
Abstract
Rodent diabetic models, used to understand the pathophysiology of diabetic cardiomyopathy (DCM), remain several limitations. Engineered cardiac tissues (ECTs) have emerged as robust 3D in vitro models to investigate structure–function relationships as well as cardiac injury and repair. Advanced glycation end-products (AGEs), produced through glycation of proteins or lipids in response to hyperglycemia, are important pathogenic factor for the development of DCM. In the current study, we developed a murine-based ECT model to investigate cardiac injury produced by AGEs. We treated ECTs composed of neonatal murine cardiac cells with AGEs and observed AGE-related functional, cellular, and molecular alterations: (1) AGEs (150 µg/mL) did not cause acute cytotoxicity, which displayed as necrosis detected by medium LDH release or apoptosis detected by cleaved caspase 3 and TUNEL staining, but negatively impacted ECT function on treatment day 9; (2) AGEs treatment significantly increased the markers of fibrosis (TGF-β, α-SMA, Ctgf, Collagen I-α1, Collagen III-α1, and Fn1) and hypertrophy (Nppa and Myh7); (3) AGEs treatment significantly increased ECT oxidative stress markers (3-NT, 4-HNE, HO-1, CAT, and SOD2) and inflammation response markers (PAI-1, TNF-α, NF-κB, and ICAM-1); and (4) AGE-induced pathogenic responses were all attenuated by pre-application of AGE receptor antagonist FPS-ZM1 (20 µM) or the antioxidant glutathione precursor N-acetylcysteine (5 mM). Therefore, AGEs-treated murine ECTs recapitulate the key features of DCM’s functional, cellular and molecular pathogenesis, and may serve as a robust in vitro model to investigate cellular structure-function relationships, signaling pathways relevant to DCM and pharmaceutical intervention strategies.
Similar content being viewed by others


Introduction
The global diabetes prevalence in 2019 was estimated to be 9.3% (463 million) and has been predicted to rise to 10.2% (578 million) by 2030 and 10.9% (700 million) by 2045 [1]. Diabetic cardiomyopathy (DCM), the main cause of heart failure in diabetic patients and one of the most lethal complications [2], is characterized by cardiac hypertrophy, fibrosis, and dysfunction [3]. In recent years, rodent diabetic models have advanced our understanding of the pathophysiology of DCM; however, these models require a long time to develop DCM and are affected by the confounding influences inherent to in vivo models [4], and in vitro cardiomyocyte culture provides the ability to study intracellular processes but does not reflect the tissue-level injury and remodeling typical of DCM.
Engineered cardiac tissues (ECTs) have emerged as robust 3D in vitro models of maturing myocardium due to the rapid structural and functional maturation of ECTs and the ability to vary ECT composition as well as mimic ECT functional evolution in in vivo models. ECTs are well suited for a variety of applications, including disease modeling [5,6,7,8], drug screening [9,10,11,12], tissue repair [13,14,15,16,17], investigating cardiac development and maturation [18,19,20,21,22], and exploration of gene-editing technology in the heart [23,24,25]. By utilizing different interventions, ECTs are well adapted for establishment of different disease models as well as drug toxicity testing. ECT patches developed for cardiac repair and regeneration through tissue implantation also showed promising results. Of note, cardiac cell isolation from neonatal murine hearts is technically very challenging, but when they are isolated successfully, these neonatal cells rapidly proliferate, undergo remodeling, and generate functioning myocardium [6].
As a highly heterogeneous group of compounds, advanced glycation end-products (AGEs) are proteins or lipids that are glycated in response to clinical hyperglycemia and can occur as the result of excessive temperature food processing. AGEs are deleterious in mammals due to their promotion of oxidative stress and inflammation and are the most important pathogenic initiator for the development of DCM [26,27,28,29,30]. The mechanistic understanding of the role of AGEs in the development of DCM has confirmed that AGEs interact with the receptor of AGEs (RAGE) to turn on intracellular pathways that generate excess reactive oxygen or nitrogen species (ROS/RNS) and cause cardiac chronic oxidative stress and damage, inflammation, and consequently cardiac cell hypertrophy and fibrosis, resulting in cardiac dysfunction [26, 28].
The goal of the present study was to develop and validate an in vitro model of engineered myocardial tissue with AGEs treatment, which may reflect the functional, cellular, and molecular features of DCM and be useful for further investigation.
Materials and methods
Animals
FVB mice were housed at the University of Louisville Research Resources Center at 22 °C with a 12-h light/dark cycle and were provided free access to standard rodent chow and tap water. All animal procedures were approved by the Institutional Animal Care and Use Committee, which is certified by the American Association for Accreditation of Laboratory Animal Care.
Isolation of neonatal mouse ventricular cardiac cells and ECT construction
We used the enzymatic dissociation method (as described in our previous work [6]) to isolate cardiomyocytes (CMs) from the hearts of pups within 3 days after birth. The cell suspensions were preplated into sterilized tissue culture plates (100 × 20 mm; Corning) and incubated at 37 °C for 45 min to enrich the CM population, which was followed by rotation at 50 rpm on a shaker (Labnet International) for an additional 4 h at 37 °C in 5% CO2 to facilitate CM cluster formation. The cardiac cells remaining in suspension were collected. Cells were stained with Trypan blue (Sigma), and the number of live CMs was determined using a hemocytometer. ECTs were constructed as previously described [6]. Briefly, 8 × 105 CMs were mixed with collagen and Matrigel to generate a total of 200 µL of cell/matrix suspension, which was poured to form 20 × 3 mm cylindrical constructs within a Flexcell Tissue TrainTM culture plate (Flexcell International) and incubated under preprogrammed conditions in a vacuum for 2 h (37 °C, 5% CO2) to form the cylindrical ECT constructs. Following initial ECT gelation, 4 mL medium was added to the 6-well culture dish. All the ECTs were cultured in medium with 15% serum during the first 3 days, and after the 3rd day, the normal serum group (NS) was maintained in the same culture medium, while the low serum group (LS) was maintained in culture medium with 10% serum. All the ECTs were maintained in culture medium for 3–15 days, with media changes every three days. AGEs (Sigma-Aldrich) and BSA (Sigma-Aldrich, as a control for comparison to AGEs) treatment started on the 4th day, and the medium with AGEs was changed every 3 days until harvest. DMSO, FPS-ZM1 (TOCRIS, 20 µM) or N-acetylcysteine (NAC, Sigma-Aldrich, 5 mM) was added to ECTs 2 h before the control or AGEs treatment.
ECT function classification
ECT function was digitally recorded for 15 s every day after ECT construction and then qualitatively classified as previously described [6]. Briefly, normal function (N) corresponded to increased ECT deformation during shortening with regional dyssynchrony (A); increased ECT deformation during shortening with global dyssynchrony (B); decreased beating rate and deformation with global dyssynchrony (C); and arrested beating (D).
Histology
ECTs were fixed in situ using 4% paraformaldehyde in 1× PBS (pH = 7.2) at room temperature for 30 min, removed from the Tissue TrainTM plate, stored in PBS at 4 °C, dehydrated with a graded alcohol series, cleared with xylene, embedded in paraffin and sectioned at a 5 µm thickness. Hematoxylin and eosin staining was performed as previously described [6]. For troponin T staining, after antigen retrieval, the ECT sections were washed in PBS with 0.05% Tween 20, blocked with 0.05% Triton-X-100 + 5% BSA in PBS for 0.5 h and then incubated with mouse anti-cTnT (1:500 dilution; Thermo Fisher Scientific, California, USA). We counterstained the nuclei with DAPI (Invitrogen) after incubation with the secondary antibody for 1 h. We randomly selected 10 fields for each ECT to count the double-stained cTnT (red) - and DAPI (blue)-positive cells, and the cTnT+/DAPI+ percentage was used to determine the CM ratio in the ECTs.
Measurement of ROS by dihydroethidium (DHE) staining
To evaluate the oxidative stress level in the different treatment groups, we analyzed superoxide and hydrogen peroxide production by using DHE staining as previously described [31]. The frozen sections were incubated at 37 °C for 30 min with DHE (10 µmol/L, Sigma) and then counterstained with DAPI (Invitrogen) after washing with PBS according to the manufacturer’s instructions. Exposure to light was avoided during the whole experiment. For quantitative analysis of the immunofluorescence intensity, the IOD and ROI were measured by ImageJ software (National Institutes of Health).
TdT-mediated dUTP nick-end labeling assay (TUNEL)
We used the DeadEnd™ Fluorometric TUNEL System (Promega, Madison, WI) to determine the apoptotic cell proportion as previously described [5]. The TUNEL-positive cells were counted in 10 fields for each ECT.
Protein extraction and Western blotting
ECTs were washed thoroughly in ice-cold PBS, rapidly homogenized in lysis buffer (100 µL/ECT) containing 50 mmol/L Tris-HCl (pH 7.4), 1× protease inhibitor (Sigma), 1 mmol/L EDTA (pH 8.0), and 1× PMSF and lysed at 4 °C for 4 h. After centrifugation at 12,000 rpm for 20 min, supernatants containing soluble proteins were subjected to protein concentration measurements using the Bradford method. Equal amounts of protein (15–30 mg) were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes by blotting after blocking with 5% nonfat milk for 1 h at room temperature. Then, the membranes were washed three times with Tris‐buffered saline containing 0.1% Tween-20 (TBST). The membranes were incubated with primary antibodies overnight at 4 °C, washed with TBST and then incubated with a secondary antibody for 1 h at room temperature. The antigen–antibody complexes were then visualized using an ECLkit (Bio-Rad). The primary antibodies included Caspase-3 (1:1000; Cell Signaling Technology, Danvers, USA), 3‐nitrotyrosine (3-NT; 1:1000; Millipore Corp, Temecula, USA), 4-hydroxynonenal (4-HNE; 1:2000; Abcam, Cambridge, MA, USA), home oxygenase-1 (HO-1; 1:1000; Cell Signaling Technology, Danvers, USA), catalase (CAT; 1:5000; Santa Cruz Biotechnology, Dallas, USA), superoxide dismutase-2 (SOD-2; 1:5000; Santa Cruz Biotechnology, Dallas, USA), plasminogen activator inhibitor type 1 (PAI-1; 1:1000; BD Biosciences, San Jose, USA), intercellular adhesion molecule-1 (ICAM-1, 1:1000; Santa Cruz Biotechnology, Dallas, USA), nuclear factor kappa-light chain enhancer of activated B cells p65 (NF-κB, 1:1000, Cell Signaling Technology, Danvers, USA), tumor necrosis factor‐α (TNF-α; 1:1000; Abcam, Cambridge, USA), connective tissue growth factor (CTGF, 1:1000, Santa Cruz Biotechnology, Dallas, USA), transforming growth factor β (TGF-β, 1:1000, Cell Signaling Technology, Danvers, USA), and α-smooth muscle actin (α-SMA, 1:1000, Cell Signaling Technology, Danvers, USA), followed by an anti-rabbit/anti-mouse HRP-linked secondary antibody (1:5000 dilution; Cell Signaling, Danvers, USA). β-Actin (1:3000 dilution, Santa Cruz, Dallas, USA) and GAPDH (1:3000 dilution; Abcam, Cambridge, USA) were used as internal controls.
Isolation of RNA from ECTs and quantitative real-time PCR
Briefly, ECT samples were homogenized by an Omni Tip Tissue homogenizer (USA Scientific, Ocala, Florida). Total RNA was isolated using TRIzol (Invitrogen)and purified with an RNeasy Mini Kit (Valencia, California). RNA quality and quantity were measured using a NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, Massachusetts). Reverse transcription was performed with a Mastercycler Gradient (Eppendorf) at 42 °C for 50 min and 95 °C for 5 min with 0.5 µg total RNA in a final volume of 20 µL (4 µL of 25 mmol/L MgCl2, 4 µL AMV reverse transcriptase 5×buffer, 2 µL dNTPs, 0.5 µL RNase inhibitor, 1 µL AMV reverse transcriptase, 1 µL dT primer and nuclease-free water). Primers targeting Fn1, Collagen I-α1, Collagen III-α1, Nppa, Ctgf, Myh7 and Gapdh were purchased from Thermo Fisher. Quantitative PCR was performed in a 20 µL volume (10 µL TaqMan Universal PCR Master Mix, 1 µL primers and 9 µL cDNA) using the ABI 7300 Real-Time PCR system. Data are expressed as the fold differences vs. those of the controls using the ΔΔCt method and Gapdh as a reference gene.
Statistical analyses
Data are presented as the normalized mean ± SD. Statistical differences were determined using 2-sided unpaired Student’s t tests or 2-way analysis of variance followed by Tukey’s multiple comparisons test. A P value < 0.05 was considered statistically significant.
Results
Basic ECT characteristics
Our previous studies used medium containing 15% serum (NS) for ECT culture, and we modified our standard protocol to utilize medium containing 10% serum (LS) for ECT culture after ECT maturation (3 days after ECT construction). Therefore, we compared the effects of 10% and 15% serum on the structural, functional and molecular maturation of ECTs. As we previously reported, ECTs start to contract rapidly after construction, and the beating rate gradually declines over time. No significant difference was found for the gel compaction and beating rates between the LS and NS groups (Supplementary material: Fig. S1a, b). Histological examination with H&E staining (Supplementary material: Fig. S1c) did not reveal any significant difference between the LS and NS groups in terms of cell density or arrangement. Considering that H&E staining cannot distinguish different cell types, immunofluorescence staining of cTnT (a CM-specific marker) was applied, which revealed no difference in the CM distributions (Supplementary material: Fig. S2a, b) and numbers (Supplementary material: Fig. S2c) between the LS and NS groups.
Of note, the LS group ECTs showed no significant differences compared to the NS group ECTs in terms of global function during culture (Supplementary material: Table S1). We also checked the expression of cleaved caspase 3/caspase 3 (marker of apoptosis), HO-1 (marker of oxidative stress), CTGF (marker of fibrosis), PAI-1 and TNFα (inflammatory markers) in the LS and NS groups (Supplementary material: Fig. S3), which were high on day 0 likely because of cell injury due to cell isolation and then decreased to relatively stable low levels from day 4 to day 14 (Supplementary material: Fig. S3). Therefore, the LS medium culture group showed molecular and functional features comparable to those of the NS medium culture group after up to 14 days in culture.
Identification of the dose range of AGEs unable to induce acute cytotoxicity but able to impair ECT function for establishing AGE-induced cardiomyopathy in an in vitro model
Since diabetes involves a chronic pathogenic process, diabetes-related pathogenic factors such as AGEs should not be significantly or acutely cytotoxic, but rather they should trigger chronic oxidative stress and inflammatory responses leading to cell hypertrophy, tissue fibrosis and eventually cardiac dysfunction. To establish a relevant in vitro model, we first determined the noncytotoxic doses of AGEs in ECTs. Through concentration gradient experiments, we found that AGEs at 75–300 µg/mL did not induce necrotic effects, as assayed by measuring LDH in cultured media for a treatment time from 24 to 72 h; at 600 µg/mL, AGEs also did not induce necrotic effects until treatment was extended for 72 h (Fig. 1a). Upon detecting cleaved caspase 3/caspase 3 in the total protein lysate of ECTs treated with AGEs for 72 h, apoptotic cell death was not observed at 75 – 150 µg/mL AGEs but was noted at 300–600 µg/mL in a dose-dependent manner (Fig. 1b, c). Furthermore, we used TUNEL staining to assess cell apoptosis in AGE-treated ECTs for 12 days and found that AGEs at 150 µg/mL did not increase apoptotic cell death, but AGEs at 300–600 µg/mL increased cell apoptosis in a dose-dependent manner (Fig. 1d, e).

a LDH assays of the medium from ECTs at different time points treated with different concentrations of AGEs revealed significant necrosis occurred only after AGEs treatment at 600 µg/mL for 72 h. n = 3 per group per time point. b, c Western blotting of c-Caspase 3/Caspase 3 in ECTs treated with different concentrations of AGEs. n = 3 per group, treatment for 72 h. d, e Representative ECT immunofluorescent staining for the terminal transferase dUTP nick end labeling assay (TUNEL, green) and of nuclei (DAPI, blue) following treatment with different concentrations of AGEs for 12 days (×40 magnification). n = 3 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus the respective control groups. Data are summarized as the normalized mean ± SD.
Next, we tested the effect of the highest dose of AGEs used in this study (150 µg/mL), which did not cause acute cytotoxic effects, on ECT function and found that treatment of ECTs with AGEs at 150 µg/mL did not impact function on day 4 but showed an effect on day 9, when all ECTs showed different degrees of dysfunction compared to the time-matched controls (Supplementary material: Table S2), which mainly manifested as grade A - C dysfunction according to the ECT functional classification.
Determination of whether AGE-induced ECT dysfunction is accompanied by typical pathogenic alterations observed in animal DCM models
AGEs triggered remodeling in ECTs
Cardiac hypertrophy and fibrosis are the typical pathological changes associated with remodeling noted during the development of DCM [32]. Therefore, the potential of AGEs to induce fibrotic and hypertrophic changes in ECTs was examined by the analysis of hypertrophic and fibrotic markers. AGEs significantly increased CTGF, α-SMA, and TGF-β protein expression (Fig. 2a–d) after treatment for 3–9 days. Expression of Collagen I-α1, Collagen III-α1, Fn1, and Ctgf transcripts also increased after treatment for 3 or 9 days (Fig. 2e). Levels of markers of cardiomyocyte hypertrophy, Nppa and Myh7, also increased after AGEs treatment for 9 days (Fig. 2f). Hence, AGEs were able to trigger profibrotic and hypertrophic remodeling as well ECT’s dysfunction (Supplementary material: Table S2), consistent with the pathological changes in the development of DCM.

a–d The fibrosis markers CTGF, α-SMA, and TGF-β were assessed by Western blotting. n = 3 per group. e The fibrosis markers Collagen I-α1, Collagen III-α1, Fn1, and Ctgf were assessed by qRT-PCR. n = 3 per group. f The cardiomyocyte hypertrophy markers Nppa and Myh7 were assessed by qRT-PCR. n = 3 per group. *P < 0.05, **P < 0.01, ***P < 0.001 versus the respective control groups. Data are summarized as the normalized mean ± SD.
AGE-induced inflammation in ECTs
AGE-induced cardiac remodeling is associated with inflammatory responses, and we noted increases in the inflammatory cascade markers NF-κB, PAI-1, TNF-α, and ICAM-1 in ECTs exposed to AGEs (Fig. 3); NF-κB was increased starting at 6 h until day 6, with peak levels observed between 12 and 72 h, whereas ICAM-1 was increased at 6 h and gradually increased throughout day 1. Both TNF-α and PAI-1 were increased at 12 h until 72 h or 24 h, respectively. These results suggest the capacity of AGEs to activate ECT inflammatory responses.

a–e The inflammatory markers ICAM-1, NF-κB, PAI-1, and TNF-α were assessed by Western blotting. n = 3 per group per time point, *P < 0.05, **P < 0.01, ***P < 0.001 versus the respective control groups. Each panel shows the normalized mean ± SD.
AGE-induced oxidative stress in ECTs
Since inflammatory responses may be derived from oxidative stress, we examined 4-HNE as an oxidized product marker (Fig. 4a, b) and 3-NT as a nitrated product marker (Fig. 4c, d). Both were found to significantly increase within 6 h after AGEs treatment and reached their peak levels at 12 or 12–24 h after AGEs treatment and then returned to control levels on day 6 after AGEs exposure.

a–h The oxidative stress markers 4-HNE, 3-NT, CAT, SOD2, and HO-1 were assessed by Western blotting. n = 3 per group per time point, *P < 0.05, **P < 0.01, ***P < 0.001 versus the respective control groups. Each panel shows the normalized mean ± SD.
Since oxidative stress is caused by an imbalance between ROS/RNS and antioxidant activity, we further examined antioxidant protein levels (CAT, SOD2, and HO-1) (Fig. 4e–h). As expected, CAT was increased at 12 h, reached a peak level at 24 h and gradually returned to the control level by day 9 after AGEs-treatment; SOD2 was increased at 12 h, showed similar levels until 72 h, and then gradually decreased but remained higher than the control level until day 9. HO-1 was increased at 24 h, reached a very high level at 72 h and remained higher than the control level until day 9.
AGE-induced pathological changes are oxidative stress-dependent and mediated by RAGE
As revealed in the above experiments, AGE-induced inflammatory, fibrotic and hypertrophic effects in ECTs are associated with oxidative stress, and we next sought to determine whether AGEs treatment promoted the generation of ROS in ECTs and whether blocking AGE-induced ROS would prevent oxidative stress and damage. Therefore, ECTs were collected 3 h after AGEs treatment and stained with DHE to identify ROS, which showed increased red fluorescence in AGEs-treated ECTs compared to that in control ECTs (Fig. 5a, b). We also confirmed the preventability of AGE-induced production of ROS by pretreatment with NAC of AGEs-treated ECTs (Fig. 5a, b).

a, b DHE staining of ROS (×10 magnification). AGEs treatment was performed for 3 h before DHE staining. DMSO, FPS-ZM1 (20 µM), or NAC (5 mM) was added to ECTs 2 h before control or AGEs treatment. n = 5 per group, ***P < 0.001 vs control. ##P < 0.01, ###P < 0.001 versus AGEs group. Each panel shows the normalized mean ± SD.
Finally, we tested the hypothesis that the prevention of AGE-induced ROS by NAC results in the prevention of AGE-induced pro-inflammatory effects (shown by the expression of NF-κB, ICAM-1, PAI-1 and TNF-α, Fig. 6a, b) and remodeling, as shown by significantly decreased expression of profibrotic (CTGF, TGF-β, α-SMA protein, and Ctgf mRNA, Fig. 6c–e), fibrotic (Col-1α1, Col-IIIα1, and Fn1 mRNA, Fig. 6e) and hypertrophic markers (Myh7 and Nppa mRNA, Fig. 6f) as well as the preservation of normal ECT function (Table 1).

a, b The inflammatory markers PAI-1, TNF-α, NF-κB, and ICAM-1 were assessed by Western blotting; n = 3 per group. c, d The fibrosis markers CTGF, α-SMA, and TGF-β were assessed by Western blotting; n = 3 per group. e The fibrosis markers collagen I-α1, collagen III-α1, Fn1, and Ctgf were assessed by qRT-PCR; n = 4 per group. f The cardiomyocyte hypertrophy markers Nppa and Myh7 were assessed by qRT-PCR; n = 4 per group. NAC, N-acetylcysteine; *P < 0.05, **P < 0.01, ***P < 0.001 vs Control. #P < 0.05, ##P < 0.01, ###P < 0.001 versus AGEs+DMSO group. Each panel shows the normalized mean ± SD.
As a demonstration that AGE-induced oxidative stress and other associated pathogenic effects are mediated by the interactions of AGEs with its receptor, we found that ECTs preincubated with the RAGE inhibitor FPS-ZM1 showed reduced ROS increases (Fig. 5a, b) and partly or completely normalized levels of markers of oxidative stress (CAT, SOD2 and HO-1, Fig. 7a, c) and inflammation (PAI-1 and TNF-α, NF-κB, and ICAM-1, Fig. 7a, b). Additionally, RAGE inhibitor treatment partly or completely prevented remodeling (Fig. 7d–g) and prevented ECT dysfunction after AGEs exposure (Table 1).

a, b The inflammatory markers ICAM-1, NF-κB, PAI-1, and TNF-α were assessed by Western blotting; n = 3 per group. a, c The oxidative stress markers CAT, SOD2, and HO-1 were assessed by Western blotting; n = 3 per group. d, e The fibrosis markers CTGF, α-SMA, and TGF-β were assessed by Western blotting; n = 3 per group. f The fibrosis markers Collagen I-α1, Collagen III-α1, Fn1, and Ctgf were assessed by qRT-PCR; n = 4 per group. g The cardiomyocyte hypertrophy markers Nppa and Myh7 were assessed by qRT-PCR; n = 4 per group. FPS-ZM1, inhibitor of RAGE; *P < 0.05, **P < 0.01, ***P < 0.001 vs control. #P < 0.05, ##P < 0.01, ###P < 0.001 versus AGEs+DMSO groups. Each panel shows the normalized mean ± SD.
Discussion
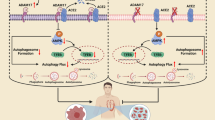
In our current study, we found that noncytotoxic doses of AGEs could induce neonatal murine ECT remodeling (hypertrophy and fibrosis) as well as dysfunction. AGE induced the production of ROS and activated the inflammation pathways via RAGE. RAGE inhibitors and antioxidants could partly attenuate the increase in ROS and the inflammatory response and rescue ECT function after long-term AGEs exposure, as shown in Fig. 8. Thus, AGE-treated neonatal murine ECTs recapitulate key cellular and molecular pathophysiological features of DCM [32].

AGEs (pathogenic factors of diabetic cardiomyopathy) interact with RAGE to induce ROS generation. Excessive oxidative stress can lead to ECT remodeling (cell hypertrophy and accumulation of fibrotic products) directly or indirectly via the inflammatory response. These alterations result in ECT functional abnormalities. ECT engineered cardiac tissue, FPS-ZM1 RAGE antagonist, NAC N-acetylcysteine.
Given the high prevalence of diabetes and the broad range of clinical complications, the continued investigation of novel, safe, and more potent therapeutics is urgently required to reduce the global disease burden of diabetes. This necessitates the continued use of animal models to discover, validate and optimize novel therapeutics prior to their testing in clinical trials [33]. Indeed, various preclinical models have advanced our understanding of the pathophysiology of DCM; however, limitations persist in both animal models and cardiomyocyte cultures [4, 34, 35]. ECTs have emerged as a robust in vitro platform to investigate tissue-level cardiac injury and recovery, including changes in electromechanical function, and are a promising and scalable preclinical model for studying DCM injury/remodeling paradigms. The nonenzymatic glycation process, which occurs inside the body (endogenous glycation, which is present in hyperglycemia) [28, 36, 37], leads to the generation and accumulation of a heterogeneous class of AGE molecules that have been shown to be significantly increased in diabetic patients and animals compared to controls [38,39,40]. Increasing evidence demonstrates that AGEs play a pivotal role in the development and progression of diabetic heart failure [27,28,29, 38, 41,42,43]. Therefore, it would be quite appealing to establish a platform to investigate the pathophysiology of DCM in ECTs by using AGEs as inducers [30].
In this study, we established that long-term administration of noncytotoxic doses of AGEs can lead to ECT dysfunction, which is mainly manifested as a decreased beating rate with regional or global dyssynchrony but not arrested beating, which was quite different from the results of our previously published toxicity study [6]. AGEs triggered a sustained increase in the markers of fibrosis and collagen accumulation as well as the markers of cardiac hypertrophy Nppa and Myh7, which is consistent with the current knowledge regarding pathological remodeling in DCM [27, 28, 32, 38].
The detailed pathobiology of the AGE-RAGE axis and the consequences of its activation play an important role in the development of DCM [38, 44]. Following the interaction with RAGE, a series of events leading to myocardial damage are elicited and sustained by AGEs, which include increases in oxidative stress [45], activation of inflammation [29, 46] and enhancement of extracellular matrix accumulation, finally resulting in cardiac dysfunction [28, 32]. Methods that involve breaking down AGEs or blocking RAGE have been proven to attenuate oxidative stress and inflammation as well as to alleviate cardiac dysfunction [38, 41, 47]. Consistent with previous studies, we demonstrate here that markers of oxidative stress and inflammation are increased at the early stage after AGEs treatment and are attenuated by RAGE inhibitor or NAC pretreatment; moreover, the ECT dysfunction caused by AGEs exposure can also be partly rescued (Fig. 8). Therefore, AGE-treated neonatal murine ECTs recapitulate key molecular pathophysiological features of DCM.
Thus, AGE-treated neonatal murine ECTs recapitulate key cellular and molecular pathophysiological features of DCM and can serve as a robust in vitro model to investigate cell-cell interaction-specific pathways relevant to cardiac injury and DCM along with pharmaceutical intervention strategies.
References
Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pr. 2019;157:107843.
Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–9.
Rubler SDJ, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:596–602.
Fuentes-Antras J, Picatoste B, Gomez-Hernandez A, Egido J, Tunon J, Lorenzo O. Updating experimental models of diabetic cardiomyopathy. J Diabetes Res. 2015;2015:656795.
Yu HT, Zhen J, Xu JX, Cai L, Leng JY, Ji HL, et al. Zinc protects against cadmium-induced toxicity in neonatal murine engineered cardiac tissues via metallothionein-dependent and independent mechanisms. Acta Pharmacol Sin. 2020;41:638–49.
Yu H, Ye F, Yuan F, Cai L, Ji H, Keller BB. Neonatal murine engineered cardiac tissue toxicology model: impact of metallothionein overexpression on cadmium-induced injury. Toxicol Sci. 2018;165:499–511.
Kawatou M, Masumoto H, Fukushima H, Morinaga G, Sakata R, Ashihara T, et al. Modelling Torsade de Pointes arrhythmias in vitro in 3D human iPS cell-engineered heart tissue. Nat Commun. 2017;8:1078.
Borchert T, Hubscher D, Guessoum CI, Lam TD, Ghadri JR, Schellinger IN, et al. Catecholamine-dependent beta-adrenergic signaling in a pluripotent stem cell model of takotsubo cardiomyopathy. J Am Coll Cardiol. 2017;70:975–91.
Feric NT, Pallotta I, Singh R, Bogdanowicz DR, Gustilo M, Chaudhary K, et al. Engineered cardiac tissues generated in the Biowire II: a platform for human-based drug discovery. Toxicol Sci. 2019;172:89–97.
Truitt R, Mu A, Corbin EA, Vite A, Brandimarto J, Ky B, et al. Increased afterload augments sunitinib-induced cardiotoxicity in an engineered cardiac microtissue model. JACC Basic Transl Sci. 2018;3:265–76.
Lee EK, Tran DD, Keung W, Chan P, Wong G, Chan CW, et al. Machine learning of human pluripotent stem cell-derived engineered cardiac tissue contractility for automated drug classification. Stem Cell Rep. 2017;9:1560–72.
Conant G, Ahadian S, Zhao Y, Radisic M. Kinase inhibitor screening using artificial neural networks and engineered cardiac biowires. Sci Rep. 2017;7:11807.
Chen J, Zhan Y, Wang Y, Han D, Tao B, Luo Z, et al. Chitosan/silk fibroin modified nanofibrous patches with mesenchymal stem cells prevent heart remodeling post-myocardial infarction in rats. Acta Biomater. 2018;80:154–68.
Valarmathi MT, FuselerJW, Davis JM, Price RL. A novel human tissue-engineered 3-D functional vascularized cardiac muscle construct. Front Cell Dev Biol. 2017;5:2.
Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, et al. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat Commun. 2017;8:1825.
Rashedi I, Talele N, Wang XH, Hinz B, Radisic M, Keating A. Collagen scaffold enhances the regenerative properties of mesenchymal stromal cells. PLoS ONE. 2017;12:e0187348.
Masumoto H, Nakane T, Tinney JP, Yuan F, Ye F, Kowalski WJ, et al. The myocardial regenerative potential of three-dimensional engineered cardiac tissues composed of multiple human iPS cell-derived cardiovascular cell lineages. Sci Rep. 2016;6:29933.
Zamani M, Karaca E, Huang NF. Multicellular interactions in 3D engineered myocardial tissue. Front Cardiovasc Med. 2018;5:147.
Jackman C, Li H, Bursac N. Long-term contractile activity and thyroid hormone supplementation produce engineered rat myocardium with adult-like structure and function. Acta Biomater. 2018;78:98–110.
Iseoka H, Miyagawa S, Fukushima S, Saito A, Masuda S, Yajima S, et al. Pivotal role of non-cardiomyocytes in electromechanical and therapeutic potential of induced pluripotent stem cell-derived engineered cardiac tissue. Tissue Eng Part A. 2018;24:287–300.
Rupert CE, Coulombe KLK. IGF1 and NRG1 enhance proliferation, metabolic maturity, and the force-frequency response in hesc-derived engineered cardiac tissues. Stem Cells Int. 2017;2017:7648409.
An M, Kwon K, Park J, Ryu DR, Shin JA, Lee Kang J, et al. Extracellular matrix-derived extracellular vesicles promote cardiomyocyte growth and electrical activity in engineered cardiac atria. Biomaterials 2017;146:49–59.
Mosqueira D, Mannhardt I, Bhagwan JR, Lis-Slimak K, Katili P, Scott E, et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J. 2018;39:3879–92.
Smith JGW, Owen T, Bhagwan JR, Mosqueira D, Scott E, Mannhardt I, et al. Isogenic pairs of hiPSC-CMs with hypertrophic cardiomyopathy/LVNC-associated ACTC1E99K mutation unveil differential functional deficits. Stem Cell Rep. 2018;11:1226–43.
Ceholski DK, Turnbull IC, Kong CW, Koplev S, Mayourian J, Gorski PA, et al. Functional and transcriptomic insights into pathogenesis of R9C phospholamban mutation using human induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol. 2018;119:147–54.
Wold LE, Ceylan-Isik AF, Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol Sin. 2005;26:908–17.
Cheng G, Wang LL, Qu WS, Long L, Cui H, Liu HY, et al. C16, a novel advanced glycation endproduct breaker, restores cardiovascular dysfunction in experimental diabetic rats. Acta Pharmacol Sin. 2005;26:1460–6.
Bodiga VL, Eda SR, Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy. Heart Fail Rev. 2014;19:49–63.
Hou J, Zheng D, Fung G, Deng H, Chen L, Liang J, et al. Mangiferin suppressed advanced glycation end products (AGEs) through NF-kappaB deactivation and displayed anti-inflammatory effects in streptozotocin and high fat diet-diabetic cardiomyopathy rats. Can J Physiol Pharmacol. 2016;94:332–40.
Wang Y, Luo W, Han J, Khan ZA, Fang Q, Jin Y, et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat Commun. 2020;11:2148.
Lu J, Wu DM, Zheng YL, Hu B, Cheng W, Zhang ZF, et al. Troxerutin counteracts domoic acid-induced memory deficits in mice by inhibiting CCAAT/enhancer binding protein beta-mediated inflammatory response and oxidative stress. J Immunol. 2013;190:3466–79.
Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020;17:585–607.
Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, et al. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol. 2018;14:140–62.
King AJ. The use of animal models in diabetes research. Br J Pharmacol. 2012;166:877–94.
King A, Bowe J. Animal models for diabetes: Understanding the pathogenesis and finding new treatments. Biochem Pharmacol. 2016;99:1–10.
Schalkwijk CG, Baidoshvili A, Stehouwer CD, van Hinsbergh VW. NiessenHW. Increased accumulation of the glycoxidation product Nepsilon-(carboxymethyl) lysine in hearts of diabetic patients: generation and characterisation of a monoclonal anti-CML antibody. Biochim Biophys Acta. 2004;1636:82–9.
Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest. 1997;99:457–68.
Ma H, Li SY, Xu P, Babcock SA, DolenceEK, Brownlee M, et al. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med. 2009;13:1751–64.
Grzebyk E, Knapik‑Kordecka M, Piwowar A. Advanced glycation end-products and cathepsin cysteine protease in type 2 diabetic patients. Pol Arch Intern Med 2013;123:364–70.
Tore J, Berg KD-J, Torjesen PeterA, Hanssen KristianF. Increased serum levels of advanced glycation end products (AGEs) in children and adolescents with IDDM. Diabetes Care. 1997;20:1006–8.
Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res. 2003;92:785–92.
Pei Z, Deng Q, Babcock SA, He EY, Ren J, Zhang Y. Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: role of autophagy and ER stress. Toxicol Lett. 2018;284:10–20.
Kranstuber AL, Del Rio C, Biesiadecki BJ, Hamlin RL, Ottobre J, Gyorke S, et al. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Front Physiol. 2012;3:292.
Senatus LM, Schmidt AM. The AGE-RAGE axis: implications for age-associated arterial diseases. Front Genet. 2017;8:187.
Tan AL, Forbes JM, Cooper ME. AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol. 2007;27:130–43.
Chavakis T, Bierhaus A, Nawroth PP. RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect. 2004;6:1219–25.
Jud P, Sourij H. Therapeutic options to reduce advanced glycation end products in patients with diabetes mellitus: a review. Diabetes Res Clin Pr. 2019;148:54–63.
Acknowledgements
This work was supported in part by the American Diabetes Association (1-18-IBS-082, to LC), the University of Louisville-China Pediatric Research Exchange Program (BBK and LC), and the National Key R&D Program of China (2016YFC0900903 to YZ). All personnel expenses and partial research-related expenses for XW and HTY were provided for by the First Hospital of Jilin University, Changchun, China. All basic science experiments were completed at the University of Louisville, Louisville, KY, USA.
Author information
Authors and Affiliations
Contributions
XW, XXC, and HTY designed and performed the experiments, analyzed the data, and prepared and reviewed the manuscript for submission. YT and QL prepared and reviewed the manuscript for submission. YZ, BBK, and LC designed the experiments, analyzed the data, and prepared and reviewed the manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, X., Chen, Xx., Yu, Ht. et al. Engineered cardiac tissues: a novel in vitro model to investigate the pathophysiology of mouse diabetic cardiomyopathy. Acta Pharmacol Sin 42, 932–941 (2021). https://doi.org/10.1038/s41401-020-00538-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00538-8
