasome [139]. MCC950 in combination with rapamycin exerts improved efficacy in MS [140]. The hydroxyl-sulfonamide analog JC-171 has been reported to modify the NLRP3/ASC interaction stimulated by LPS/ATP and attenuate the progression of EAE [141]. Similarly, the selective NLRP3 inhibitor OLT1177 significantly decreases the levels of TNF-α, IL-6, and IL-1β in the spinal cord in EAE mice [142]. In addition, prednisone has been shown to exert protective effects on demyelinating diseases by inhibiting NLRP3 and relevant inflammatory cytokines and chemokines [143]. Bai et al. have demonstrated that tetramethylpyrazine might be a promising candidate for MS treatment by reducing NLRP3 inflammasome expression and modulating the Th1/Th17/Th2 response [144]. Recently, Koo et al. suggested that the LRR domain of the NLRX1 protein reduced the number of infiltrating T cells producing inflammatory cytokines. A fusion protein of the LRR and the blood-brain barrier (BBB)-permeable peptide dNP2 could regulate CNS inflammation and significantly ameliorate EAE disease severity [145].
NLRPs in other autoimmune diseases therapy
Recently, Wu et al. showed that low-methoxyl pectin (LMP) decreased activation of the NLRP3 inflammasome by increasing cecal bacterial species to augment the production of short-chain fatty acids, thus reshaping the pancreatic immune environment in T1D [146]. In addition, ginsenoside Rg1 has been shown to be a promising drug to prevent the development of streptozotocin-induced T1D by attenuating the functions of NLRP3 in the mouse liver and pancreas, as well as inhibiting the secretion of IL-1β and IL-18 [147]. At present, there are still no effective treatment for AIH, and the disease survival rate can only be improved by a combination of corticosteroids and azathioprine. In 2017, Chen et al. reported that miR223-containing exosomes derived from bone marrow stem cells (BMSCs) exhibited protective effects on AIH by downregulating the expression of NLRP3 and caspase-1 [148] (Table 2).
Other NLR subfamilies in autoimmune diseases therapy
Recently, CIITA and NLRX1 have been shown to participate in the pathogenesis of autoimmune diseases. As a consequence, some compounds and active ingredients targeting these proteins directly have been developed. Midura-Kiela et al. showed that curcumin exerted protective effects on colonic epithelial cells by suppressing the IFN-γ-induced transcription of MHCII and CIITA [149]. In 2019, Liu et al. showed that the ERβ selective agonist diarylpropionitrile abrogated CIITA expression and inhibited IFN-γ regulatory factor 1 (IRF-1) translocation into the nucleus, ameliorating the inflammatory response and symptoms in an EAE model [150]. In addition, Leber and colleagues demonstrated that NX-13, which specifically targets the binding pocket of NLRX1 in the colon, decreased the differentiation of CD4+ T cells into Th1 and Th17 subsets, as well as the overexpression of proinflammatory cytokines, thus attenuating the severity of IBD [151] (Table 2). In 2020, NX-13 was approved to enter a phase I clinical trial for the treatment of IBD.
Perspectives
Accumulating evidence shows that NLRs play key roles in the development and functions of immune cells through multiple canonical immunomodulatory mechanisms. For example, NOD1 and NOD2 are two primary members of the NLRC subfamily and act on the downstream serine/threonine kinase RIP2, resulting in the activation of various signaling cascades, including the MAPK and NF-κB pathways. Other NLRs, such as NLRP1, NLRP3, NLRC4, and NLRC6, have been reported to regulate the secretion of proinflammatory cytokines through the formation of inflammasomes. However, in recent years, many studies in the field of NLRs have shown that the functions of these proteins may be more diverse than originally thought. In addition, the biochemical processes that regulate the activation and downstream functions of NLRs need to be further investigated.
To date, the functions of NLRs have been controversial, with protective and harmful effects on a variety of autoimmune diseases. For example, NOD2 is a critical regulator of immune homeostasis, as mutations in NOD2 are associated with several autoinflammatory diseases. Napier et al. reported that NOD2 suppressed arthritis in SKG mice and that Nod2−/− SKG mice had a worsened form of arthritis through augmented Th17 responses [152]. However, other experiments showed that NOD2/RIP2 signaling was upregulated in the immune cells of RA patients, and inhibiting the NOD2/RIP2 signaling pathway could reduce the proliferation and inflammation of FLSs [61]. Similarly, inflammasomes consisting of NLRs, the adapter protein ASC, and the effector molecule pro-caspase-1 are highlighted in the current research. Increasing evidence has shown that the hyperactivation of inflammasomes plays a pathological role in colitis. However, Hirota et al. found that Nlrp3−/− mice had increased susceptibility to experimental colitis, accompanied by a reduction in colonic antimicrobial secretions and the production of a unique intestinal microbiota, which confirmed an essential role for the NLRP3 inflammasome in the regulation of intestinal homeostasis [54]. A recent report by Yao et al. also suggested that balanced inflammasome activity was critical for maintaining intestinal homeostasis [55]. Collectively, there is an urgent need to further explore the exact mechanisms of NLR signaling pathways in complex in vivo conditions to find appropriate inhibitors.
Considerable compounds targeting NLRs have been developed and validated in cellular and animal models of autoimmune diseases, and various botanicals with fewer side effects than conventional drugs have attracted the attention of many researchers. However, the nonspecific effects of these compounds have limited their clinical potential. Broad anti-inflammatory activity might cause unwanted immunosuppression and increase the risk of infection. In addition, agents that act on the upstream of NLRs, such as K+ efflux, or on the downstream, such as IL-1β production, also have other unavoidable biological consequences.
Fortunately, a few compounds with high selectivity and specificity have been identified, including MCC950, NX-13, JC-171, and OLT1177, all of which have shown strong inhibitory activity on NLR signaling pathways and beneficial effects on experimental mouse models of NLR-related autoimmune diseases. There are currently no NLR-related drugs on the market, and we ought to focus on investigating whether the protective effects of these drugs observed in cellular and animal models could be replicated in human clinical trials. In addition, drugs targeting NLRs in the treatment of autoimmune diseases mainly focus on the NOD1, NOD2, and NLRP3 signaling pathways, but other rarely studied members of the NLR family, such CIITA and NLRX, also widely participate in the pathogenesis of diseases. These findings prompt us to discover and develop a greater variety of NLR inhibitors for the treatment of autoimmune diseases.
References
Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16:35–50.
Zhu G, Xu Y, Cen X, Nandakumar KS, Liu S, Cheng K. Targeting pattern-recognition receptors to discover new small molecule immune modulators. Eur J Med Chem. 2018;144:82–92.
Michallet MC, Rota G, Maslowski K, Guarda G. Innate receptors for adaptive immunity. Curr Opin Microbiol. 2013;16:296–302.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20.
Macleod C, Bryant CE. Visualising pattern recognition receptor signalling. Biochem Soc Trans. 2017;45:1077–85.
Elias T, Zahava V. Innate immune-responses and their role in driving autoimmunity. Autoimmun Rev. 2019;18:306–11.
Saferding V, Bluml S. Innate immunity as the trigger of systemic autoimmune diseases. J Autoimmun. 2020;110:1023.
Neerincx A, Castro W, Guarda G, Kufer TA. NLRC5, at the heart of antigen presentation. Front Immunol. 2013;4:397.
Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol. 2019;10:276.
Meunier E, Broz P. Evolutionary convergence and divergence in NLR function and structure. Trends Immunol. 2017;38:744–57.
Heim VJ, Stafford CA, Nachbur U. NOD signaling and cell death. Front Cell Dev Biol. 2019;7:208.
Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. 2015;278:369–95.
Coronel-Restrepo N, Posso-Osorio I, Naranjo-Escobar J, Tobon GJ. Autoimmune diseases and their relation with immunological, neurological and endocrinological axes. Autoimmun Rev. 2017;16:684–92.
Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol. 2018;194:231–43.
Guo CH, Fu R, Zhou MJ, Wang S, Huang YF, Hu HQ, et al. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J Autoimmun. 2019;103:102286.
Tervaniemi MH, Katayama S, Skoog T, Siitonen HA, Vuola J, Nuutila K, et al. NOD-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci Rep. 2016;6:22745.
Deerhake ME, Biswas DD, Barclay WE, Shinohara ML. Pattern recognition receptors in multiple sclerosis and its animal models. Front Immunol. 2019;10:2644.
Costa FRC, Francozo MCS, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. 2016;213:1223–39.
Guo QL, Wu Y, Hou YY, Liu YP, Liu TT, Zhang H, et al. Cytokine secretion and pyroptosis of thyroid follicular cells mediated by enhanced NLRP3, NLRP1, NLRC4, and AIM2 inflammasomes are associated with autoimmune thyroiditis. Front Immunol. 2018;9:1197.
Luan J, Zhang X, Wang S, Li Y, Fan J, Chen W, et al. NOD-like receptor protein 3 inflammasome-dependent IL-1beta accelerated ConA-induced hepatitis. Front Immunol. 2018;9:758.
Liu P, Lu Z, Liu L, Li R, Liang Z, Shen M, et al. NOD-like receptor signaling in inflammation-associated cancers: from functions to targeted therapies. Phytomedicine. 2019;64:152925.
Accolla RS, Ramia E, Tedeschi A, Forlani G. CIITA-driven MHC class II expressing tumor cells as antigen presenting cell performers: toward the construction of an optimal anti-tumor vaccine. Front Immunol. 2019;10:1806.
Davoodi J, Ghahremani MH, Es-haghi A, Mohammad-gholi A, MacKenzie A. Neuronal apoptosis inhibitory protein, NAIP, is an inhibitor of procaspase-9. Int J Biochem Cell B. 2010;42:958–64.
Correa RG, Milutinovic S, Reed JC. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep. 2012;32:597–608.
Kim YK, Shin JS, Nahm MH. NOD-like receptors in infection, immunity, and diseases. Yonsei Med J. 2016;57:5–14.
Velloso FJ, Trombetta-Lima M, Anschau V, Sogayar MC, Correa RG. NOD-like receptors: major players (and targets) in the interface between innate immunity and cancer. Biosci Rep. 2019;39:BSR20181709.
Davis BK, Wen HT, Ting JPY. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35. 29
Stokman G, Kors L, Bakker PJ, Rampanelli E, Claessen N, Teske GJD, et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J Exp Med. 2017;214:2405–20.
Girardin SE, Boneca IG, Carneiro LAM, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science. 2003;300:1584–7.
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72.
Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–9.
Nembrini C, Kisielow J, Shamshiev AT, Tortola L, Coyle AJ, Kopf M, et al. The kinase activity of Rip2 determines its stability and consequently Nod1-and Nod2-mediated immune responses. J Biol Chem. 2009;284:19183–8.
Hu ZH, Chai JJ. Structural mechanisms in NLR inflammasome assembly and signaling. Curr Top Microbiol. 2016;397:23–42.
Motta V, Soares F, Sun T, Philpott DJ. Nod-like receptors: versatile cytosolic sentinels. Physiol Rev. 2015;95:149–78.
Di Virgilio F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol Rev. 2013;65:872–905.
Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun. 2016;7:11929.
Mathur A, Hayward JA, Man SM. Molecular mechanisms of inflammasome signaling. J Leukoc Biol. 2018;103:233–57.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Liu X, Zhang ZB, Ruan JB, Pan YD, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14.
He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–21.
Yang XD, Li WG, Zhang SY, Wu DD, Jiang XL, Tan R, et al. PLK4 deubiquitination by Spata2-CYLD suppresses NEK7-mediated NLRP3 inflammasome activation at the centrosome. EMBO J. 2020;39:e102201.
Huang LM, Luo RH, Li J, Wang D, Zhang YN, Liu LP, et al. Beta-catenin promotes NLRP3 inflammasome activation via increasing the association between NLRP3 and ASC. Mol Immunol. 2020;121:186–94.
Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–24.
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–8.
Mukherjee T, Hovingh ES, Foerster EG, Abdel-Nour M, Philpott DJ, Girardin SE. NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys. 2019;670:69–81.
Jiang W, Wang XQ, Zeng BH, Liu L, Tardivel A, Wei H, et al. Recognition of gut microbiota by NOD2 is essential for the homeostasis of intestinal intraepithelial lymphocytes. J Exp Med. 2013;210:2465–76.
Natividad JMM, Petit V, Huang XX, de Palma G, Jury J, Sanz Y, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−;Nod2−/− Mice. Inflamm Bowel Dis. 2012;18:1434–46.
Jamontt J, Petit S, Clark N, Parkinson SJ, Smith P. Nucleotide-binding oligomerization domain 2 signaling promotes hyperresponsive macrophages and colitis in IL-10-deficient mice. J Immunol. 2013;190:2948–58.
Corridoni D, Rodriguez-Palacios A, Di Stefano G, Di Martino L, Antonopoulos DA, Chang EB, et al. Genetic deletion of the bacterial sensor NOD2 improves murine Crohn’s disease-like ileitis independent of functional dysbiosis. Mucosal Immunol. 2017;10:971–82.
Lazaridis LD, Pistiki A, Giamarellos-Bourboulis EJ, Georgitsi M, Damoraki G, Polymeros D, et al. Activation of NLRP3 inflammasome in inflammatory bowel disease: differences between crohn’s disease and ulcerative colitis. Dig Dis Sci. 2017;62:2348–56.
Ranson N, Veldhuis M, Mitchell B, Fanning S, Cook AL, Kunde D, et al. NLRP3-dependent and -independent processing of interleukin (IL)-1beta in active ulcerative colitis. Int J Mol Sci. 2018;20:57.
Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-kappaB signaling. Cell Death Dis. 2019;10:906.
Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17:1359–72.
Yao X, Zhang C, Xing Y, Xue G, Zhang Q, Pan F, et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat Commun. 2017;8:1896.
Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol. 2015;194:3369–80.
Tye H, Yu CH, Simms LA, de Zoete MR, Kim ML, Zakrzewski M, et al. NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat Commun. 2018;9:3728.
Kempster SL, Belteki G, Forhead AJ, Fowden AL, Catalano RD, Lam BY, et al. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am J Physiol-Gastr L. 2011;300:G253–G63.
Eike MC, Skinningsrud B, Ronninger M, Stormyr A, Kvien TK, Joner G, et al. CIITA gene variants are associated with rheumatoid arthritis in Scandinavian populations. Genes Immun. 2012;13:431–6.
Yokota K, Miyazaki T, Hemmatazad H, Gay RE, Kolling C, Fearon U, et al. The pattern-recognition receptor nucleotide-binding oligomerization domain-containing protein 1 promotes production of inflammatory mediators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum-Us. 2012;64:1329–37.
Franca RFO, Vieira SM, Talbot J, Peres RS, Pinto LG, Zamboni DS, et al. Expression and activity of NOD1 and NOD2/RIPK2 signalling in mononuclear cells from patients with rheumatoid arthritis. Scand J Rheumatol. 2016;45:8–12.
Kim HW, Kwon YJ, Park BW, Song JJ, Park YB, Park MC. Differential expressions of NOD-like receptors and their associations with inflammatory responses in rheumatoid arthritis. Clin Exp Rheumatol. 2017;35:630–7.
Liu YR, Yan X, Yu HX, Yao Y, Wang JQ, Li XF, et al. NLRC5 promotes cell proliferation via regulating the NF-kappaB signaling pathway in rheumatoid arthritis. Mol Immunol. 2017;91:24–34.
Jenko B, Praprotnik S, Tomsic M, Dolzan V. NLRP3 and CARD8 polymorphisms influence higher disease activity in rheumatoid arthritis. J Med Biochem. 2016;35:319–23.
Zhao C, Gu Y, Zeng X, Wang J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin Immunol. 2018;197:154–60.
Jager E, Murthy S, Schmidt C, Hahn M, Strobel S, Peters A, et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat Commun. 2020;11:4243.
Wang T, Zhu CL, Wang S, Mo LW, Yang GD, Hu J, et al. Role of NLRP3 and NLRP1 inflammasomes signaling pathways in pathogenesis of rheumatoid arthritis. Asian Pac J Trop Med. 2014;7:827–31.
Yang XL, Hu ZD, Wu Q, Liu X, Liu QJ, Zhang YC, et al. Association of polymorphisms in SPARC and NLRP2 genes with rheumatoid arthritis in a Chinese Han population. Mod Rheumatol. 2015;25:67–71.
Lin Y, Luo ZQ. NLRP6 facilitates the interaction between TAB2/3 and TRIM38 in rheumatoid arthritis fibroblast-like synoviocytes. FEBS Lett. 2017;591:1141–9.
Prado DS, Veras FP, Ferreira RG, Damasceno LEA, Melo PH, Zamboni DS, et al. NLRP12 controls arthritis severity by acting as a checkpoint inhibitor of Th17 cell differentiation. FASEB J. 2020;34:10907–19.
Kufer TA, Nigro G, Sansonetti PJ. Multifaceted functions of NOD-like receptor proteins in myeloid cells at the intersection of innate and adaptive immunity. Microbiol Spectrum. 2016;4:1–9.
Yu SL, Wong CK, Wong PTY, Chen DP, Szeto CC, Li EK, et al. Down-regulated NOD2 by immunosuppressants in peripheral blood cells in patients with SLE reduces the muramyl dipeptide-induced IL-10 production. PLoS ONE. 2011;6:e23855.
Li J, Wu S, Wang MR, Wang TT, Li BK, Zhu JM. Potential roles of nucleotide-binding oligomerization domain 2 in the pathogenesis of systemic lupus erythematosus. Rheumatol Int. 2014;34:1339–44.
Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, et al. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity. 2012;45:271–8.
Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190:1217–26.
Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, et al. Self double-stranded (ds)DNA induces IL-1β production from human monocytes by activating NLRP3 inflammasome in the presence of anti-dsDNA antibodies. J Immunol. 2013;190:1407–15.
Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, et al. Podocyte activation of NLRP3 inflammasomes contributes to the development of proteinuria in lupus nephritis. Arthritis Rheumatol. 2017;69:1636–46.
Yang QR, Yu CC, Yang ZW, Wei Q, Mu K, Zhang Y, et al. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J Rheumatol. 2014;41:444–52.
Tang LL, Cheng YY, Zhu CH, Yang C, Liu L, Zhang YJ, et al. Integrative methylome and transcriptome analysis to dissect key biological pathways for psoriasis in Chinese Han population. J Dermatol Sci. 2018;91:285–91.
Jordan CT, Cao L, Roberson EDO, Pierson KC, Yang CF, Joyce CE, et al. PSORS2 is due to mutations in CARD14. Am J Hum Genet. 2012;90:784–95.
Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, Nair RP, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet. 2012;90:796–808.
Wang MC, Zhang SS, Zheng GX, Huang JJ, Zhou SY, Zhao XQ, et al. Gain-of-function mutation of Card14 leads to spontaneous psoriasis-like skin inflammation through enhanced keratinocyte response to IL-17A. Immunity. 2018;49:66.
Carlstrom M, Ekman AK, Petersson S, Soderkvist P, Enerback C. Genetic support for the role of the NLRP3 inflammasome in psoriasis susceptibility. Exp Dermatol. 2012;21:932–7.
Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi HB, Mak TW, et al. Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity. 2011;34:75–84.
Natarajan C, Yao SY, Zhang FL, Sriram S. Activation of NOD2/RIPK2 pathway induces mitochondrial injury to oligodendrocyte precursor cells in vitro and CNS demyelination in vivo. J Neuroimmunol. 2013;265:51–60.
Pereira VCS, Fontes-Dantas FL, Paradela ER, Malfetano FR, Scherpenhuijzen SDB, Mansur LF, et al. Polymorphisms in the CIITA-168A/G (rs3087456) and CIITA+1614G/C (rs4774) may influence severity in multiple sclerosis patients. Arq Neuro-Psiquiat. 2019;77:166–73.
Bronson PG, Goldstein BA, Ramsay PP, Beckman KB, Noble JA, Lane JA, et al. The rs4774 CIITA missense variant is associated with risk of systemic lupus erythematosus. Genes Immun. 2011;12:667–71.
Maver A, Lavtar P, Ristic S, Stopinsek S, Simcic S, Hocevar K, et al. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci Rep. 2017;7:3715.
Gris D, Ye ZM, Iocca HA, Wen HT, Craven RR, Gris P, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–81.
Malhotra S, Costa C, Eixarch H, Keller CW, Amman L, Martinez-Banaclocha H, et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain. 2020;143:1414–30.
Hou BH, Zhang YH, Liang PY, He Y, Peng BW, Liu WH, et al. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 2020;11:377.
Gharagozloo M, Gris KV, Mahvelati T, Amrani A, Lukens JR, Gris D. NLR-dependent regulation of inflammation in multiple sclerosis. Front Immunol. 2017;8:2012.
Braga TT, Brandao WN, Azevedo H, Terra FF, Melo ACL, Pereira FV, et al. NLRP3 gain-of-function in CD4+ T lymphocytes ameliorates experimental autoimmune encephalomyelitis. Clin Sci (Lond). 2019;133:1901–16.
Gharagozloo M, Mahmoud S, Simard C, Mahvelati TM, Amrani A, Gris D. The dual immunoregulatory function of Nlrp12 in T cell-mediated immune response: lessons from experimental autoimmune encephalomyelitis. Cells. 2018;7:119.
Fu Y, Zhan X, Wang Y, Jiang X, Liu M, Yang Y, et al. NLRC3 expression in dendritic cells attenuates CD4+ T cell response and autoimmunity. EMBO J. 2019;38:e101397.
Gharagozloo M, Mahmoud S, Simard C, Yamamoto K, Bobbala D, Ilangumaran S, et al. NLRX1 inhibits the early stages of CNS inflammation and prevents the onset of spontaneous autoimmunity. PLoS Biol. 2019;17:e3000451.
Li YY, Pearson JA, Chao C, Peng J, Zhang X, Zhou Z, et al. Nucleotide-binding oligomerization domain-containing protein 2 (Nod2) modulates T1DM susceptibility by gut microbiota. J Autoimmun. 2017;82:85–95.
Gyllenberg A, Asad S, Piehl F, Swanberg M, Padyukov L, Van Yserloo B, et al. Age-dependent variation of genotypes in MHC II transactivator gene (CIITA) in controls and association to type 1 diabetes. Genes Immun. 2012;13:632–40.
Hu CY, Ding HY, Li YY, Pearson JA, Zhang XJ, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. P Natl Acad Sci USA. 2015;112:11318–23.
Marie J, Kovacs D, Pain C, Jouary T, Cota C, Vergier B, et al. Inflammasome activation and vitiligo/nonsegmental vitiligo progression. Br J Dermatol. 2014;170:816–23.
Li SL, Kang P, Zhang WF, Jian Z, Zhang Q, Yi XL, et al. Activated NLR family pyrin domain containing 3 (NLRP3) inflammasome in keratinocytes promotes cutaneous T-cell response in patients with vitiligo. J Allergy Clin Immun. 2020;145:632–45.
Jia C, Zhang J, Chen HW, Zhuge YZ, Chen HQ, Qian FY, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10:778.
Castro MFG, Miculan E, Herrera MG, Ruera C, Perez F, Prieto ED, et al. p31-43 gliadin peptide forms oligomers and induces NLRP3 inflammasome/caspase 1-dependent mucosal damage in small intestine. Front Immunol. 2019;10:31.
Pietz G, De R, Hedberg M, Sjoberg V, Sandstrom O, Hernell O, et al. Immunopathology of childhood celiac disease-Key role of intestinal epithelial cells. PLoS ONE. 2017;12:e0185025.
Hollenbach E, Neumann M, Vieth M, Roessner A, Malfertheiner P, Naumann M. Inhibition of p38 MAP kinase- and RICK/NF-kappa B-signaling suppresses inflammatory bowel disease. FASEB J. 2004;18:1550.
Haile PA, Casillas LN, Bury MJ, Mehlmann JF, Singhaus R Jr., Charnley AK, et al. Identification of quinoline-based RIP2 kinase inhibitors with an improved therapeutic index to the hERG ion channel. ACS Med Chem Lett. 2018;9:1039–44.
Huang Z, Wang J, Xu X, Wang H, Qiao Y, Chu WC, et al. Antibody neutralization of microbiota-derived circulating peptidoglycan dampens inflammation and ameliorates autoimmunity. Nat Microbiol. 2019;4:766–73.
White M, Webster G, O’Sullivan D, Stone S, La Flamme AC. Targeting innate receptors with MIS416 reshapes Th responses and suppresses CNS disease in a mouse model of multiple sclerosis. PLoS One. 2014;9:e87712.
Yu H, Ding C, Dai S, Sun J, Wang S, Zhang Z. Long noncoding RNA FER1L4 regulates rheumatoid arthritis via targeting NLRC5. Clin Exp Rheumatol. 2020;38:713–23.
Liu YR, Yang L, Xu QQ, Lu XY, Ma TT, Huang C, et al. Long noncoding RNA MEG3 regulates rheumatoid arthritis by targeting NLRC5. J Cell Physiol. 2019;234:14270–84.
Sun Y, Zhao Y, Yao J, Zhao L, Wu Z, Wang Y, et al. Wogonoside protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting NF-kappaB and NLRP3 inflammasome activation. Biochem Pharmacol. 2015;94:142–54.
He X, Wei Z, Wang J, Kou J, Liu W, Fu Y, et al. Alpinetin attenuates inflammatory responses by suppressing TLR4 and NLRP3 signaling pathways in DSS-induced acute colitis. Sci Rep. 2016;6:28370.
Zhou W, Liu X, Zhang X, Tang J, Li Z, Wang Q, et al. Oroxylin A inhibits colitis by inactivating NLRP3 inflammasome. Oncotarget. 2017;8:58903–17.
Cao H, Liu J, Shen P, Cai J, Han Y, Zhu K, et al. Protective effect of naringin on DSS-induced ulcerative colitis in mice. J Agric Food Chem. 2018;66:13133–40.
Guo W, Liu W, Jin B, Geng J, Li J, Ding H, et al. Asiatic acid ameliorates dextran sulfate sodium-induced murine experimental colitis via suppressing mitochondria-mediated NLRP3 inflammasome activation. Int Immunopharmacol. 2015;24:232–8.
Liu X, Zhou W, Zhang X, Lu P, Du Q, Tao L, et al. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem Pharm. 2016;112:37–49.
Neudecker V, Haneklaus M, Jensen O, Khailova L, Masterson JC, Tye H, et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J Exp Med. 2017;214:1737–52.
Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8:8618.
Wang Z, Chen T, Yang CR, Bao T, Yang XL, He F, et al. Secoisolariciresinol diglucoside suppresses dextran sulfate sodium salt-induced colitis through inhibiting NLRP1 inflammasome. Int Immunopharmacol. 2020;78:105931.
Radulovic K, Normand S, Rehman A, Delanoye-Crespin A, Chatagnon J, Delacre M, et al. A dietary flavone confers communicable protection against colitis through NLRP6 signaling independently of inflammasome activation. Mucosal Immunol. 2018;11:811–9.
Chen J, Wu W, Zhang M, Chen C. Taraxasterol suppresses inflammation in IL-1beta-induced rheumatoid arthritis fibroblast-like synoviocytes and rheumatoid arthritis progression in mice. Int Immunopharmacol. 2019;70:274–83.
Nagoor Meeran MF, Azimullah S, Laham F, Tariq S, Goyal SN, Adeghate E, et al. alpha-Bisabolol protects against beta-adrenergic agonist-induced myocardial infarction in rats by attenuating inflammation, lysosomal dysfunction, NLRP3 inflammasome activation and modulating autophagic flux. Food Funct. 2020;11:965–76.
Pang ZQ, Wang GQ, Ran N, Lin HQ, Wang ZY, Guan XW, et al. Inhibitory effect of methotrexate on rheumatoid arthritis inflammation and comprehensive metabolomics analysis using ultra-performance liquid chromatography-quadrupole time of flight-mass spectrometry (UPLC-Q/TOF-MS). Int J Mol Sci. 2018;19:2894.
Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci. 2017;130:3955–63.
Su B, Ye H, You X, Ni H, Chen X, Li L. Icariin alleviates murine lupus nephritis via inhibiting NF-kappaB activation pathway and NLRP3 inflammasome. Life Sci. 2018;208:26–32.
Li X, Wang M, Hong H, Luo C, Liu Z, Yang R. Sophocarpine attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-kappaB activation. Immunol Res. 2018;66:521–7.
Peng X, Yang T, Liu G, Liu H, Peng Y, He L. Piperine ameliorated lupus nephritis by targeting AMPK-mediated activation of NLRP3 inflammasome. Int Immunopharmacol. 2018;65:448–57.
Zhao J, Wang J, Zhou M, Li M, Li M, Tan H. Curcumin attenuates murine lupus via inhibiting NLRP3 inflammasome. Int Immunopharmacol. 2019;69:213–6.
He J, Sun M, Tian S. Procyanidin B2 prevents lupus nephritis development in mice by inhibiting NLRP3 inflammasome activation. Innate Immun. 2018;24:307–15.
Bonomini F, Dos Santos M, Veronese FV, Rezzani R. NLRP3 inflammasome modulation by melatonin supplementation in chronic pristane-induced lupus nephritis. Int J Mol Sci. 2019;20:3466.
Li D, Shi G, Wang J, Zhang D, Pan Y, Dou H, et al. Baicalein ameliorates pristane-induced lupus nephritis via activating Nrf2/HO-1 in myeloid-derived suppressor cells. Arthritis Res Ther. 2019;21:105.
Wang L, Law HK. The role of autophagy in lupus nephritis. Int J Mol Sci. 2015;16:25154–67.
Lin TJ, Wu CY, Tsai PY, Hsu WH, Hua KF, Chu CL, et al. Accelerated and severe lupus nephritis benefits from M1, an active metabolite of ginsenoside, by regulating NLRP3 inflammasome and T cell functions in mice. Front Immunol. 2019;10:1951.
Tan W, Gu Z, Leng J, Zou X, Chen H, Min F, et al. Let-7f-5p ameliorates inflammation by targeting NLRP3 in bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Biomed Pharmacother. 2019;118:109313.
Irrera N, Vaccaro M, Bitto A, Pallio G, Pizzino G, Lentini M, et al. BAY 11-7082 inhibits the NF-kappaB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin Sci (Lond). 2017;131:487–98.
Deng G, Chen W, Wang P, Zhan T, Zheng W, Gu Z, et al. Inhibition of NLRP3 inflammasome-mediated pyroptosis in macrophage by cycloastragenol contributes to amelioration of imiquimod-induced psoriasis-like skin inflammation in mice. Int Immunopharmacol. 2019;74:105682.
Yang BY, Cheng YG, Liu Y, Liu Y, Tan JY, Guan W, et al. Datura Metel L. ameliorates imiquimod-induced psoriasis-like dermatitis and inhibits inflammatory cytokines production through TLR7/8-MyD88-NF-B-NLRP3 inflammasome pathway. Molecules. 2019;24:2157.
Luo Q, Zeng JX, Li W, Lin L, Zhou X, Tian X, et al. Silencing of miR-155 suppresses inflammatory responses in psoriasis through inflammasome NLRP3 regulation. Int J Mol Med. 2018;42:1086–95.
Coll RC, Robertson AAB, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248.
Xu L, Zhang CL, Jiang N, He D, Bai Y, Xin Y. Rapamycin combined with MCC950 to treat multiple sclerosis in experimental autoimmune encephalomyelitis. J Cell Biochem. 2019;120:5160–8.
Guo CQ, Fulp JW, Jiang YQ, Li X, Chojnacki JE, Wu JD, et al. Development and characterization of a hydroxyl-sulfonamide analogue, 5-chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. Neuroscience. 2017;8:2194–201.
Sanchez-Fernandez A, Skouras DB, Dinarello CA, Lopez-Vales R. OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front Immunol. 2019;10:2578.
Yu H, Wu MF, Lu G, Cao TT, Chen N, Zhang YJ, et al. Prednisone alleviates demyelination through regulation of the NLRP3 inflammasome in a C57BL/6 mouse model of cuprizone-induced demyelination. Brain Res. 2018;1678:75–84.
Bai XY, Wang XF, Zhang LS, Du PC, Cao Z, Hou Y. Tetramethylpyrazine ameliorates experimental autoimmune encephalomyelitis by modulating the inflammatory response. Biochem Bioph Res Commun. 2018;503:1968–72.
Koo JH, Kim DH, Cha D, Kang MJ, Choi JM. LRR domain of NLRX1 protein delivery by dNP2 inhibits T cell functions and alleviates autoimmune encephalomyelitis. Theranostics. 2020;10:3138–50.
Wu CF, Pan LL, Niu WY, Fang X, Liang WJ, Li JH, et al. Modulation of gut microbiota by low methoxyl pectin attenuates type 1 diabetes in non-obese diabetic mice. Front Immunol. 2019;10:1733.
Gao Y, Li J, Chu S, Zhang Z, Chen N, Li L, et al. Ginsenoside Rg1 protects mice against streptozotocin-induced type 1 diabetic by modulating the NLRP3 and Keap1/Nrf2/HO-1 pathways. Eur J Pharmacol. 2020;866:172801.
Chen L, Lu FB, Chen DZ, Wu JL, Hu ED, Xu LM, et al. BMSCs-derived miR-223-containing exosomes contribute to liver protection in experimental autoimmune hepatitis. Mol Immunol. 2018;93:38–46.
Midura-Kiela MT, Radhakrishnan VM, Larmonier CB, Laubitz D, Ghishan FK, Kiela PR. Curcumin inhibits interferon-gamma signaling in colonic epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2012;302:G85–96.
Liu X, Deng J, Li R, Tan C, Li H, Yang Z, et al. ERβ-selective agonist alleviates inflammation in a multiple sclerosis model via regulation of MHC II in microglia. Am J Transl Res. 2019;11:4411–24.
Leber A, Hontecillas R, Zoccoli-Rodriguez V, Bienert C, Chauhan J, Bassaganya-Riera J. Activation of NLRX1 by NX-13 alleviates inflammatory bowel disease through immunometabolic mechanisms in CD4+ T cells. J Immunol. 2019;203:3407–15.
Napier RJ, Lee EJ, Vance EE, Snow PE, Samson KA, Dawson CE, et al. Nod2 deficiency augments Th17 responses and exacerbates autoimmune arthritis. J Immunol. 2018;201:1889–98.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant Nos. 81903882 and 81871240), the National Science & Technology Major Project “New Drug Creation and Manufacturing Program” (Grant No. 2018ZX09711002-014-001), and the “Personalized Medicines—Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA12020369).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Chen, L., Cao, Sq., Lin, Zm. et al. NOD-like receptors in autoimmune diseases. Acta Pharmacol Sin 42, 1742–1756 (2021). https://doi.org/10.1038/s41401-020-00603-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00603-2
Keywords
This article is cited by
-
NLRP inflammasomes in health and disease
Molecular Biomedicine (2024)
-
Exploring the mechanism of Celastrol in the treatment of rheumatoid arthritis based on systems pharmacology and multi-omics
Scientific Reports (2024)
-
Linking NLRP3 inflammasome and pulmonary fibrosis: mechanistic insights and promising therapeutic avenues
Inflammopharmacology (2024)
-
Inflammasomes in rheumatoid arthritis: a pilot study
BMC Rheumatology (2023)
-
Macrophage DCLK1 promotes obesity-induced cardiomyopathy via activating RIP2/TAK1 signaling pathway
Cell Death & Disease (2023)
