
Abstract
Recent evidence shows that the expression levels of histamine receptor H3 (Hrh3) are upregulated in several types of cancer. However, the role of Hrh3 in non-small cell lung cancer (NSCLC) has not been elucidated. In the present study, we showed that the expression levels of Hrh3 were significantly increased in NSCLC samples, and high levels of Hrh3 were associated with poor overall survival (OS) in NSCLC patients. In five human NSCLC cell lines tested, Hrh3 was significantly upregulated. In NSCLC cell lines H1975, H460, and A549, Hrh3 antagonist ciproxifan (CPX, 10–80 μM) exerted moderate and concentration-dependent inhibition on the cell growth and induced apoptosis, whereas its agonist RAMH (80 μM) reversed these effects. Furthermore, inhibition of Hrh3 by CPX or siRNA retarded the migration and invasion of NSCLC cells through inhibiting epithelial-mesenchymal transition (EMT) progression via reducing the phosphorylation of PI3K/Akt/mTOR and MEK/ERK signaling pathways. In nude mice bearing H1975 cell xenograft or A549 cell xenograft, administration of CPX (3 mg/kg every other day, intraperitoneal) significantly inhibited the tumor growth with increased E-cadherin and ZO-1 expression and decreased Fibronectin expression in tumor tissue. In conclusion, this study reveals that Hrh3 plays an important role in the growth and metastasis of NSCLC; it might be a potential therapeutic target against the lung cancer.
Similar content being viewed by others
Introduction
Lung cancer is a major public health problem worldwide and was the first leading cause of death among both men (24%) and women (23%) in the U.S. in 2019 [1]. One-quarter of all cancer deaths are due to lung cancer. Approximately 85% of clinical lung cancer cases are non-small cell lung cancer (NSCLC) [2]. Surgical resection combined with chemotherapy and radiation therapy is the gold standard for the treatment of lung cancer [3]. However, the overall survival (OS) of this disease remains disappointing. Therefore, it is critical to elucidate the molecular mechanism and biochemical pathway involved in lung cancer for the development of novel therapeutic approaches to improve clinical efficacy.
Histamine is a biogenic amine with endogenous physiological activity. Over the past decades, accumulating preclinical and clinical evidence has indicated that histamine is closely correlated with cancer-associated biological processes during cancer development in multiple cell types. It is well known that the effects of histamine are mediated through its binding to four different G protein-coupled receptors (histamine receptors H1, H2, H3, and H4), leading to the activation of different signal transduction pathways. Different histamine receptors (HRs) have been found in several cancer cell lines and human neoplastic lesions, including glioblastoma [4], colorectal cancer [5], lung cancer [6], cholangiocarcinoma [7], hepatoma cells [8], gastric carcinoma [9] and breast cancer [10]. Depending on the binding of histamine to each receptor, the induced physiological response differs. Histamine receptor H3 (Hrh3) was first noted as a presynaptic autoreceptor on brain histamine neurons, although it was also found on other cells [10]. Hrh3 participates in regulating neurotransmitter release [11], but its role in carcinogenesis has not yet been elucidated. A report by Tanaka et al. showed that Hrh3 antagonists suppressed proliferation and induced caspase-dependent apoptotic death in breast cancer cells [10]. Other findings also indicate that Hrh3 exhibits a neuroprotective effect on cortical neurons under hypoxic conditions through the ERK1/2 signaling pathway [12]. In addition, some studies have reported the modulation of cell proliferation in tissues by Hrh3 ligands [13]. These data suggest that Hrh3 may be an effective therapeutic target against cancers.
Human Hrh3 has more than 40% homology with Hrh4 [14]. The high degree of homology between Hrh3 and Hrh4 leads to their similar pharmacological behaviors [15]. The receptor Hrh4 plays an important role in the invasion and metastasis of various tumors [16]. Meng and colleagues demonstrated that an Hrh4 antagonist (clobenpropit) suppressed cholangiocarcinoma progression by disrupting tumor metastasis [7]. Notably, clobenpropit also has a high affinity for Hrh3, and the study could not exclude the possible involvement of Hrh3 in the effect of clobenpropit against epithelial-mesenchymal transition (EMT) [17]. We proposed that Hrh3 might also participate in the progression of NSCLC, including EMT. Therefore, to better understand the roles of Hrh3 in the progression of NSCLC and their underlying mechanisms, we performed both in vitro and in vivo experiments using pharmacologic and molecular approaches to gain insight into the function of Hrh3 in the present study. Our results revealed that Hrh3 could modulate metastatic behavior by regulating EMT progression via the PI3K/Akt/mTOR and MEK/ERK signaling pathways.
Materials and methods
Cells and cell cultures
The NSCLC cell lines H460 (human large cell carcinoma), A549 (human adenocarcinoma), H1703 (human squamous carcinoma), PC-9 (human adenocarcinoma), H1975 (human adenocarcinoma), and BEAS-2B (human normal lung epithelial cell) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (Gibco, NY, USA) and 1% penicillin/streptomycin at 37 °C in a 5% CO2 humidified atmosphere.
Oncomine and in silico data analysis
Oncomine, a cancer microarray database, facilitates various methods of analysis, including molecular concepts analysis and meta-analysis [18]. In this study, the Oncomine database was used to analyze the gene expression level of Hrh3, with a fold change in expression of 1.5, P value of 0.05, and top 10% gene rank as threshold criteria. Correlations between lung cancer patient survival and Hrh3 expression (probe: 221663_x_at) were analyzed by KM plotter (http://kmplot.com). “Auto select best cutoff” was chosen in the analysis.
Cell viability assay
The inhibitory effects of treatment with the Hrh3 antagonist ciproxifan (CPX, purchased from Selleck, Houston, TX, USA) or its agonist, (R)-(α)-(-)-methylhistamine dihydrobromide (RAMH, purchased from Tocris, Bristol, U.K.) on tumor cell viability were determined by CCK-8 assay. H460, A549, H1975, and BEAS-2B cells were seeded onto 96-well plates at 1×104 cells/well. The cells were treated with CPX at different concentrations. After 48 h of treatment, 10 µL of CCK-8 solution (MedChem Express, NJ, USA) was added to each well, and the cells were cultured for an additional 1–3 h at 37 °C. The cell viability was measured at 450 nm using a Multiskan Spectrum spectrophotometer (Thermo Scientific, Rockford, IL, USA).
Colony-formation assay
For each cell line, cells were seeded in 6-well plates at 800 cells/well and cultured for 10–14 days with or without 40 μM CPX. After visible colonies had formed, the cells were fixed with methanol and stained with a 0.1% crystal violet (Sigma-Aldrich, St Louis, MO, USA) solution.
Apoptosis detection by flow cytometry
For each cell line, 2 × 105 cells/well were seeded in a 6-well plate. After treatment with CPX for 48 h, the cells were harvested, stained with reagent from the Annexin V-FITC Apoptosis Kit (BD Biosciences, Franklin Lakes, NY, USA) according to the manufacturer’s instructions and analyzed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
Wound healing migration assay/cell scratch injury
The cells were seeded in 6-well dishes. After the cells had grown to confluence, mechanical injury was performed using a sterile pipette tip (200 μL), and the debris was washed away with PBS. Cells were incubated in culture medium with or without CPX for 24 h. Images were captured at 0 and 24 h after scratching.
Transwell matrigel invasion assays
The invasion assays were performed to investigate cell invasion. The cells (5 × 105 cells/well in 100 μl of serum-free RPMI-1640 medium) were seeded in Matrigel-coated cell culture inserts and then treated with CPX at different concentrations. Complete growth medium containing 10% FBS was placed outside the chambers, and cells were allowed to penetrate through the Matrigel to the lower surface of the filters. Cells were incubated at 37 °C in a 5% CO2 humidified atmosphere for 24 h. Invasive cells were visualized and counted under a light microscope (Olympus, Tokyo, Japan).
Western blot analysis
Cells and tumor tissues were harvested, and proteins were extracted and quantified using a BCA protein kit (Thermo Fisher Scientific). Equal amounts (40 μg) of tissue lysates were separated by 10% SDS-PAGE, transferred to a PVDF membrane (Bio-Rad, Hercules, CA, USA), and then incubated with primary antibody overnight at 4 °C. The following antibodies were used: anti-phosphorylated mTOR (Cell Signaling Technology, Danvers, MA, USA), anti-mTOR (Cell Signaling Technology), anti-phosphorylated ERK1/2 (Cell Signaling Technology), anti-ERK1/2 (Cell Signaling Technology), anti-phosphorylated PI3K (Abcam, Cambridge USA), anti-PI3K (Cell Signaling Technology), anti-phosphorylated AKT (Cell Signaling Technology), anti-AKT (Cell Signaling Technology), and anti-GAPDH (Cell Signaling Technology). The membranes were incubated with secondary antibodies (anti-rabbit IgG and anti-mouse IgG from Santa Cruz Biotechnology, Santa Cruz, CA, USA) at room temperature for 2 h. Protein bands were visualized using an ECL kit (WESTAR SUPERNOVA, Cyanagen, Bologna, Italy). Images were captured with an Odyssey infrared imaging system (LI-COR Biosciences, USA). All experiments were repeated at least three times.
Quantitative polymerase chain reaction(Q-PCR)
Total RNA was isolated using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Quantitative real-time PCR analysis was carried out using SYBR Green (Takara, Kyoto, Japan) according to the manufacturer’s protocol with an ABI7500 (Applied Biosystems, USA) real-time PCR system. The ΔΔCt method of quantification was used to calculate relative mRNA expression. The primer sequences are listed in Table 1.
Hrh3 knockdown and overexpression
For Hrh3 knockdown, cells were transiently transfected with a human siRNA targeting Hrh3. A specific siRNA sequence targeting Hrh3 was designed and synthesized by Gene Pharma (Shanghai, China). The siRNA sequence was 5′-CAUACACGCUGCUGAUGAUTT-3′. Briefly, cells were seeded in 6-well plates and incubated with transfection mixture containing RPMI Opti-MEM, Lipofectamine 3000 (Invitrogen, NY, USA) reagent and siRNA targeting human Hrh3 (siHrh3) or siRNA with a nonspecific sequence (siNC) according to the manufacturer’s instructions. The medium was replaced appropriately with fresh medium after transfection for 6 h, and the cells were then incubated for an additional 48–72 h.
To establish a stable cell line with Hrh3 overexpression, H1975 and H460 cells were transfected with a lentivirus vector containing complementary DNA (cDNA) encoding Hrh3 (Vigene Bioscience, Shandong, China). Cells were transfected with polybrene for 48 h. For the selection of stably transfected cells with Hrh3 overexpression, 2 µg/mL puromycin (Santa Cruz Biotechnology) was used. After continuous selection with puromycin for ~3 weeks, the cells were used for the following experiments. The study on Hrh3 overexpression was performed as described in the Supplementary Material section.
In vivo experiment
All experimental procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and in accordance with the Experimental Animal Welfare Ethics Committee of Zhejiang Academy of Medical Sciences. Immunodeficient nude mice weighing 20–24 g were bred in the animal center at 20–22 °C and 50% to 60% humidity with standard 12 h light/dark cycles. H1975 cells and A549 cells (2 × 106 cells) were injected subcutaneously into the right oxter of the mice. Tumor dimensions and body weight were measured every other day, and tumor volume was calculated as follows: tumor volume (mm3) = length (mm) × width (mm) × width (mm)/2. After the tumor volume reached ~50 mm3, the mice were randomly divided into two groups (n = 6 in each group). The control group received an intraperitoneal (I.P.) injection of vehicle (0.9% NaCl), while the treatment group received CPX I.P. at a dosage of 3 mg/kg every other day. At the end of the treatment schedule, the tumors were excised and fixed in formalin for immunohistochemical (IHC) analysis or lysed for Western blotting.
IHC analysis
Before IHC staining, sections were treated with 0.01 M sodium citrate buffer at 98 °C in a water bath for 30 min and then placed in a bathing solution of 3% H2O2 for 5–10 min. After blocking with 5% bovine serum albumin in PBS, sections were incubated with primary antibodies at the recommended dilution overnight at 4 °C. The following antibodies were used: anti-PCNA (1:100 dilution, BD Biosciences, San Jose, CA, USA), anti-E-cadherin, and anti-Vimentin (1:100 dilution, Cell Signaling Technology). This was followed by incubation with biotinylated goat anti-mouse IgG or biotinylated goat anti-rabbit IgG secondary antibody (1:400 dilution, Vector Laboratories, Burlingame, CA, USA). Images were captured using a 20× objective on a Nikon microscope. At least 4–5 images were captured per tumor section. Quantification was performed by counting the number of positive cells or positive areas per high power field using ImageJ software.
Statistical analyses
Data are presented as the means ± SDs. All analyses represent at least triplicate experiments in vitro. Student’s t test was used for single comparisons, and one-way ANOVA followed by Tukey’s post hoc test was used for multiple comparisons using GraphPad Prism version 5.0 software (GraphPad Software Inc., Philadelphia, USA). P values less than 0.05 were used to indicate statistical significance.
Results
Histamine H3 receptor expression in human non-small cell lung cancer and normal lung cells
To date, the evidence of Hrh3 expression in lung cancer progression has remained insufficient. Using Oncomine analysis (http://www.oncomine.org) with certain criteria (a fold change in expression of 1.5 fold, P value = 0.05, and top 10% gene rank), we surveyed mRNA levels of the Hrh3 gene in human NSCLC. In the lung cancer data set reported by Hou [19], Hrh3 expression was significantly increased in NSCLC samples (n = 91) compared with normal human lung tissues (n = 65, P = 0.028, Fig. 1a). The mRNA levels of Hrh3 in human large-cell cancer samples (LCC, n = 19) were higher than those in normal human lung tissues (n = 65, P = 0.005, Fig. 1b) and slightly increased in lung adenocarcinoma (ADC, n = 45, P = 0.058, Fig. 1c) and squamous cell carcinoma (SCC, n = 27, P = 0.134, Fig. 1d) samples compared to normal human lung tissues. To illustrate the potential relationship between Hrh3 and NSCLC progression, we further used the Kaplan–Meier plotter database (http://www.kmplot.com) to evaluate the survival of NSCLC patients (probe 221663_x_at for Hrh3). The result indicated that a high level of Hrh3 was associated with poor OS among NSCLC patients (HR 1.16, P = 0.022, Fig. 1e). In addition, high Hrh3 mRNA expression was related to poor OS in ADC patients (HR 1.41, P = 0.0038, Fig. 1f) but not in SCC patients (HR 0.9, P = 0.37, Fig. 1g). Thus, Hrh3 may play a crucial role in the progression of NSCLCs, especially ADC.

a–d Scatter diagram validating the expression of Hrh3 in different lung cancer and healthy control groups based on data from the Oncomine database. e–g Survival analysis was used to evaluate the prognostic value of Hrh3 in all NSCLC patients. h The expression of Hrh3 in five NSCLC cell lines was assessed via Western blot analysis in vitro. *P < 0.05, ***P < 0.001 versus the control group.
To confirm the potential role of Hrh3 in NSCLC cancers, we assessed its expression in NSCLC cell lines in vitro. The protein expression of Hrh3 was increased in all five NSCLC cell lines compared with normal lung epithelial cells, as shown by Western blot analysis (Fig. 1h). Relatively higher endogenous Hrh3 receptor expression was observed in H1975 cells, H460 cells, and A549 cells among the five cell lines tested. Therefore, the H1975, H460, and A549 cell lines were selected for the following experiments.
The histamine receptor H3 regulates NSCLC cell viability and apoptosis
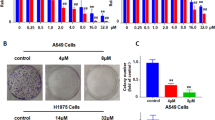
It has been reported that Hrh3 is involved in cancer progression. However, it is unclear whether Hrh3 regulates cell viability in NSCLC cells. To test the role of Hrh3 in tumor cell growth, H1975, H460, A549, and normal lung epithelial (BEAS-2B) cells were incubated with CPX (an Hrh3-specific antagonist [12, 20]) or RAMH (an Hrh3-specific agonist [21]). After 48 h, the CCK-8 assay was used to evaluate cell viability. As shown in Fig. 2a, CPX inhibited cell proliferation in a dose-dependent manner, except in BEAS-2B cells. In contrast, the stimulation of Hrh3 activity with RAMH at 80 μM induced the proliferation of H1975, H460, and A549 cells (Fig. 2b). As shown in Fig. 2c, CPX (40 μM) markedly suppressed colony formation in NSCLC cell lines, suggesting that CPX strongly inhibited the proliferation of NSCLC cells. Notably, BEAS-2B cells with relatively low Hrh3 expression showed less sensitivity to CPX treatment, and CPX seemed to be much less toxic to normal cells than to NSCLC cells.

a,b Cell viability was measured by CCK-8 assay in H1975, H460, A549, and BEAS-2B cells treated with the Hrh3 antagonist CPX or Hrh3 agonist RAMH for 48 h. c H1975, H460, A549, and BEAS-2B cells were plated at 800 cells/well in 6-well plates and treated with or without CPX for colony-formation assays. After 10–14 days, the colonies were fixed, stained with a 0.1% crystal violet solution and then photographed. d H1975, H460, and A549 cells were treated with CPX for 48 h, and the cells were collected. Cell apoptosis was determined by flow cytometry. e, g, i The results of quantitative analyses of Annexin-FITC-positive H1975, H460, and A549 cells are shown. **P < 0.01, ***P < 0.001 versus the control group, &P < 0.05 versus the CPX (40 μM) group. f, h, j The expression of Caspase-3 was detected by Western blot analysis. The quantitative results are also illustrated. *P < 0.05, **P < 0.01 versus the control group. The data represent the average of three independent experiments.
To determine whether the type of cell death observed was apoptosis, the effect of CPX treatment on apoptosis was observed. Total apoptotic cells included early apoptotic cells (Annexin V+/PI-) and late apoptotic cells (Annexin V+/PI+). The results showed that the total number of apoptotic cells among NSCLC cells was significantly higher following treatment with 40 μM CPX (Fig. 2d, e, g, i). Western blot analysis showed that the expression of Caspase-3 was increased upon stimulation with CPX in a dose-dependent manner (Fig. 2f, h, j). These results suggest that the inhibitory effect of the Hrh3 antagonist on NSCLC cell survival was partially related to apoptosis.
Pharmacological inhibition of Hrh3 regulates cell metastatic behavior and EMT progression in NSCLC cells
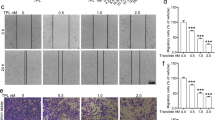
To investigate whether Hrh3 plays an essential role in NSCLC progression, wound-healing migration and Transwell invasion assays were used. We found that CPX inhibited the wound-healing process by suppressing migration in H1975, H460, and A549 cells (Fig. 3a, c, e). Furthermore, the results of Transwell invasion assays indicated that the metastasis potential of CPX-treated cells was considerably decreased compared with that of control cells (Fig. 3b, d, f). To better understand the molecular mechanism of Hrh3-mediated modulation of cell migration and invasion, major EMT-related molecules were analyzed. Pharmacological inhibition of Hrh3 by CPX resulted in significant upregulation of the epithelial markers ZO-1 and E-cadherin and downregulation of the mesenchymal marker Fibronectin in a dose-dependent manner (Fig. 3g–i). Hrh3 inhibitions slightly decreased the protein expression of Vimentin. These results suggest that the activation of Hrh3 plays a role in NSCLC cell metastatic progression.

H1975 (a), H460 (c), and A549 (e) cells were treated with CPX at different concentrations for 24 h. Cell migration was determined via a wound-healing assay. *P < 0.05, **P < 0.01 versus the control group. Magnification: ×200. b, d, f Cell invasion ability after CPX treatment was measured via the Transwell assay. ***P < 0.001 versus the control group. Magnification: ×400. g, h, i Western blotting was performed to analyze the protein levels of ZO-1, E-cadherin, Fibronectin and Vimentin in H1975 cells after treatment with an Hrh3 antagonist. The quantitative results are also illustrated. *P < 0.05, **P < 0.01 versus the control group. The data represent the average of three independent experiments.
Inhibition of the expression of Hrh3 repressed cellular motility, invasion and EMT progression
To further identify the role of endogenous Hrh3 in the progression of NSCLC, we performed H3 receptor gene knockdown in H1975, H460, and A549 cells. A specific siRNA targeting Hrh3 was generated to knockdown Hrh3 expression. Q-PCR and Western blot analysis indicated that the mRNA and protein levels of Hrh3 were significantly decreased in the NSCLC cell lines upon siRNA transfection, indicating a good knockdown efficiency (Fig. 4a–c). Next, the wound-healing migration and Transwell Matrigel invasion assays were used to examine the metastatic potential of NSCLC cells upon the modulation of Hrh3 expression. As expected, the results revealed that the motility and invasiveness of NSCLC cells were dramatically hampered by the knockdown of Hrh3 (Fig. 4d–f). Western blot analyses confirmed that the downregulation of Hrh3 significantly increased the protein expression of epithelial markers (ZO-1, P < 0.05, and E-cadherin, P < 0.01) but decreased expression of the mesenchymal marker Fibronectin (P < 0.05, Fig. 4g–j).

a–c H1975, H460, and A549 cells were transfected with siRNA to knockdown Hrh3 expression. The Hrh3 expression at the mRNA level was confirmed by Q-PCR, and that at the protein level was confirmed by Western blotting. **P < 0.01, ***P < 0.001 versus the siNC group. d, e, f Cell migration ability was determined via a wound-healing assay. Cell invasion ability was measured via the Transwell assay. *P < 0.05, **P < 0.01, ***P < 0.001 versus the siNC group. Magnification: ×100 and ×200. g The protein expression levels of ZO-1, E-cadherin, Fibronectin and Vimentin in Hrh3-knockdown cells were analyzed by Western blot analyses. h, j, k Relative changes in ZO-1, E-cadherin, Fibronectin and Vimentin protein expression in various groups are shown. *P < 0.05, **P < 0.01, ***P < 0.001.
Furthermore, we transfected a lentiviral vector containing cDNA encoding Hrh3 into H1975 and H460 cells (Supplementary Fig. S1a, b). We found that the wounds in Hrh3-overexpressing NSCLC cells healed much faster than those in GFP-overexpressing control cells. The Transwell Matrigel assay revealed that the number of cancer cells that crossed the membrane was significantly higher among Hrh3-overexpressing cells than among the GFP-overexpressing control group (Supplementary Fig. S1c, d).
Several transcription factors (TFs), such as Snail, Slug, and Twist, are well known to play important roles during EMT progression [22, 23]. To gain a deeper understanding of the mechanism by which Hrh3 affects EMT in lung cancer cells, the mRNA levels of EMT-TFs upon the modulation of Hrh3 expression were examined using real-time qPCR. Overexpression of Hrh3 upregulated the mRNA expression levels of slug and twist in H1975 and H460 cells (Supplementary Fig. S1e, f). However, Hrh3 stimulation had no significant effect on the mRNA expression of Snail. Upregulation of Hrh3 markedly suppressed the expression of ZO-1 and E-cadherin but increased the expression of Fibronectin (Supplementary Fig. S1g, h). However, the expression of Vimentin was not affected by Hrh3 knockdown or overexpression in NSCLC cells (Fig. 4g–j and Supplementary Fig. S1g, h).
Hrh3 participates in regulating EMT progression via the PI3K/Akt/mTOR and ERK/MEK signaling pathways
Numerous signaling pathways, including the Akt and ERK signaling pathways, play important roles in regulating cell invasion and EMT progression [24, 25]. Thus, the activation of these two intracellular signaling pathways was investigated (Fig. 5). We discovered that the levels of phosphorylated PI3K, Akt, mTOR, MEK, and ERK were significantly reduced by siRNA targeting Hrh3, but their total protein levels were not decreased in NSCLC cells (Fig. 5a–c). As expected, overexpression of the Hrh3 protein increased the levels of phosphorylated PI3K, Akt, mTOR, MEK, and ERK. The downregulation of total protein expression at different levels was observed (Supplementary Fig. S2a, b). These data revealed that Hrh3 is involved in regulation of the PI3K/Akt/mTOR and MEK/ERK signaling pathways, thereby inhibiting EMT progression.

a The protein expression of PI3K, p-PI3K, Akt, p-Akt, mTOR, p-mTOR, ERK, p-ERK, MEK, and p-MEK in H1975 cells in which Hrh3 had been knocked down was detected by Western blotting. The quantitative results are illustrated. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the siNC group. b The protein expression of PI3K, p-PI3K, Akt, p-Akt, mTOR, p-mTOR, ERK, p-ERK, MEK, and p-MEK in H460 cells in which Hrh3 had been knocked down was detected by Western blotting. The quantitative results are also illustrated. *P < 0.05, **P < 0.01. c The protein expression of PI3K, p-PI3K, Akt, p-Akt, mTOR, p-mTOR, ERK, p-ERK, MEK, and p-MEK in A549 cells in which Hrh3 had been knocked down was detected by Western blotting. The quantitative results are also illustrated. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the GFP control group. The data represent the average of three independent experiments.
Inhibition of Hrh3 suppressed tumor progression in vivo
To verify the inhibitory effect of Hrh3 on lung tumorigenesis in vivo, an H1975 cell xenograft tumor model was established. The length and width of each tumor were measured every other day. The results demonstrated that the inhibition of Hrh3 by CPX significantly inhibited tumor growth (Fig. 6a, b). A similar phenomenon was observed in a H460 cell xenograft tumor model (Fig. 6d, e). There was no difference in body weight between the N.S. control group and CPX treatment group (Fig. 6c, f). Furthermore, IHC analysis revealed that the positive expression of Ki67 (a proliferation marker) was significantly reduced in the CPX-treated group compared with the N.S. group (P < 0.001) of H1975 cells (Fig. 6g, h). Compared with that of the N.S.-treated mice, the mean integral optical density (IOD) indicating cells immunoreactive for E-cadherin and ZO-1 was greatly increased in the CPX-treated groups (P < 0.001 and P < 0.001, respectively), while the expression of Fibronectin was markedly decreased (Fig. 6g, h). There was no difference in the expression of Vimentin between the two groups. Western blot analysis indicated that CPX treatment increased the expression of E-cadherin (P < 0.05) and ZO-1 (P < 0.01) and decreased the expression of Fibronectin (P < 0.01) in the H460 cell xenograft tumor model (Supplementary Fig. S3). Thus, these results demonstrated that the receptor Hrh3 is involved in the progression of lung cancer tumors in vivo and that the mechanism may be related to the inhibition of EMT.

a, d Differences in tumor volume in mice treated with CPX compared with the control group are shown. b, e Tumor volume growth curves are shown. *P < 0.05 vs. the control group. c, f The body weights of mice treated with or without CPX are shown. g Representative photographs from IHC analyses are shown (magnification: 200×). h The results of quantitative analyses of Ki67, ZO-1, E-cadherin, Fibronectin and Vimentin expression are shown. Data are the means ± SDs; *P < 0.05, ***P < 0.001 vs. the control group.
Discussion
Histamine and HRs have attracted scientific attention for their role in physiological and pathological processes [26] and may be factors in tumorigenesis and tumor progression [27]. The regulation of HRs has been reported to contribute to tumor progression and metastasis in pathologies such as cholangiocarcinoma [7]. However, among the HR subtypes, no data showing that Hrh3 inactivation has a tumor-suppressive role in lung cancer cells have been reported. Herein, we discovered that the downregulation of Hrh3 suppressed proliferation and decreased invasion through the inhibition of EMT in NSCLC. Furthermore, the possible mechanism for these effects may be related to decreases in the phosphorylation of two signaling pathways that trigger EMT, the MEK/ERK and PI3K/AKT/mTOR pathways. Our study provides an experimental basis for the functional study of this type of histamine receptor.
Lung cancer, the leading type of malignant tumor, has a poor prognosis and very low long-term survival [28]. Among the pathological types of lung cancer, NSCLC, which easily invades, migrates, and relapses, accounts for approximately 80% of lung cancer cases [29]. At present, the specific molecular mechanisms of lung cancer occurrence and progression, especially lung cancer metastasis, are unclear. More recent studies have reported the expression of Hrh3 in several cancer cells, including breast cancer [13], prostate cancer [30] and hepatocellular carcinoma [31] cells. Nevertheless, the function of Hrh3 in tumor progression is controversial. In vitro studies demonstrated that Hrh3 antagonists could inhibit histamine-induced breast cancer growth and progression, indicating that Hrh3 may mediate breast cancer growth and progression [13]. In agreement with these results, it was recently reported that the upregulation of Hrh3 facilitated the growth and metastasis of hepatocellular carcinoma cells [31]. On the other hand, several investigators have shown that the activation of Hrh3 inhibited tumor growth. Activation of Hrh3 could inhibit cholangiocarcinoma growth by ERK1/2 dephosphorylation [32]. Hrh3 antagonists increased the proliferation of McA-RH7777 hepatoma cells [8]. In addition, Hegyesi H and colleagues [33] found that Hrh3 activation was not able to modulate proliferation in human melanoma cells. These studies offer evidence that Hrh3 may have different effects in different cell types. To the best of our knowledge, the function of Hrh3 in NSCLC has not been elucidated. In the present work, we found that the mRNA levels of Hrh3 were significantly increased in NSCLC samples (P = 0.028) and marginally increased in ADC samples (P = 0.058) compared with normal lung tissues. A similar increase in Hrh3 gene expression was not observed in SCC (P = 0.134). In addition, high Hrh3 mRNA expression was found to be correlated with a significantly poorer OS in all NSCLC patients, including all ADC patients, but not SCC patients (Fig. 1e–g). Our results indicated that the expression level of Hrh3 might not be associated with the clinicopathological features of NSCLC patients, such as histology type. Notably, our current data obtained from Oncomine are mainly focused on the mRNA expression of Hrh3, and examination of the protein expression of Hrh3 from large clinical samples in further studies would be useful to verify the results of our current data analysis. A comprehensive assessment of the expression and prognostic value of Hrh3 would be beneficial for understanding the heterogeneity and complexity of the molecular biology of NSCLC, assist in more accurately predicting the prognosis of NSCLC and facilitate the discovery of potential drug targets for NSCLC.
According to the authors of a study, histamine and RAMH increased MDA-MB-231 cell proliferation via Hrh3 [13]. Thus, we hypothesized that an antagonist of Hrh3 would have an antitumor effect in NSCLCs. Our results revealed that CPX inhibited the viability of NSCLC cells, but the Hrh3 agonist RAMH had the opposite effect on the viability of NSCLC cells. The knockdown of Hrh3 by siRNA significantly inhibited cell proliferation (Supplementary Fig. S4). This anti-proliferative effect might be related to changes in apoptosis via the Caspase-3 signaling pathway. Furthermore, the Hrh3 antagonist CPX did not affect cell proliferation or colony formation in normal lung epithelial cells. Furthermore, CPX had no adverse health effect on mice, as shown by monitoring their body weight, in an in vivo experiment, making CPX a potentially attractive therapeutic compound in the treatment of NSCLC.
From a clinical point of view, the invasion and metastasis of a malignant tumor are more serious than the growth of the tumor [34]. EMT plays a crucial role in the process of tumor migratory and invasive capacity [35]. During EMT, epithelial cells lose epithelial markers, such as E-cadherin and ZO-1, but the expression of mesenchymal markers, such as Vimentin, Fibronectin and matrix metalloproteinase (MMP)-9, is upregulated [36]. In a previous study on the roles of Hrh4 in EMT progression, clobenpropit was used as an Hrh4 agonist [7]. In fact, the affinity of clobenpropit for human Hrh3 was shown to be much higher than that for human Hrh4 [17], but the findings of this study still need to be further clarified. Therefore, we hypothesized that Hrh3 might be involved in the regulation of EMT. As expected, we discovered that CPX could significantly inhibit migration as well as invasion. The expression of epithelial morphology hallmarks, such as E-cadherin, was increased, while that of interstitial cell markers, such as Fibronectin, was decreased in CPX-treated cells compared to control cells. The tight junction protein ZO-1, an epithelial marker, was found to be upregulated by CPX treatment. Furthermore, we found that Hrh3 siRNA-mediated knockdown inhibited the expression of EMT-TFs (data not shown) and increased the expression of epithelial cell markers (E-cadherin and ZO-1) but downregulated the expression of a mesenchymal cell marker (Fibronectin) in NSCLC cells. The effect of Hrh3 activation in promoting EMT in NSCLC cells was confirmed by the overexpression of Hrh3. The results of further animal experimental confirmed the effects of Hrh3 on the proliferation and EMT progression of H1975 and A549 cells. In accordance with the in vitro data, the results of a xenograft experiment indicated that the inhibition of Hrh3 by CPX also had obvious inhibitory effects on tumor growth and EMT progression. Interestingly, we found that the modulation of Hrh3 expression had no significant effects on Vimentin expression in vitro or in vivo. This finding is inconsistent with other reports. Hrh3 downregulation resulted in the downregulation of mesenchymal markers, including Vimentin [4, 7]. We speculate that the regulation of Hrh3 can induce EMT suppression in NSCLC and that this process does not necessarily require cells to lose all mesenchymal features, such as a decrease in Vimentin. This can be explained by the different expression levels of EMT markers in cancer cells [37]. Similar results showing that the change in mesenchymal markers after the administration of clobenpropit (a specific H3 antagonist and H4 agonist) differed according to the cell type observed [38]. Vimentin was decreased in only AsPC-1 cells (and not in Panc-1 or MiaPaCa-2 cells) [38]. However, the comprehensive changes in cellular properties and multiple molecular markers observed in this study clearly indicated disruption of the EMT process by the inhibition of Hrh3.
Several GPCRs utilize a variety of signaling mechanisms to perform their functions. GPCR/Gi signaling has been reported to control the migration and invasion of breast cancers [39]. Furthermore, inactivation of the PI3K/Akt/mTOR signaling pathway retards cell migration induced by growth factors [40]. Hrh3 can activate multiple intracellular signaling pathways, such as the PI3K pathway and mitogen-activated protein kinase (MAPK) pathway, and intracellular Ca2+ concentrations to exert physiological and pathological functions [41]. In the present study, we found that the downregulation of Hrh3 expression could significantly suppress EMT via inactivation of the PI3K/Akt/mTOR and MEK/ERK signaling pathways, while the upregulation of Hrh3 expression could activate both signaling pathways and promote EMT progression. These results are in accordance with previous research that revealed the effect of Hrh3 on glioblastoma tumor invasion [4]. These data raise the possibility that Hrh3 might be involved in regulating cell growth and EMT progression through modulating these intracellular signaling pathways in human NSCLC cell lines. In addition, we found that the overexpression of Hrh3 affected the total expression of ERK, AKT, and mTOR at the mRNA and protein levels. Ono et al. demonstrated that silencing PTK6 reduced ERK1/2 activation by detecting the phosphorylation of ERK1/2 but not total ERK1/2, while PTK6 overexpression increased ERK1/2 activation and downregulated the total expression of ERK [42]. This model may be consistent with our observations. Whether Hrh3 directly interacts with ERK, AKT and mTOR is unknown, and the mechanism by which Hrh3 overexpression affects total protein levels awaits further study.
In conclusion, our present study provides the first evidence that expression of the histamine H3 receptor is upregulated in NSCLC. We found that Hrh3 plays important roles in cell growth and EMT progression in NSCLC through both in vitro and in vivo experiments. The underlying mechanism was shown to be related to regulating the expression of E-cadherin, ZO-1, and Fibronectin and inactivating the PI3K/Akt/mTOR and MEK/ERK phosphorylation signaling pathways. Our in vitro and in vivo findings indicate that Hrh3 might be a potential molecular target for the prevention and treatment of NSCLC progression and metastasis.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Dai L, Smith CD, Foroozesh M, Miele L, Qin Z. The sphingosine kinase 2 inhibitor ABC294640 displays anti-non-small cell lung cancer activities in vitro and in vivo. Int J Cancer. 2018;142:2153–62.
Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19.
Lin JJ, Zhao TZ, Cai WK, Yang YX, Sun C, Zhang Z, et al. Inhibition of histamine receptor 3 suppresses glioblastoma tumor growth, invasion, and epithelial-to-mesenchymal transition. Oncotarget. 2015;6:17107–20.
Fang Z, Yao W, Xiong Y, Li J, Liu L, Shi L, et al. Attenuated expression of HRH4 in colorectal carcinomas: a potential influence on tumor growth and progression. BMC Cancer. 2011;11:1–11.
Cai WK, Hu J, Li T, Meng JR, Ma X, Yin SJ, et al. Activation of histamine H4 receptors decreases epithelial-to-mesenchymal transition progress by inhibiting transforming growth factor-beta1 signalling pathway in non-small cell lung cancer. Eur J Cancer. 2014;50:1195–206.
Meng F, Han Y, Staloch D, Francis T, Stokes A, Francis H. The H4 histamine receptor agonist, clobenpropit, suppresses human cholangiocarcinoma progression by disruption of epithelial mesenchymal transition and tumor metastasis. Hepatology. 2011;54:1718–28.
Davenas E, Rouleau A, Morisset S, Arrang JM. Autoregulation of McA-RH7777 hepatoma cell proliferation by histamine H3 receptors. J Pharmacol Exp Ther. 2008;326:406–13.
Zhang C, Xiong Y, Li J, Yang Y, Liu L, Wang W, et al. Deletion and down-regulation of HRH4 gene in gastric carcinomas: a potential correlation with tumor progression. PLoS One. 2012;7:e31207.
Tanaka S, Sakaguchi M, Yoneyama H, Usami Y, Harusawa S. Histamine H3 receptor antagonist OUP-186 attenuates the proliferation of cultured human breast cancer cell lines. Biochem Biophys Res Commun. 2016;480:479–85.
Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM, et al. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–78.
Lai X, Ye L, Liao Y, Jin L, Ma Q, Lu B, et al. Agonist-induced activation of histamine H3 receptor signals to extracellular signal-regulated kinases 1 and 2 through PKC-, PLD-, and EGFR-dependent mechanisms. J Neurochem. 2016;137:200–15.
Medina V, Croci M, Crescenti E, Mohamad N, Sanchez-Jimenez F, Massari N, et al. The role of histamine in human mammary carcinogenesis: H3 and H4 receptors as potential therapeutic targets for breast cancer treatment. Cancer Biol Ther. 2008;7:28–35.
Hough LB. Genomics meets histamine receptors: new subtypes, new receptors. Mol Pharmacol. 2001;59:415–9.
Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–241.
He GH, Lu J, Shi PP, Xia W, Yin SJ, Jin TB, et al. Polymorphisms of human histamine receptor H4 gene are associated with breast cancer in Chinese Han population. Gene. 2013;519:260–5.
He GH, Xu GL, Cai WK, Xia W. H3 or H4 histamine receptors: that which contributes to suppressing human cholangiocarcinoma progression still remains to be clarified. Hepatology. 2012;56:1182–3.
Wang T, Liu H, Pei L, Wang K, Song C, Wang P, et al. Screening of tumor-associated antigens based on Oncomine database and evaluation of diagnostic value of autoantibodies in lung cancer. Clin Immunol. 2019;210:108262.
Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312.
Morisset S, Rouleau A, Ligneau X, Gbahou F, Tardivel-Lacombe J, Stark H, et al. High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature. 2000;408:860–4.
Aquino-Miranda G, Escamilla-Sánchez J, González-Pantoja R, Bueno-Nava A, Arias-Montaño JA. Histamine H3 receptor activation inhibits dopamine synthesis but not release or uptake in rat nucleus accumbens. Neuropharmacology. 2016;106:91–101.
Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7:re8.
Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45.
Singh M, Yelle N, Venugopal C, Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther. 2018;182:80–94.
Zhang ZQ, Chen J, Huang WQ, Ning D, Liu QM, Wang C, et al. FAM134B induces tumorigenesis and epithelial-to-mesenchymal transition via Akt signaling in hepatocellular carcinoma. Mol Oncol. 2019;13:792–810.
Misto A, Provensi G, Vozella V, Passani MB, Piomelli D. Mast cell-derived histamine regulates liver ketogenesis via oleoylethanolamide signaling. Cell Metab. 2019;29:91–102 e5.
Massari NA, Nicoud MB, Medina VA. Histamine receptors and cancer pharmacology: an update. Br J Pharmacol. 2020;177:516–38.
Yusen W, Xia W, Shengjun Y, Shaohui Z, Hongzhen Z. The expression and significance of tumor associated macrophages and CXCR4 in non-small cell lung cancer. J BUON. 2018;23:398–402.
Fan S, Zou Y, Wang Y, Fang F, Song W. An observational study of apatinib mesylate in treating advanced non-small cell lung cancer with unknown driving gene(s). J BUON. 2018;23:654–8.
Chen J, Hu XY. Inhibition of histamine receptor H3R suppresses prostate cancer growth, invasion and increases apoptosis via the AR pathway. Oncol Lett. 2018;16:4921–8.
Yu D, Zhao J, Wang Y, Hu J, Zhao Q, Li J, et al. Upregulated histamine receptor H3 promotes tumor growth and metastasis in hepatocellular carcinoma. Oncol Rep. 2019;41:3347–54.
Francis H, Onori P, Gaudio E, Franchitto A, DeMorrow S, Venter J, et al. H3 histamine receptor-mediated activation of protein kinase Calpha inhibits the growth of cholangiocarcinoma in vitro and in vivo. Mol Cancer Res. 2009;7:1704–13.
Hegyesi H, Horvath B, Pallinger E, Pos Z, Molnar V, Falus A. Histamine elevates the expression of Ets-1, a protooncogen in human melanoma cell lines through H2 receptor. FEBS Lett. 2005;579:2475–9.
Boer K, Helinger E, Helinger A, Pocza P, Pos Z, Demeter P, et al. Decreased expression of histamine H1 and H4 receptors suggests disturbance of local regulation in human colorectal tumours by histamine. Eur J Cell Biol. 2008;87:227–36.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96.
Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74.
Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–8.
Paik WH, Ryu JK, Jeong K-S, Park JM, Song BJ, Lee SH, et al. Clobenpropit enhances anti-tumor effect of gemcitabine in pancreatic cancer. World J Gastroenterol. 2014;20:8545–57.
Li H, Yang L, Fu H, Yan J, Wang Y, Guo H, et al. Association between Galphai2 and ELMO1/Dock180 connects chemokine signalling with Rac activation and metastasis. Nat Commun. 2013;4:1706.
Caggia S, Chunduri H, Millena AC, Perkins JN, Venugopal SV, Vo BT, et al. Novel role of Gialpha2 in cell migration: downstream of PI3-kinase-AKT and Rac1 in prostate cancer cells. J Cell Physiol. 2018;234:802–15.
Bongers G, Bakker RA, Leurs R. Molecular aspects of the histamine H3 receptor. Biochem Pharmacol. 2007;73:1195–204.
Ono H, Basson MD, Ito H. PTK6 promotes cancer migration and invasion in pancreatic cancer cells dependent on ERK signaling. PLoS One. 2014;9:e96060.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation (81602555, 81803042), Natural Science Foundation of Zhejiang Province (LQ17H160003, LQ19H280004), Medical and Health Technology project of Zhejiang Province (2018KY580), the Traditional Chinese Medicine Science and Technology Plan of Zhejiang Province (2018ZQ012, 2018ZQ013), Social Development Application Project of Hangzhou (20180533B41). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
YYZ, SRZ, and SLM designed the experiments; YYZ, SRZ, and SLM were involved in project administration; YYZ and SRZ wrote and edited the manuscript; YYZ, JJ, JJZ, YPX, SJX, JFL, HGG, and JZZ performed experiments and analyzed the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhao, Yy., Jia, J., Zhang, Jj. et al. Inhibition of histamine receptor H3 suppresses the growth and metastasis of human non-small cell lung cancer cells via inhibiting PI3K/Akt/mTOR and MEK/ERK signaling pathways and blocking EMT. Acta Pharmacol Sin 42, 1288–1297 (2021). https://doi.org/10.1038/s41401-020-00548-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00548-6
Keywords
This article is cited by
-
Expression pattern and prognostic potential of histamine receptors in epithelial ovarian cancer
Journal of Cancer Research and Clinical Oncology (2023)
