
Abstract
Group I metabotropic glutamate receptors (mGlu1 and mGlu5) are promising targets for multiple psychiatric and neurodegenerative disorders. Understanding the subtype selectivity of mGlu1 and mGlu5 allosteric sites is essential for the rational design of novel modulators with single- or dual-target mechanism of action. In this study, starting from the deposited mGlu1 and mGlu5 crystal structures, we utilized computational modeling approaches integrating docking, molecular dynamics simulation, and efficient post-trajectory analysis to reveal the subtype-selective mechanism of mGlu1 and mGlu5 to 10 diverse drug scaffolds representing known negative allosteric modulators (NAMs) in the literature. The results of modeling identified six pairs of non-conserved residues and four pairs of conserved ones as critical features to distinguish the selective NAMs binding to the corresponding receptors. In addition, nine pairs of residues are beneficial to the development of novel dual-target NAMs of group I metabotropic glutamate receptors. Furthermore, the binding modes of a reported dual-target NAM (VU0467558) in mGlu1 and mGlu5 were predicted to verify the identified residues that play key roles in the receptor selectivity and the dual-target binding. The results of this study can guide rational structure-based design of novel NAMs, and the approach can be generally applicable to characterize the features of selectivity for other G-protein-coupled receptors.
Similar content being viewed by others
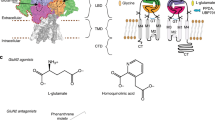

Introduction
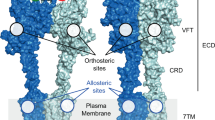
G-protein-coupled receptors (GPCRs) constitute a large superfamily of membrane proteins that transduce endogenous signals across the cell membrane. Metabotropic glutamate receptors (mGlus) are class C GPCRs, and are distinguished by a characteristically large extracellular (VFT) domain that is linked to the seven-transmembrane (7TM) domain by a cysteine-rich domain (Supplementary Fig. S1) [1, 2]. To date, eight subtypes of these receptors (mGlu1–8) have been identified and can be divided into three groups according to their biochemical, structural and pharmacological properties (Supplementary Table S1) [3]. Group I receptors (mGlu1 and mGlu5) are considered promising targets for the treatment of many psychiatric and neurodegenerative disorders including chronic pain, schizophrenia, Alzheimer’s disease, depression, and anxiety [4,5,6,7,8,9,10,11,12]. In the VFT domain of mGlus, a distinctive ligand-binding site (orthosteric site) exists for endogenous compounds. An allosteric site is present in the 7TM domain, and the binding of modulators results in a conformational change, which affects the binding properties of orthosteric ligands. Based on their functional effects, these modulators can be divided into three types: positive allosteric modulators (PAMs), negative allosteric modulators (NAMs), and silent allosteric modulators (SAMs) [1, 2]. Furthermore, allosteric modulators have attracted more attention than traditional orthosteric mGlu1/5 compounds. This is because allosteric modulators are safer than orthosteric ligands in drug development, which can reduce the risk of receptor oversensitization by modulating the natural response to the endogenous ligand instead of directly activating the receptor [13,14,15,16,17,18].
Dipraglurant, a selective NAM of mGlu5, received orphan drug designation from the US FDA in 2016 for Parkinson’s disease levodopa-induced dyskinesia [19]. In addition, several alkyne-based mGlu5 NAMs (mavoglurant, basimglurant, and STX107) and non-alkyne-based NAMs (fenobam, HTL14242, RGH-618, and VU0424238) are progressing to clinical studies (Fig. 1a and Supplementary Table S2) [14, 20,21,22,23,24,25,26,27]. For the mGlu1 receptor, although a number of selective NAMs with high efficacies have been reported for many neurological diseases (Fig. 1b), so far, none of them has been marketed or entered a clinical study, and the binding mode of NAMs in mGlu1 remains elusive [28,29,30,31].

a The structures of clinical mGlu5 NAMs, b the structures of four selective mGlu1 NAMs and one dual-target NAM (VU0467558).
As in other GPCR-based drug discovery programs, subtype selectivity is an important requirement for the drug candidates acting at mGlu receptors [13, 32,33,34]. Research from the Gloriam group demonstrates that the difficulty in achieving subtype-selective compounds with the desired properties is a main factor limiting the approval of a marketed drug for mGlu receptors [34]. mGlu1 and mGlu5 have a high sequence identity of 78.5% between the 7TM domain of the two receptors (Supplementary Fig. S2) [16, 35]. There are only six non-conserved residues in the two allosteric sites [2]. Moreover, the literature reports the use of mGlu1 NAM to produce motor and cognitive side effects, as well as shows concerns about the psychotomimetic effects of certain mGlu5 NAM [15, 36, 37]. Therefore, it is important to develop NAMs with excellent subtype-selectivity profiles for mGlu1 and mGlu5. On the other hand, according to research by Emmitte group, dual-target mGlu1/5 NAMs may achieve an improved safety profile by reducing the occupancy at each receptor required for efficacy [38]. Although a new lead series of dual-target mGlu1/5 NAMs have been discovered, active compounds that are selective versus the other six members of the mGlu family and that exhibit the properties required for use in vivo are lacking. Therefore, exploring the subtype-selective mechanism of mGlu1 and mGlu5 NAMs will provide a considerable insight into the rational design of novel NAMs with single- or dual-target mechanisms of action.
The crystal structures of the TM domain of mGlu1 and mGlu5 have been successfully resolved with several NAMs [1, 2, 24, 28, 39, 40], providing good start points for computational modeling to accurately predict the ligand–receptor binding mode [41,42,43,44,45,46] and provide further valuable insights into the drug-selective mechanism of targets with high homology [47,48,49,50,51,52]. In our previous study, the shared common features of five clinical NAMs (mavoglurant, dipraglurant, basimglurant, STX107, and fenobam) interacting with 11 residues in the allosteric site of mGlu5 were explored by molecular dynamics (MD) simulations [53]. However, except for fenobam, the other four compounds are alkyne-based scaffolds.
In this study, three new non-alkyne-based clinical candidates (HTL14242, RGH-618, and VU0424238) of mGlu5 were selected to improve the shared binding mode of NAMs in mGlu5 by combining molecular docking and MD simulations [14, 23, 24, 54]. The binding mode of fenobam was further optimized based on the co-crystalized structure (PDB ID: 6FFH) reported in 2018 [39]. For the mGlu1 receptor, four selective NAMs (FITM, BDBM50301534, EMQMCM, and JNJ16259685) with high efficacies were selected to predict the elusive binding modes of NAMs in mGlu1 [28,29,30,31]. Based on the equilibrated MD trajectories, the ligand–protein binding free energy and interaction fingerprint were analyzed. As a result, the subtype-selective features underlying the binding modes of 12 different NAMs in mGlu1 or mGlu5 were identified. Furthermore, the binding modes of a unique dual-target NAM (VU0467558) [38] in mGlu1 and mGlu5 (Fig. 1b) were predicted to verify the subtype-selective features and to provide information for the design of novel dual-target NAMs. This study has positive implications for the treatment of psychiatric and neurodegenerative disorders that seriously affect the quality of human life [44, 55].
Materials and methods
Molecular docking
Molecular docking is one of the most frequently used methods to determine the predominant binding pose of a ligand within a protein binding site [56]. Information about the mGlu1 and mGlu5 crystal structures is summarized in Supplementary Table S3. The allosteric sites studied in this work are located in the 7TM domain of mGlus, far from the VFT and RCD domains (Fig. S1). For mGlu1, only one crystal structure of the receptor’s 7TM domain complexed with a selective NAM FITM (PDB ID: 4OR2) was selected for the docking study. For mGlu5, seven crystal structures are available in the PDB database, including five 7TM domain structures complexed with different NAMs and two apo full structures. To explore the selective binding mechanism of NAMs at the allosteric sites, the high-resolution crystal structure of the first representative NAM (mavoglurant) bound receptor (PDB ID: 4OO9) was selected for docking other NAMs into mGlu5. The extracellular and intracellular loops (ECL2 and ICL2) in 4OO9 and 4OR2 are missing. They are far from the allosteric site, but ECL2 is involved in ligand entry [2, 13]. Therefore, the missing structures were modeled and refined by means of Prime in Schrödinger software [57].
In this work, the standard precision (SP) Glide Docking and Induced Fit Docking (IFD) [58] were employed to predict the initial conformations of NAMs binding in the allosteric sites of group I mGlus. To guarantee the reliability of the docking method, re-docking and cross-docking studies were performed based on crystal structures 4OR2, 4OO9, 5CGD, and 6FFH (bound with FITM, mavoglurant, HTL14242 and fenobam) [2, 24, 28, 39, 40, 59]. During docking, the Protein Preparation Wizard tool was used to prepare the receptor structures and the size of the grid boxes was set by selecting the original ligands in the allosteric binding site. The 3D structures of the studied NAMs retrieved from the PDB or PubChem database were prepared using LigPrep [60] with the OPLS-2005 force field for energy minimization and with the ionized state assigned by Epik [61] at a pH value of 7.0 ± 2.0. Finally, SP Glide Docking was applied to search the ligand–receptor binding pose and IFD was used to refine the interaction between the ligand and the binding site residues (within 5.0 Å of the ligand).
MD simulations
Preparation for MD simulation
Initial conformations of NAMs binding to mGlu1 and mGlu5 from docking or re-docking were prepared for the MD simulations. First, OPM [62] was used to calculate the spatial orientations of the NAM–mGlu1/5 complexes with respect to the membrane normal defined by the Z-axis. Second, membrane builder in CHARMM-GUI [63,64,65] was employed to build the solvent box: each of the complex was inserted into the explicit POPC lipid bilayer and immersed in TIP3P water with a 20.0 Å thickness, and NaCl was added with a concentration of 0.15 mol/L according to the physiological concentration [43, 66]. As a result, each system contained ~60,140 atoms per periodic cell with a box size of 74 × 74 × 115 Å. Then, the HF/6-31G* level of Gaussian09 suite [67] was used to implement the geometric optimization and the electrostatic potential calculation of NAMs. The LEaP module in AMBER16 [68, 69] was used to assign the force field parameters for each of the complex, with ff14SB [70] and Lipid14 [71] were used for protein and lipids, respectively. Meanwhile, the conserved disulfide bonds between cysteine residues (Cys644-Cys733 in mGlu5 and Cys657-Cys746 in mGlu1) were considered by LEaP. Finally, the force field parameters for NAMs were created by use of the Antechamber program, using gaff [72] atom types and RESP [73] partial charges.
Implementation of MD simulation
All MD simulations were carried out in four stages with GPU-accelerated NAMD (version 2.12) [74]. Energy minimization and equilibration simulations were conducted before the production simulations to eliminate bad contacts formed by solute and solvent water molecules. Firstly, except for the lipid tail all atoms were fixed, 0.1 ns minimization and 0.5 ns equilibration were carried out. Then, ligand heavy atoms, protein Cα atoms and ions were fastened, and the system was further minimized for 0.1 ns and equilibrated for 0.5 ns. Next, a 5 ns equilibration MD simulation was carried out with all of the atoms free. Finally, a 100 ns production MD simulation was conducted in the NPT ensemble at a temperature of 310.0 K and a pressure of 1 bar. The integration step size was 2 fs and the coordinates of the trajectory were saved every 5000 steps. Moreover, periodic boundary condition was employed and electrostatic calculation was based on the particle-mesh Ewald (PME) method with a 10.0 Å nonbonded cutoff. For each complex, multiple parallel trajectories were conducted to ensure the convergence of the simulation. After the computational simulation, all of the MD trajectory analyses were based on CPPTRAJ [75,76,77], such as the root means square deviation (RMSD) calculation and the extraction of representative snapshots.
Binding free energy analysis
The binding free energies (ΔGcal) of the NAM–mGlu1/5 complexes were calculated by the molecular mechanics/generalized Born surface area (MM/GBSA) method implemented in AMBER16 [78, 79]. For each complex, 500 snapshots were extracted from the last 50 ns equilibrium trajectory, and the binding free energy was calculated by:
The energy terms ΔEvdW, ΔEele, ΔGpol, and ΔGnonpol in the equation represent the van der Waals interaction, electrostatic contribution in the gas phase, polar and nonpolar solvent interaction energies, respectively. Besides, ΔEvdW and ΔEele were calculated using the AMBER force field ff14SB, the modified GB model (igb = 2) was used to calculate the electrostatic free energy of solvation (ΔGpol), and ΔGnonpol was estimated via the solvent accessible surface area (ΔSASA) using the linear combination of pairwise overlaps (LCPO) method calculated by ΔGnonpol = 0.0072 × SASA with 1.4 Å Probe radii. And in the binding free energy calculation, solute and solvent dielectric constants were set to 1 and 80.
Then, per-residue decomposition energy (\(\Delta {{G}}_{{\mathrm{cal}}}^{{\mathrm{per}} - {\mathrm{residue}}}\)) was determined to quantitatively analyze the energy contribution of per residue by decomposing the total binding free energy into each residue according to the following equation:
\(\Delta {{E}}_{{\mathrm{vdW}}}^{{\mathrm{per}} - {\mathrm{residue}}}\), \(\Delta {{E}}_{{\mathrm{ele}}}^{{\mathrm{per}} - {\mathrm{residue}}}\) and \(\Delta {{G}}_{{\mathrm{pol}}}^{{\mathrm{per}} - {\mathrm{residue}}}\) are the same terms as depicted in Eq. (1). However, ΔSASA was obtained from the icosahedron (ICOSA) method, rather than the LCPO method, which was employed to calculate \(\Delta {{G}}_{{\mathrm{nonpol}}}^{{\mathrm{per}} - {\mathrm{residue}}}\).
Hierarchical clustering analysis of per-residue binding free energy
The hierarchical clustering analysis was performed based on the per-residue energy contribution to recognize the common features of the ligand–receptor complexes. Additionally, a clustering tree was generated using R statistical analysis software [80] with the similarity levels among the vectors measured by the Manhattan distance:
where i represents each dimension of per-residue energies a and b. the Cluster algorithm applied here was based on the Ward’s minimum variance method, which was developed to minimize the total within-cluster variance. The hierarchical tree graph was generated by the online tree generator iTOL [81].
Computational alanine scanning analysis
In order to verify the credibility of the critical residues identified by MD simulations, the computational alanine scanning (CAS) was calculated in this study [82, 83]. The last 50 ns trajectories were also used to produce mutated snapshots, and then the binding free energies of 500 mutated snapshots were calculated. More specifically, the alanine mutation was generated by cuting off the selected residue at Cγ atom and replacing it with a hydrogen atom at a 1.09 Å distance from Cß along the direction of Cγ–Cß bond. And the topology files for all the mutation complexes were regenerated by the LEaP module in AMBER16 [68, 69]. Next, the MM/GBSA method was also used to calculate the mutated binding free energy, and the relative binding free energy (ΔΔGCAS) was used to describe the difference between the WT and MUT systems, as shown below:
where ΔGWT and ΔGMUT in Eq. (4) are the binding free energies of the wild-type and mutated systems, respectively.
Molecular interaction fingerprint analysis
Decomposition analysis of the binding free energy (\(\Delta {{G}}_{{\mathrm{cal}}}^{{\mathrm{per}} - {\mathrm{residue}}}\)) describes the energy contribution of per residue, and then molecular interaction fingerprint analysis was conducted to further clarify the interactions between ligand and receptor in this study. In each simulation system, 500 snapshots extracted from the last 50 ns MD simulation trajectories were used to implement the molecular interaction fingerprints based on IChem with the default parameters [79, 84, 85]. Residues within 5.0 Å of the ligand mass center were extracted from trajectories to form MOL2 files. Then, molecular interaction fingerprints were calculated, and seven interactions (hydrophobic, aromatic face-to-face, aromatic edge-to-face, hydrogen bond accepted by ligand, hydrogen bond donated by ligand, ionic bond with ligand negatively charged, ionic bond with ligand positively charged) were successively considered in every snapshot [84]. If a particular interaction between receptor and ligand was detected, “1” was output, otherwise “0” was recorded. Finally, the calculated results were displayed in a radar chart to clearly represent the ligand–receptor interactions.
Results and discussion
Initial conformations of NAMs binding to mGlu1 and mGlu5
In this work, molecular docking was carried out to predict the initial binding poses of NAMs to the mGlu1 and mGlu5 receptor. To guarantee the reliability of the docking protocol, re-docking and cross-docking were first performed based on four crystal structures (PDB ID: 4OR2, 4OO9, 6FFH, and 5CGD) (Supplementary Fig. S3a). The RMSD values between the crystal conformations and re-docking poses for FITM, mavoglurant, fenobam and HTL14242 are 0.48, 0.17, 0.64, and 0.35 Å, respectively. The obtained cross-docking poses of mavoglurant (binding to 5CGD) and HTL14242 (binding to 4OO9) are also consistent with the crystal conformations, and the RMSD values are 0.46 and 1.09 Å, indicating that the docking protocol is suitable for obtaining the receptor–ligand complexes in this study.
The 20 top-ranked poses were generated from SP Glide Docking and IFD refinement for each studied NAM. According to their conformations in the binding pockets, they were defined as two clusters (cluster 1 and cluster 2 in Supplementary Fig. S3b). The docking results are consistent with the previous studies showing that NAMs represent two completely different conformations when binding to mGlu receptors [35, 39, 86,87,88]. The docking poses of FITM and its analog BDBM50301534 were selected according to the binding mode of FITM reported by the Gloriam group, in which the N-methylamide moiety is flipped compared with the co-crystalized structure 4OR2 (Supplementary Fig. S3a) [28, 34]. For the other NAMs (EMQMCM, JNJ16259685, VU0467558, RGH-618, and VU0424238), as none of them show significant chemical similarity to crystal ligands (the Tanimoto coefficients of the NAMs were calculated and the values are shown in Supplementary Table S4), two representative poses from two docking conformation clusters for each ligand (Supplementary Fig. S3b) were selected as the initial poses for further MD simulation and MM/GBSA analysis.
Molecular mechanism of NAMs binding to mGlu1 and mGlu5
MD simulation of NAM–mGlu1 and NAM–mGlu5 complexes
For each NAM–mGlu1/5 complex, multiple 100 ns MD simulations with different initial velocities were carried out (4.5 μs in total), starting from the initial pose picked out from molecular docking. The simulation stabilities were assessed by the RMSD values of protein backbone atoms, backbone atoms of binding site residues (within 5.0 Å of ligand mass center) and ligand heavy atoms as a function of time (Supplementary Fig. S4). As shown, the complexes were stable during the simulation inferring from a small variation of the RMSD values for binding sites and ligands. Compared with the initial pose, the calculated RMSD values indicate that the NAM and receptor have undergone appropriate conformational changes to accommodate each other during the simulation and maintain the equilibration state.
Interaction and binding free energy analysis of mGlu1–NAM complexes
To interpret the molecular mechanism of NAM binding to mGlu1, the representative structure of each complex was extracted from the equilibrated MD trajectory, and the MM/GBSA method [89, 90] was applied to calculate the total binding free energies (ΔGcal) (Table 1, Supplementary Table S5 and Fig. 2). As Fig. 2 depicts, the nonpolar residues distributed around the binding pocket, such as L6482.56, V6643.36, V7535.40, P7565.43, L7575.44, I7976.49, W7986.50, F8016.53, I8127.29, A8187.35, and V8197.36 interact with the NAMs through hydrophobic contacts. In the binding pocket, residues Q6603.32, R6613.33, T748ECL2, G7525.39, N7605.47, and Y8056.57 form hydrogen bonds with NAMs directly or mediated through water molecules. For instance, all four NAMs contain an aldehyde that forms a hydrogen bond with N7605.47, and the aldehyde connects to the monocyclic and polycyclic substituents on both sides of the molecule, respectively. In addition, water mediated polar interactions have also been found in the binding modes of NAM to the members of mGlu family [13, 35, 39, 91, 92]. Water networks play an important role in understanding binding modes, exploring selective features and designing new ligands targeting mGlu members.

a, b, c, c1, d, and d1 are the binding conformations of FITM, BDBM50301534, EMQMCM (pose 1), EMQMCM (pose 2), JNJ16259685 (pose 1), and JNJ16259685 (pose 2), respectively. The important residues and NAMs are shown in sticks. The hydrogen bonds are depicted as red dotted lines, and the water molecules are displayed as red balls.
In FITM and BDBM50301534 binding complexes, the N-methylamide moiety flips and forms a hydrogen bond with N7605.47, which is consistent with the optimized binding mode reported by the Gloriam group (Fig. 2) [34]. The calculated binding free energies of FITM and BDBM50301534 in mGlu1 are −49.49 and −39.05 kcal/mol (Table 1). The higher binding affinity of FITM can be explained by the additional hydrogen bonds (with T748ECL2 and Y8056.57) and the stronger hydrophobic interactions (with L6482.56 and I8127.29) to some extent.
For EMQMCM and JNJ16259685, two docking poses representing cluster 1 (pose 1) and cluster 2 (pose 2) were used for MD simulations. The calculated binding free energies for EMQMCM (pose 1), EMQMCM (pose 2), JNJ16259685 (pose 1), and JNJ16259685 (pose 2) are −48.01, −44.82, −52.51, and −49.30 kcal/mol, respectively (Fig. 2 and Supplementary Table S5). Compared to pose 2 of EMQMCM, the pose 1 participates in stronger hydrophobic interactions with residues V6643.36, V7535.40, and it also forms additional π–π interaction with F8016.53. Likewise, stronger interactions between JNJ16259685 (pose 1) and residues V6643.36, V7535.40, T8157.32 are also observed. Therefore, the equilibrated conformations from MD simulation of pose 1 (Fig. 2c, d) were proposed as the binding modes of EMQMCM–mGlu1 and JNJ16259685–mGlu1 complexes.
“Hot Spots” and “Warm Spots” in mGlu1 for NAM binding
To quantify the contribution of each residue in mGlu1 to NAM binding, the total binding free energy was decomposed on a per-residue basis. Fig. 3 shows a collection of 19 residues whose absolute energy contribution value is ≥0.5 kcal/mol in any complex of the four selective NAMs binding in mGlu1. Among them, five residues V6643.36, V7535.40, L7575.44, N7605.47, and F8016.53 contributed significantly to the binding of NAMs (absolute energy contribution value ≥ 1.5 kcal/mol) and were defined as “Hot Spots”. In addition, ten residues (G6653.37, S6683.40, T748ECL2, P7565.43, I7976.49, W7986.50, Y8056.57, T8157.32, A8187.35, and V8197.36) that were necessary for at least two binding complexes (0.5 kcal/mol ≤ absolute energy contribution value < 1.5 kcal/mol), were defined as “Warm Spots”. As a result, 15 residues including 5 “Hot Spots” and 10 “Warm Spots” were considered as the major energy contributors shared by the four selective NAM–mGlu1 complexes, and they are mainly located in the TM3, TM5, TM6, and TM7 domains of mGlu1. It should be noted that P7565.43, N7605.47, F8016.53, Y8056.57, and T8157.32 have been implicated in mutagenesis-based studies, and the mutations can reduce the binding affinity of FITM [28, 93].

Five residues V6643.36, V7535.40, L7575.44, N7605.47, and F8016.53 contributed significantly to the binding of NAMs (absolute energy. contribution value ≥1.5 kcal/mol) were defined as “Hot Spots”, and ten residues (G6653.37, S6683.40, T748ECL2, P7565.43, I7976.49, W7986.50, Y8056.57, T8157.32, A8187.35, and V8197.36), which were necessary at least two binding complexes (0.5 kcal/mol ≤ absolute energy contribution value < 1.5 kcal/mol), were defined as “Warm Spots”.
Interaction and binding free energy analysis of mGlu5–NAM complexes
In our previous study, the binding mechanism of five clinical NAMs (mavoglurant, dipraglurant, basimglurant, STX107, and fenobam) in the mGlu5 receptor was explored by the same computational approach. In this study, three new non-alkyne-based clinical candidates (RGH-618, VU0424238, and HTL14242) of mGlu5 were picked out to improve the shared binding mode of NAMs in mGlu5. Furthermore, the binding mode of fenobam in mGlu5 was optimized based on the crystal structure (PDB ID: 6FFH) reported in 2018 [39]. For each complex, the representative structures extracted from the equilibrated MD trajectories and the estimated ΔGcal values are shown and labeled in Fig. 4. The results of MD simulation show that the novel non-alkyne-based clinical candidates (fenobam, RGH-618, VU0424238, and HTL14242) of mGlu5 located in the same binding pocket as alkyne-based clinical NAMs [53] defined by residues (G6242.45, I6252.46, I6513.36, S6543.39, P6553.40, S6583.43, Y6593.44, V7405.40, L7445.44, N7475.47, T7816.46, W7856.50, F7886.53, M8027.32, S8057.35, V8067.36, S8097.39, and A8107.40) from the TM2, TM3, TM5, TM6, and TM7 domains. The aromatic ring of non-alkyne-based NAMs (corresponding to the alkyne group of alkyne-based NAMs) binds to the “narrow linker” in a similar way for the side instead of lying flat. Moreover, the conformations of HTL14242 and fenobam in the binding pocket are similar to the crystal structures (PDB ID: 5CGD and 6FFH) [24, 39], making direct or water mediated hydrogen bonds with residues I6513.36, S6543.39, Y6593.44, N7475.47, T7816.46, W7856.50, and S8097.39. For the fenobam–mGlu5 complex, a difference from the crystal structure is that the distance between the ligand and S8097.39 decreases from 2.9 to 2.7 Å, resulting in additional hydrogen bonds between Y6593.44 and W7856.50 with fenobam. In 2017, a novel NAM RGH-168 was discovered with its preclinical characterization. Galambos et al. reported that the phenyl rings and one sulfone oxygen of RGH-168 form aromatic interactions and hydrogen bond with W7856.50 and S8057.35 [23]. In this work, two docking poses of RGH-168 were sampled by MD simulation (Fig. 4c, c1). The better binding conformation (Fig. 4c) shows that, except for the aromatic interactions with W7856.50, an additional interaction is observed between the sulfone oxygen and residue N7475.47, and another sulfone oxygen forms hydrogen bond with S8057.35 through a water molecule. For the VU0424238, among its two binding conformations in Fig. 4d, d1, the former is proposed to be the better one. In Fig. 4d, VU0424238 has a higher binding affinity and forms an interactional network of hydrogen bonds with several residues (R6483.33, Y6593.44, T7816.46, and S8057.35) and water molecules.

a, b, c, c1, d, and d1 are the binding conformations of HTL14242, fenobam, RGH-618 (pose 1), RGH-618 (pose 2), VU0424238 (pose 1), and VU0424238 (pose 2), respectively. The important residues and NAMs are shown as sticks. The hydrogen bonds are depicted as red dotted lines, and the water molecules are displayed as red balls.
Common features shared by NAM–mGlu5 recognition
The hierarchical clustering analysis of per-residue binding free energy was exploited to characterize the common features shared by clinical NAMs (fenobam, HTL14242, RGH-618, VU0424238, mavoglurant, basimglurant, dipraglurant, STX107) in the mGlu5 receptor allosteric site (Fig. 5). A total of 149 residues were picked out for cluster analysis, and these residues have energy contributions in at least one NAM–mGlu5 system. As shown, the per-residue energy contributions of residues in group A (I6252.46, I6513.36, S6543.39, P6553.40, L7445.44, W7856.50, F7886.53, M8027.32, V8067.36, S8097.39 and A8107.40) are consistently higher than those of residues in other groups, and are characterized as the common features shared by the eight clinical NAM–mGlu5 complexes with different chemical structures. Furthermore, several residues with destabilization contribution to the NAM–mGlu5 complexes were also identified, which were harmful to ligands binding. Among them, residue G624 in the HTL14242–mGlu5 complex has the maximum energy contribution of 0.07 kcal/mol.

Energy contributions favoring ligand binding are displayed in red, with the highest contribution set as exact red and lower contributions gradually fading towards white. Energy contributions hampering ligand binding are shown in blue, with the highest contribution set as exact blue and lower contributions gradually fading towards white.
Verifying the computational models of MD simulation
The ranking ability of the estimated binding free energy ΔGcal is usually considered to be important evidence for evaluating the accuracy of the computational model [89, 90, 94]. To verify the models of MD simulation in our study, the experimental binding free energies (ΔGexp) of the NAMs in mGlu1 and mGlu5 were calculated using reported activity values by ΔGexp = RTln(IC50) or ΔGexp = RTln(Ki) (Table 1) [10, 14, 23, 24, 28,29,30,31, 38, 86]. The correlation coefficient (R2) between the values of ΔGcal and ΔGexp for five mGlu1 NAMs (FITM, BDBM50301534, EMQMCM, JNJ16259685, and the dual-target NAM VU0467558) and eight mGlu5 NAMs (fenobam, HTL14242, RGH-618, VU0424238, mavoglurant, basimglurant, MPEP, and 51D) are 0.71 and 0.69, respectively (Fig. 6). Therefore, the computational model is capable of evaluating the relative binding affinities of NAMs to mGlu1 and mGlu5. Nevertheless, the ΔGcal for each complex is overestimated compared to the experimental results (ΔGexp), which is caused by omitting the entropy effect from the formula. In this work, the calculation of free energy was used to rank the ability of NAMs binding to mGlu1 and mGlu5 and then to identify the key residues, rather than calculating the absolute binding free energy of the ligand–receptor complex. To further ensure the reliability of our computational models, multiple parallel trajectories were conducted, and the results indicate that the computational binding free energy can be reproduced by the parallel trajectories of each NAM–mGlu1/5 complex (Supplementary Table S5).

a is the correlation between the calculated (ΔGcal) and experimental (ΔGexp) binding free energies for NAM–mGlu1 complexes, and b is the corresponding value for NAM–mGlu5 complexes. The circle, square, and triangle indicate the NAMs targeting mGlu1, mGlu5, and mGlu1/5, respectively. c is the result of computational alanine scanning based on five “Hot Spots” in four NAMs–mGlu1 complexes.
On the other hand, in silico site-directed mutagenesis analysis has also been conducted on the MPEP–mGlu5 complex based on seven residues (P6553.40, S6583.43, Y6593.44, L7445.44, T7816.46, and Y7926.57), and the calculated results showed a good correlation (R2 = 0.90) with the experimental values in our previous study [53]. Herein, to verify the identified important residues, the CAS analysis was performed with the five “Hot Spots” (V6643.36, V7535.40, L7575.44, N7605.47, and F8016.53) in NAM–mGlu1 complexes. As shown in Fig. 6c, the ΔΔGCAS values of the five “Hot Spots” range from 1.72 to 4.11 kcal/mol, which implies that they are the major energy contributors, and further indicates that the computational model can recognize key residues of NAMs–mGlu1/5 complexes. In summary, the computational models are reliable for exploring the subtype-selective mechanism of NAMs in mGlu1 and mGlu5.
Subtype-selective mechanism of mGlu1 and mGlu5 to NAMs
As mGlu1 and mGlu5 are transmembrane proteins, NAMs from the outside of membrane enter the allosteric cavity through a narrow entrance [2, 28]. Structural alignment of mGlu1 and mGlu5 shows that the allosteric binding pockets of the two receptors do not completely coincide (Fig. 7). The two pockets of mGlu1 and mGlu5 are divided into three subsites: the “upper chamber”, “lower chamber”, and “narrow linker”, of which only the upper chamber is an area where the two pockets overlap [13, 24, 92]. The large difference between the two binding pockets may result in significant conformational and energetic variations of the corresponding residues to ligand binding, and the completely different chemical structures of selective mGlu1 and mGlu5 NAMs (the Tanimoto coefficients between NAMs are shown in Supplementary Table S4) also allow them to selectively bind to different receptor subtypes with high sequence identity. Based on the per-residue energy analysis, 15 important energy contributors (5 “Hot Spots” and 10 “Warm Spots”) shared by four NAM–mGlu1 complexes were identified. Sixteen residues in mGlu5 were identified as important residues for eight clinical NAMs’ binding, including 11 major energy contributors and 5 important residues (S6583.43, Y6593.44, N7475.47, T7816.46, and S8057.35). Therefore, a total of 18 pairs of residues, including 12 conserved ones and 6 non-conserved ones were picked out and listed in Table 2. All of them were mapped onto the structures of mGlu1 and mGlu5 shown in Fig. 7. Comparison of the structural and energetic information of mGlu1 and mGlu5 interacting with diverse NAMs, revealed the subtype-selective mechanism.

a shows the allosteric binding pockets and 18 pairs of important residues highlighted in orange (mGlu1) and green (mGlu5), and b is the image rotated 180° viewed parallel to the membrane.
Different properties of non-conserved residues in mGlu1 and mGlu5 determining the binding modes of NAMs
In Fig. 7, the upper chamber residues V6643.36, T8157.32, and A8187.35 of mGlu1 have short side chains that can provide a larger space than that of residues I6513.36, M8027.32, S8057.35 in the corresponding position of mGlu5, allowing the FITM to sit in the upper chamber. In 2019, Cong et al. speculated that M8027.32 may be an important residue for the subtype selectivity of PAMs in mGlus [35]. Our results are consistent with previous speculation and indicate that M8027.32 directly affects the space of upper chamber by larger side chains in the NAMs–mGlu5 complexes. For the narrow linker and lower chamber sites, residues S6583.43, P6553.40, and A8107.40 of mGlu5 form a broad entrance into the deeper part and provide a binding pocket with more space to accommodate the NAMs, while residues C6713.43, S6683.40, and V8237.40 in mGlu1 hinder the deep binding of NAMs (Fig. 7). The structural differences between the two binding pockets result in energy contribution variations of the corresponding residues, for instance in Fig. 8 the energy contribution of S6683.40 in mGlu1 is obviously different from that of P6553.40 in mGlu5. Furthermore, ligand–receptor interactions can also be quantitatively calculated based on the presence or absence of interactions by interaction fingerprints, and the NAMs–mGlu1/5 interaction fingerprints are depicted in Fig. 9. As shown, the key non-conserved residues in mGlu1 and mGlu5 determining the selectivity also present different numbers of interactions with NAMs during the simulation.

a–d are four NAM–mGlu1 complexes (FITM, BDBM50301534, EMQMCM, and JNJ16259685) shown in orange; e–l are eight NAM–mGlu5 complexes (fenobam, HTL14242, RGH-618, VU0424238, mavoglurant, dipraglurant, basimglurant, and STX107) shown in green.

a–d The FITM, BDBM50301534, EMQMCM, and JNJ16259685 bind to mGlu1, e–l the fenobam, HTL14242, RGH-618, VU0424238, mavoglurant, dipraglurant, basimglurant, and STX107 bind to mGlu5. The residues within 5 Å of the ligand mass center were extracted from the last 50 ns MD trajectory labeled in the radar charts, and the numbers from 0 to 1.0 indicate the probabilities of interactions between the NAM and the residues based on 500 snapshots in each complex.
In 2015, the Marshall group proposed that the substitution of residues S6683.40 and C6713.43 in mGlu1 with P6553.40 and S6583.43 in mGlu5 can be used to rationalize the subtype selectivity [13]. And the other residues (mGluR1: V6643.36, T8157.32, A8187.35; mGlu5: S8057.35, A8107.40) were considered key factors in subtype selectivity by mutagenesis studies, and their effects were summarized in the GPCRpd database [28, 34, 93, 95]. In our models, NAMs of mGlu1 bind in the upper chamber of the pocket, while the NAMs of mGlu5 further deeply integrate into the narrow linker and lower chamber (Fig. 10). Therefore, it is proposed that the unequal positions of NAMs located in the two receptors determined by the different properties of non-conserved residues in the binding pockets are critical factors in the subtype-selective mechanism of mGlu1 and mGlu5. Except the residues reported by previous research, a total of six pairs of non-conserved residues were recognized, and their differences in terms of energy and conformation were clarified to guide further experimental study.

The NAMs FITM(orange), BDBM50301534 (pink), EMQMCM (magenta), and JNJ16259685 (read) bind in the upper chamber of the mGlu1 allosteric site, while fenobam (green), HTL14242 (yellow), RGH-618 (cyan), VU0424238 (blue), mavoglurant (lime), dipraglurant (pea), basimglurant (wheat), and STX107 (gray) further deeply integrate into the narrow linker and lower chamber of the mGlu5 binding pocket.
Different roles of conserved residues in mGlu1 and mGlu5 stabilizing the binding modes of NAMs
There are five pairs of conserved residues (mGlu1: L7575.44, N7605.47, W7986.50, F8016.53, and V8197.36; mGlu5: L7445.44, N7475.47, W7856.50, F7886.53, and V8067.36) that play important roles in the binding of NAMs to both mGlu1 and mGlu5 (Fig. 8). In NAM–mGlu1 complexes, L7575.44 and N7605.47 (mGlu5: L7445.44, N7475.47) fix the NAMs in the upper chamber by strongly hydrophobic and hydrogen interactions (Figs. 2 and 8). The residue W7986.50 (W7856.50) has attracted a lot attention because of its obvious conformational differences in NAM–mGlu1/5 complexes according to the structural specificity of the NAMs (Figs. 2 and 4). In addition to the important role in ligand binding revealed in this work, W7986.50 was considered a gatekeeper of water flux in the research of Cong et al. [35]. Residues F8016.53 and V8197.36 (mGlu5: F7886.53 and V8067.36) are also related to the selective binding of NAMs to mGlu1 and mGlu5. Compared with the NAM–mGlu5 complexes, tight interactions between F8016.53 and four selective mGlu1 NAMs were observed (Fig. 8). In contrast, V8067.36 facilitates the binding of NAMs to mGlu5, whose energy contribution is significantly higher than the corresponding residues V8197.36 in mGlu1 (Figs. 8 and 9). Therefore, the enhancement of the interaction between NAMs and conserved residues L7575.44, N7605.47, and F8016.53 in mGlu1 (mGlu5: L7445.44, N7475.47, and F7886.53) promotes the binding of NAMs to mGlu1, while NAMs with better selectivity for mGlu5 should form tight interaction with V8067.36 (V8197.36 in mGlu1). In 2015, the Gloriam group reported that G6282.49 is a selective hotspot in mGlu5. Unfortunately, it was not identified in this study, due to the ordinary energy contribution and the similar conformation in mGlu1 and mGlu5 (a conserved residue in mGlu1 and mGlu5). However, compared with the other mGlu members in groups II and III, Gly628 is a non-conserved residue, which seems to be a key factor of selectivity between group I and other receptors.
Binding mechanism of the dual-target NAM VU0467558
VU0467558 is a novel NAM that exhibited near equipotent activity against both mGlu1 and mGlu5 [38]. The binding mechanism of VU0467558 was predicted to verify the selective features identified in this study and provide information for the design of dual-target NAMs. Two docking poses of VU0467558 binding in mGlu1 and mGlu5 have been studied by the same approach, respectively. As shown in Supplementary Fig. S5, the binding conformations of VU0467558 in pose 1 (cluster 1) have significant energy differences over those of pose 2 (cluster 2) (6.21 and 4.65 kcal/mol). The positions of VU0467558 in the two allosteric sites are obviously different. In detail, VU0467558 binds in the upper chamber of the mGlu1 pocket, and is located deeper in the mGlu5 pocket. The large differences between the two binding pockets result in significant energetic variations of the corresponding residues to VU0467558 binding, such as I6382.46, S6673.39, and S8227.39 in mGlu1 with the corresponding residues I6252.46, S6543.39, and V8097.39 in mGlu5. The six identified pairs of non-conserved residues (mGlu1: V6643.36, S6683.40, C6713.43, T8157.32, A8187.35, and V8237.40; mGlu5: I6513.36, P6553.40, S6583.43, M8027.32, S8057.35, and A8107.40) shaping the pockets for specific NAM binding were confirmed by structural superimposition of VU0467558 in complex with mGlu1 and mGlu5 (Fig. 11). Notably, the conformation of residues V6643.36 and T8157.32 in mGlu1 can provide a larger space than that of residues I6513.36 and M8027.32 in the corresponding position of mGlu5, allowing the tricyclic group of VU0467558 to sit in the upper chamber. The conformation of S8057.35 moved to the outside of the binding pocket to accommodate the binding of VU0467558 in mGlu5, compared to A8187.35 in mGlu1. On the other hand, S6583.43, P6553.40, and A8107.40 in mGlu5 form a broad entrance allowing the small moiety of VU0467558 to insert into the deeper part of the binding site and provide the binding pocket more space to accommodate the NAM, but residues C6713.43, S6683.40, and V8237.40 in mGlu1 hinder this deep binding. In addition, the energy contributions of several residues also demonstrated the binding difference of VU0467558 in mGlu1 and mGlu5. The roles of the conserved residues L7575.44, N7605.47, F8016.53, and V8197.36 in mGlu1 (corresponding to L7445.44, N7475.47, F7886.53, and V8067.36 in mGlu5) were observed in the predicted binding modes of VU0467558 binding to the two receptors. As shown in Fig. 11c, residues L7575.44, N7605.47, F8016.53, and V8197.36 contributed −3.09, −1.85, −1.71, and −1.06 kcal/mol for VU0467558 binding in mGlu1, while the energy contributions of the corresponding residues in mGlu5 were −1.06, −0.59, −1.19, and −2.06 kcal/mol, respectively. Moreover, the energy contributions of nine pairs of residues (mGlu1: V6643.36, S6683.40, V7535.40, L7575.44, N7605.47, W7986.50, F8016.53, T8157.32, and V8197.36; mGlu5: I6513.36, P6553.40, V7405.40, L7445.44, N7475.47, W7856.50, F7886.53, M8027.32, and V8067.36) are both important in mGlu1 and mGlu5, which are the key factors in designing new dual-target NAMs.

a is the representation of VU0467558 binding in mGlu1 (orange) and mGlu5 (green), and b is the image rotated 180°. The important residues identified as critical features of the subtype-selective mechanism are shown as sticks. c is the per-residue energy contribution of VU0467558 binding to mGlu1 (orange) and mGlu5 (green).
Conclusion
In this work, a computational modeling approach of molecular docking and MD simulation were utilized to explore the binding mechanism of different single- and dual-target NAMs in group I mGlus and clarify the subtype-selective mechanism in detail. The selective mGlu1 NAMs bind in the upper chamber of the allosteric site, and the binding pocket partially overlaps with mGlu5 in the upper chamber. In the mGlu5 receptor, the novel non-alkyne compounds are located in the same binding pocket as the alkyne NAMs, deeply inserted into the narrow linker and lower chamber. By comparing the structural and energetic profiles underlying the binding of twelve NAMs with diverse scaffolds and different activities to mGlu1 and mGlu5, it was found that ten pairs of residues including six non-conserved residues (mGlu1: V6643.36, S6683.40, C6713.43, T8157.32, A8187.35, and V8237.40; mGlu5: I6513.36, P6553.40, S6583.43, M8027.32, S8057.35, and A8107.40) and four conserved residues (mGlu1: L7575.44, N7605.47, F8016.53, and V8197.36; mGlu5: L7445.44, N7475.47, F7886.53, and V8067.36), arounding the binding pockets are essential to receptors subtype selectivity. The non-conserved residues mainly affect ligand binding through spatial conformation, and the four conserved residues enhance the selective ligands binding by participating in strong interactions. This information should be considered in the design of selective NAMs. In addition, the same dual-target NAM (VU0467558) adopted completely different conformations in the two receptors verifying the results of our computational model. Moreover, the nine pairs of residues (mGlu1: V6643.36, S6683.40, V7535.40, L7575.44, N7605.47, W7986.50, F8016.53, T8157.32, and V8197.36; mGlu5: I6513.36, P6553.40, V7405.40, L7445.44, N7475.47, W7856.50, F7886.53, M8027.32, and V8067.36) were recognized as the key factors in designing new dual-target NAMs of mGlu1 and mGlu5. The results of this research can provide theoretical guidance for the development of novel NAMs and also provide valuable information on the mechanisms of class C GPCR allosteric inhibition.
References
Koehl A, Hu H, Feng D, Sun B, Zhang Y, Robertson MJ, et al. Structural insights into the activation of metabotropic glutamate receptors. Nature. 2019;566:79–84.
Dore AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–62.
Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–37.
Urwyler S. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol Rev. 2011;63:59–126.
Litim N, Morissette M, Di Paolo T. Metabotropic glutamate receptors as therapeutic targets in Parkinson’s disease: an update from the last 5 years of research. Neuropharmacology. 2017;115:166–79.
Murrough JW, Abdallah CG, Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov. 2017;16:472–86.
Maksymetz J, Moran SP, Conn PJ. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol Brain. 2017;10:15.
Ramos-Prats A, Kolldorfer J, Paolo E, Zeidler M, Schmid G, Ferraguti F. An appraisal of the influence of the metabotropic glutamate 5 (mGlu5) receptor on sociability and anxiety. Front Mol Neurosci. 2019;12:30.
Li YH, Yu CY, Li XX, Zhang P, Tang J, Yang Q, et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2018;46:D1121–7.
Lesage A, Steckler T. Metabotropic glutamate mGlu1 receptor stimulation and blockade: therapeutic opportunities in psychiatric illness. Eur J Pharmacol. 2010;639:2–16.
Li B, Tang J, Yang Q, Li S, Cui X, Li Y, et al. NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017;45:W162–70.
Yang QX, Wang YX, Li FC, Zhang S, Luo YC, Li Y, et al. Identification of the gene signature reflecting schizophrenia’s etiology by constructing artificial intelligence-based method of enhanced reproducibility. CNS Neurosci Ther. 2019;25:1054–63.
Bennett KA, Dore AS, Christopher JA, Weiss DR, Marshall FH. Structures of mGluRs shed light on the challenges of drug development of allosteric modulators. Curr Opin Pharmacol. 2015;20:1–7.
Felts AS, Rodriguez AL, Blobaum AL, Morrison RD, Bates BS, Thompson Gray A, et al. Discovery of N-(5-fluoropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (VU0424238): a novel negative allosteric modulator of metabotropic glutamate receptor subtype 5 selected for clinical evaluation. J Med Chem. 2017;60:5072–85.
Owen DR. Recent advances in the medicinal chemistry of the metabotropic glutamate receptor 1 (mGlu(1)). ACS Chem Neurosci. 2011;2:394–401.
Feng Z, Ma S, Hu G, Xie XQ. Allosteric binding site and activation mechanism of class C G-protein coupled receptors: metabotropic glutamate receptor family. AAPS J. 2015;17:737–53.
Yang Q, Wang Y, Zhang Y, Li F, Xia W, Zhou Y, et al. NOREVA: enhanced normalization and evaluation of time-course and multi-class metabolomic data. Nucleic Acids Res. 2020;48:W436–48.
Tang J, Fu J, Wang Y, Li B, Li Y, Yang Q, et al. ANPELA: analysis and performance assessment of the label-free quantification workflow for metaproteomic studies. Brief Bioinform. 2020;21:621–36.
Emmitte KA. mGlu5 negative allosteric modulators: a patent review (2013–2016). Expert Opin Ther Pat. 2017;27:691–706.
Vranesic I, Ofner S, Flor PJ, Bilbe G, Bouhelal R, Enz A, et al. AFQ056/mavoglurant, a novel clinically effective mGluR5 antagonist: identification, SAR and pharmacological characterization. Bioorg Med Chem. 2014;22:5790–803.
Quiroz JA, Tamburri P, Deptula D, Banken L, Beyer U, Rabbia M, et al. Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiatry. 2016;73:675–84.
Fuxe K, Borroto-Escuela DO. Basimglurant for treatment of major depressive disorder: a novel negative allosteric modulator of metabotropic glutamate receptor 5. Expert Opin Investig Drugs. 2015;24:1247–60.
Galambos J, Bielik A, Krasavin M, Orgovan Z, Domany G, Nogradi K, et al. Discovery and preclinical characterization of 3-((4-(4-chlorophenyl)-7-fluoroquinoline-3-yl)sulfonyl)benzonitrile, a novel non-acetylenic metabotropic glutamate receptor 5 (mGluR5) negative allosteric modulator for psychiatric indications. J Med Chem. 2017;60:2470–84.
Christopher JA, Aves SJ, Bennett KA, Dore AS, Errey JC, Jazayeri A. et al. Fragment and structure-based drug discovery for a class C GPCR: discovery of the mGlu5 negative allosteric modulator HTL14242 (3-chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). J Med Chem. 2015;58:6653–64.
Wang Y, Zhang S, Li F, Zhou Y, Zhang Y, Wang Z, et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020;48:D1031–41.
Yin J, Sun W, Li F, Hong J, Li X, Zhou Y, et al. VARIDT 1.0: variability of drug transporter database. Nucleic Acids Res. 2020;48:D1171.
Li YH, Li XX, Hong JJ, Wang YX, Fu JB, Yang H, et al. Clinical trials, progression-speed differentiating features and swiftness rule of the innovative targets of first-in-class drugs. Brief Bioinform. 2020;21:649–62.
Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64.
Satoh A, Nagatomi Y, Hirata Y, Ito S, Suzuki G, Kimura T. et al. Discovery and in vitro and in vivo profiles of 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzam ide as novel class of an orally active metabotropic glutamate receptor 1 (mGluR1) antagonist. Bioorg Med Chem Lett. 2009;19:5464–8.
Mabire D, Coupa S, Adelinet C, Poncelet A, Simonnet Y, Venet M, et al. Synthesis, structure-activity relationship, and receptor pharmacology of a new series of quinoline derivatives acting as selective, noncompetitive mGlu1 antagonists. J Med Chem. 2005;48:2134–53.
Lavreysen H, Wouters R, Bischoff F, Nobrega Pereira S, Langlois X, Blokland S, et al. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology. 2004;47:961–72.
Lane JR, Abdul-Ridha A, Canals M. Regulation of G protein-coupled receptors by allosteric ligands. ACS Chem Neurosci. 2013;4:527–34.
Weiss DR, Karpiak J, Huang XP, Sassano MF, Lyu J, Roth BL, et al. Selectivity challenges in docking screens for GPCR targets and antitargets. J Med Chem. 2018;61:6830–45.
Harpsoe K, Isberg V, Tehan BG, Weiss D, Arsova A, Marshall FH, et al. Selective negative allosteric modulation of metabotropic glutamate receptors—a structural perspective of ligands and mutants. Sci Rep. 2015;5:13869.
Cong X, Cheron JB, Golebiowski J, Antonczak S, Fiorucci S. Allosteric modulation mechanism of the mGluR5 transmembrane domain. J Chem Inf Model. 2019;59:2871–8.
Emmitte KA. mGlu5 negative allosteric modulators: a patent review (2010–2012). Expert Opin Ther Pat. 2013;23:393–408.
Newell KA, Matosin N. Rethinking metabotropic glutamate receptor 5 pathological findings in psychiatric disorders: implications for the future of novel therapeutics. BMC Psychiatry. 2014;14:23.
Felts AS, Rodriguez AL, Morrison RD, Venable DF, Blobaum AL, Byers FW, et al. N-Alkylpyrido[1’,2’:1,5]pyrazolo-[4,3-d]pyrimidin-4-amines: a new series of negative allosteric modulators of mGlu1/5 with CNS exposure in rodents. Bioorg Med Chem Lett. 2016;26:1894–900.
Christopher JA, Orgovan Z, Congreve M, Dore AS, Errey JC, Marshall FH, et al. Structure-based optimization strategies for G protein-coupled receptor (GPCR) allosteric modulators: a case study from analyses of new metabotropic glutamate receptor 5 (mGlu5) X-ray structures. J Med Chem. 2019;62:207–22.
Koehl A, Hu H, Feng D, Sun B, Zhang Y, Robertson MJ, et al. Author correction: structural insights into the activation of metabotropic glutamate receptors. Nature. 2019;567:E10.
Xue W, Wang P, Li B, Li Y, Xu X, Yang F, et al. Identification of the inhibitory mechanism of FDA approved selective serotonin reuptake inhibitors: an insight from molecular dynamics simulation study. Phys Chem Chem Phys. 2016;18:3260–71.
Zheng G, Xue W, Wang P, Yang F, Li B, Li X, et al. Exploring the inhibitory mechanism of approved selective norepinephrine reuptake inhibitors and reboxetine enantiomers by molecular dynamics study. Sci Rep. 2016;6:26883.
Wang P, Zhang X, Fu T, Li S, Li B, Xue W, et al. Differentiating physicochemical properties between addictive and nonaddictive ADHD drugs revealed by molecular dynamics simulation studies. ACS Chem Neurosci. 2017;8:1416–28.
Zheng G, Xue W, Yang F, Zhang Y, Chen Y, Yao X, et al. Revealing vilazodone’s binding mechanism underlying its partial agonism to the 5-HT1A receptor in the treatment of major depressive disorder. Phys Chem Chem Phys. 2017;19:28885–96.
Yang F, Zheng G, Fu T, Li X, Tu G, Li YH, et al. Prediction of the binding mode and resistance profile for a dual-target pyrrolyl diketo acid scaffold against HIV-1 integrase and reverse-transcriptase-associated ribonuclease H. Phys Chem Chem Phys. 2018;20:23873–84.
Hong J, Luo Y, Mou M, Fu J, Zhang Y, Xue W, et al. Convolutional neural network-based annotation of bacterial type IV secretion system effectors with enhanced accuracy and reduced false discovery. Brief Bioinform. 2020; 21:1825–36.
Xue W, Wang P, Tu G, Yang F, Zheng G, Li X, et al. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys Chem Chem Phys. 2018;20:6606–16.
Wang P, Fu T, Zhang X, Yang F, Zheng G, Xue W, et al. Differentiating physicochemical properties between NDRIs and sNRIs clinically important for the treatment of ADHD. Biochim Biophys Acta Gen Subj. 2017;1861:2766–77.
Zhang Y, Ying JB, Hong JJ, Li FC, Fu TT, Yang FY, et al. How does chirality determine the selective inhibition of histone deacetylase 6? A lesson from trichostatin A enantiomers based on molecular dynamics. ACS Chem Neurosci. 2019;10:2467–80.
Zhang Y, Fu T, Ren Y, Li F, Zheng G, Hong J, et al. Selective inhibition of HDAC1 by macrocyclic polypeptide for the treatment of glioblastoma: a binding mechanistic analysis based on molecular dynamics. Front Mol Biosci. 2020;7:41.
Xue W, Fu T, Zheng G, Tu G, Zhang Y, Yang F, et al. Recent advances and challenges of the drugs acting on monoamine transporters. Curr Med Chem. 2020;27:3830–76.
Xue W, Yang F, Wang P, Zheng G, Chen Y, Yao X, et al. What contributes to serotonin-norepinephrine reuptake inhibitors’ dual-targeting mechanism? the key role of transmembrane domain 6 in human serotonin and norepinephrine transporters revealed by molecular dynamics simulation. ACS Chem Neurosci. 2018;9:1128–40.
Fu T, Zheng G, Tu G, Yang F, Chen Y, Yao X, et al. Exploring the binding mechanism of metabotropic glutamate receptor 5 negative allosteric modulators in clinical trials by molecular dynamics simulations. ACS Chem Neurosci. 2018;9:1492–502.
Graziani D, Caligari S, Callegari E, De Toma C, Longhi M, Frigerio F, et al. Evaluation of amides, carbamates, sulfonamides, and ureas of 4-prop-2-ynylidenecycloalkylamine as potent, selective, and bioavailable negative allosteric modulators of metabotropic glutamate receptor 5. J Med Chem. 2019;62:1246–73.
Global Burden of Disease Study C. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800.
Morris GM, Lim-Wilby M. Molecular docking. Methods Mol Biol. 2008;443:365–82.
Prime v. 2.0, Schrödinger, LLC, New York. 2009.
Maestro v. 9.0, Schrödinger, LLC, New York. 2009.
Tang J, Fu J, Wang Y, Luo Y, Yang Q, Li B, et al. Simultaneous improvement in the precision, accuracy, and robustness of label-free proteome quantification by optimizing data manipulation chains. Mol Cell Proteom. 2019;18:1683–99.
LigPrep v. 2.3, Schrödinger, LLC, New York. 2009.
Epik v. 2.0, Schrödinger, LLC, New York. 2009.
Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–6.
Qi Y, Cheng X, Lee J, Vermaas JV, Pogorelov TV, Tajkhorshid E, et al. CHARMM-GUI HMMM builder for membrane simulations with the highly mobile membrane-mimetic model. Biophys J. 2015;109:2012–22.
Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J Comput Chem. 2014;35:1997–2004.
Yang Q, Li B, Tang J, Cui X, Wang Y, Li X, et al. Consistent gene signature of schizophrenia identified by a novel feature selection strategy from comprehensive sets of transcriptomic data. Brief Bioinform. 2020;21:1058–68.
Bai Q, Yao X. Investigation of allosteric modulation mechanism of metabotropic glutamate receptor 1 by molecular dynamics simulations, free energy and weak interaction analysis. Sci Rep. 2016;6:21763.
Gaussian 09 v. D.01. Gaussian, Inc, Wallingford CT. 2009.
AMBER, v. 16, University of California, San Francisco. 2016.
Yang L, Skjevik AA, Han Du WG, Noodleman L, Walker RC, Gotz AW. Data for molecular dynamics simulations of B-type cytochrome c oxidase with the Amber force field. Data Brief. 2016;8:1209–14.
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–713.
Dickson CJ, Madej BD, Skjevik AA, Betz RM, Teigen K, Gould IR, et al. Lipid14: The Amber Lipid Force Field. J Chem Theory Comput. 2014;10:865–79.
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–74.
Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97:10269–80.
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802.
Roe DR, Cheatham TE 3rd. Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. J Comput Chem. 2018;39:2110–7.
Roe DR, Cheatham TE 3rd. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–95.
Yang Q, Hong J, Li Y, Xue W, Li S, Yang H, et al. A novel bioinformatics approach to identify the consistently well-performing normalization strategy for current metabolomic studies. Brief Bioinform. 2019; bbz137. https://doi.org/10.1093/bib/bbz137.
Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZH, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev. 2019;119:9478–508.
Zheng G, Yang F, Fu T, Tu G, Chen Y, Yao X, et al. Computational characterization of the selective inhibition of human norepinephrine and serotonin transporters by an escitalopram scaffold. Phys Chem Chem Phys. 2018;20:29513–27.
Tippmann S. Programming tools: adventures with R. Nature. 2015;517:109–10.
Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47:W256–9.
Boukharta L, Gutierrez-de-Teran H, Aqvist J. Computational prediction of alanine scanning and ligand binding energetics in G-protein coupled receptors. PLoS Comput Biol. 2014;10:e1003585.
Ramadoss V, Dehez F, Chipot C. AlaScan: a graphical user interface for alanine scanning free-energy calculations. J Chem Inf Model. 2016;56:1122–6.
Da Silva F, Desaphy J, Rognan D. IChem: a versatile toolkit for detecting, comparing, and predicting protein-ligand interactions. Chem Med Chem. 2018;13:507–10.
Desaphy J, Raimbaud E, Ducrot P, Rognan D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J Chem Inf Model. 2013;53:623–37.
Anighoro A, Graziani D, Bettinelli I, Cilia A, De Toma C, Longhi M, et al. Insights into the interaction of negative allosteric modulators with the metabotropic glutamate receptor 5: discovery and computational modeling of a new series of ligands with nanomolar affinity. Bioorg Med Chem. 2015;23:3040–58.
Rovira X, Malhaire F, Scholler P, Rodrigo J, Gonzalez-Bulnes P, Llebaria A, et al. Overlapping binding sites drive allosteric agonism and positive cooperativity in type 4 metabotropic glutamate receptors. FASEB J. 2015;29:116–30.
Hong J, Luo Y, Zhang Y, Ying J, Xue W, Xie T, et al. Protein functional annotation of simultaneously improved stability, accuracy and false discovery rate achieved by a sequence-based deep learning. Brief Bioinforma. 2020;21:1437–47.
Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10:449–61.
Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51:69–82.
Jazayeri A, Marshall F. Implications of metabotropic glutamate receptor structures for drug discovery in neurotherapeutics. Expert Rev Neurother. 2015;15:123–5.
Llinas Del Torrent C, Casajuana-Martin N, Pardo L, Tresadern G, Perez-Benito L. Mechanisms underlying allosteric molecular switches of metabotropic glutamate receptor 5. J Chem Inf Model. 2019;59:2456–66.
Pandy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpsoe K, Hauser AS, et al. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 2018;46:D440–6.
Dong L, Shen S, Xu Y, Wang L, Feng R, Zhang J, et al. Computational studies on the potency and selectivity of PUGNAc derivatives against GH3, GH20, and GH84 beta-N-acetyl-D-hexosaminidases. Front Chem. 2019;7:235.
Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, et al. Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol. 2003;64:823–32.
Acknowledgements
The project was supported by funding from the National Natural Science Foundation of China (21505009, 81872798), the Technology Innovation and Application Demonstration Project of Chongqing (cstc2018jscx-msybX0287), and the Fundamental Research Funds for Central Universities (10611CDJXZ238826, 2018CDQYSG0007, 2019CDYGYB005).
Author information
Authors and Affiliations
Contributions
WWX and FZ designed the research. TTF, GT, and MP performed the research. TTF, GXZ, FYY, JYY, YZ, XJY, and WWX, analyzed the data. TTF, WWX, and FZ wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Fu, Tt., Tu, G., Ping, M. et al. Subtype-selective mechanisms of negative allosteric modulators binding to group I metabotropic glutamate receptors. Acta Pharmacol Sin 42, 1354–1367 (2021). https://doi.org/10.1038/s41401-020-00541-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00541-z
