
Abstract
Type 2 inositol 1,4,5-trisphosphate receptor (IP3R2) regulates the intracellular Ca2+ release from endoplasmic reticulum in human embryonic stem cells (hESCs), cardiovascular progenitor cells (CVPCs), and mammalian cardiomyocytes. However, the role of IP3R2 in human cardiac development is unknown and its function in mammalian cardiomyocytes is controversial. hESC-derived cardiomyocytes have unique merits in disease modeling, cell therapy, and drug screening. Therefore, understanding the role of IP3R2 in the generation and function of human cardiomyocytes would be valuable for the application of hESC-derived cardiomyocytes. In the current study, we investigated the role of IP3R2 in the differentiation of hESCs to cardiomyocytes and in the hESC-derived cardiomyocytes. By using IP3R2 knockout (IP3R2KO) hESCs, we showed that IP3R2KO did not affect the self-renewal of hESCs as well as the differentiation ability of hESCs into CVPCs and cardiomyocytes. Furthermore, we demonstrated the ventricular-like myocyte characteristics of hESC-derived cardiomyocytes. Under the α1-adrenergic stimulation by phenylephrine (10 μmol/L), the amplitude and maximum rate of depolarization of action potential (AP) were slightly affected in the IP3R2KO hESC-derived cardiomyocytes at differentiation day 90, whereas the other parameters of APs and the Ca2+ transients did not show significant changes compared with these in the wide-type ones. These results demonstrate that IP3R2 has minimal contribution to the differentiation and function of human cardiomyocytes derived from hESCs, thus provide the new knowledge to the function of IP3R2 in the generation of human cardiac lineage cells and in the early cardiomyocytes.
Similar content being viewed by others


Introduction
Ca2+ signals participate in various aspects of life processes [1, 2], including development [3, 4] and cardiac function [5]. Dysregulated Ca2+ signaling correlates with heart diseases [6, 7]. Thus, the elucidation of Ca2+ regulatory mechanisms will provide new knowledge in the understanding of heart development, functional maintenance, and disease control.
The cardiac differentiation system of human embryonic stem cells (hESCs) mimics the early developmental process of human hearts [8]. Differentiated cardiomyocytes are structurally and functionally similar to human fetal cardiomyocytes [9, 10]. These properties confer the model of cardiac differentiation of hESCs and derived cardiac lineage cells as a unique model/source for the study of human heart development, heart disease, drug development, and cell therapy [11,12,13,14,15]. However, the contributions of Ca2+ signals and Ca2+ handling proteins in the cardiac differentiation of hESCs and derived cardiac lineage cells have not yet been fully clarified.
The endoplasmic reticulum (ER) is a major intracellular Ca2+ storage site in eukaryotic cells. It plays an important role in balancing intracellular Ca2+ homeostasis through Ca2+ channels and pumps located in the ER membrane [16,17,18,19,20]. Inositol 1,4,5-trisphosphate receptors (IP3Rs), which include three subtypes (IP3R1, IP3R2, and IP3R3), and ryanodine receptors, are the two types of Ca2+ release channels located on the ER membrane. However, IP3Rs are the predominant Ca2+ release channels expressed in ESCs and early differentiating cells from ESCs, since ryanodine receptors are hardly detected at these stages [17, 20,21,22]. These properties of IP3Rs suggest that they might participate in early cell fate decisions. Accordingly, we found that IP3R3 deficiency inhibits the cardiac differentiation of mouse (m)ESCs by increasing apoptosis in mesoderm cells [23]. However, in vivo studies have revealed that mice with a single deletion of either Itpr1, Itpr2, or Itpr3 display normal cardiogenesis [24]. On the other hand, mice with double deletions of Itpr1 and Itpr2 die in utero with defects in the ventricles and atrioventricular canal at embryonic day 11.5 [24, 25]. In addition, triple knockout of all three types of Itpr genes enhances cardiomyocyte differentiation but suppresses hematopoietic differentiation of mESCs [26]. These findings suggest that the role of ip3rs in mouse cardiac development is complicated. However, most of our knowledge on the function of IP3Rs in cardiac development and cardiomyocytes comes from animal models. The precise function of Ca2+ signaling and IP3Rs in early cardiac development and fetal cardiomyocytes in humans remains largely unknown.
The function of IP3Rs in adult cardiomyocytes is also ambiguous. IP3R2 has been proposed to be the predominant isoform among the three subtypes of IP3Rs in working cardiomyocytes [27, 28]. It has also been shown that IP3R2 regulates the firing rate in rabbit ventricular cardiomyocytes [29] and mouse pacemaker cells [30]. However, deletion of IP3R2 in mice did not cause obvious changes in baseline cardiac function [24, 31]. The positive inotropic effect of IP3R2 in mouse cardiomyocytes by activation of Gq protein [32, 33] raises the possibility that IP3R2 is involved in stress-induced heart disease. This is supported by the upregulation of IP3R2 in the hypertrophic and failing heart [33,34,35]. However, deletion of IP3R2 in mice does not alter the progression of dilated cardiomyopathy or pressure overload-induced hypertrophy [31].
IP3R2 is expressed in human cardiomyocytes as detected by RNA-seq [36] and Western blot [33], but the roles of IP3Rs in human cardiac development and heart function are unclear. We recently found that IP3R2 knockout (IP3R2KO) significantly inhibits the increase in the intracellular concentration of Ca2+ stimulated by ATP or UTP in both hESCs and hESC-derived cardiovascular progenitor cells (hCVPCs) [37], suggesting that IP3R2 might contribute to the cardiac differentiation of hESCs and the function of hESC-derived cardiomyocytes. In addition, endothelin-1 (ET-1) is highly expressed in ISL1+ cardiac progenitors in human embryos, and the expansion of ISL1+ cardiac progenitors derived from hESCs is dependent on ET-1 [38]. Given the direct activation of IP3Rs by ET-1 through the Gq protein [39, 40], it is intriguing to determine the role of IP3R2 in the cardiac differentiation of hESCs and in hESC-derived cardiomyocytes.
In the present study, using the in vitro cardiomyocyte differentiation model of hESCs combined with IP3R2KO hESCs, we examined (i) the role of IP3R2 in the generation of CVPCs and cardiomyocytes from hESCs and (ii) the role of IP3R2 in the function of ventricular-like cardiomyocytes derived from hESCs. These results increase our knowledge of the contribution of Ca2+ handling proteins to early cardiac development in humans and to the functional maintenance of early-developing cardiomyocytes.
Materials and methods
hESC culture and in vitro differentiation
hESC culture was carried out as previously described [37, 41, 42]. Briefly, the hESC H7 cell line (WiCell Research Institute, Madison, WI, USA) was maintained in mTeSR1 medium (Stem Cell Technologies, Vancouver, Canada) on Matrigel (Corning, New York, NY, USA)-coated dishes.
For cardiomyocyte differentiation, hESCs were induced following a modified monolayer differentiation protocol as reported previously [43, 44]. Briefly, hESCs were seeded onto Matrigel-coated 12-well plates at a density of 2.5 × 104 cells/cm2 in mTeSR1 with 10 μmol/L Y-27632 (a ROCK inhibitor, Stem Cell Technologies), and then the medium was changed to one without Y-27632. After the hESCs reached 100% confluence, cardiac differentiation medium (CDM3) containing RPMI-1640 (Gibco, Carlsbad, CA, USA), 213 μg/mL L-ascorbic acid 2-phosphate (Sigma-Aldrich, Carlsbad, USA), and 2 mg/mL bovine serum albumin (Sigma-Aldrich) was used to induce cardiomyocyte differentiation. For the first 2 days of cardiac differentiation, CHIR99021 (a glycogen synthase kinase-3β inhibitor, Stem Cell Technologies) at 6 μmol/L was added to CDM3. Then, the medium was changed to CDM3 supplemented with the Wnt signaling inhibitor IWR-1 (Sigma-Aldrich) at 5 μmol/L on day 3 and day 4, followed by CDM3 alone until differentiation day 90 (Fig. S1).
For CVPC induction, hESCs were seeded onto Matrigel-coated 6-well plates at a density of 3.5 × 104 cells/cm2 in CVPC induction medium (CIM) for 3 days as reported previously [37, 41, 45]. The CIM contained DMEM/F12, 1×B27 supplement without vitamin A, 1% L-glutamine, 1% penicillin/streptomycin (Life Technologies, Carlsbad, CA, USA), and supplemented with 400 μmol/L L-thioglycerol (Sigma-Aldrich), 50 μg/mL ascorbic acid (Sigma-Aldrich), 25 ng/mL bone morphogenetic protein 4 (R&D Systems, Minneapolis, MN, USA), and 3 μmol/L CHIR99021 (Stem Cell Technologies).
Flow cytometry analysis
For the cell cycle analysis, cells were stained with 50 μg/mL propidium iodide (Sigma-Aldrich) before analysis. The cells were then analyzed by flow cytometry (Gallios, Beckman Coulter, Brea, CA, USA). The data were analyzed by ModFit software.
For the characterization of CTNT-positive cells, the cells were digested with 0.05% trypsin (Gibco). Then, the cells were fixed and permeabilized by a Foxp3 Staining Buffer kit (Invitrogen). Unconjugated CTNT antibody (1:200, Abcam, Cambridge, UK) was used, followed by staining with PE-Cy7-conjugated secondary antibody (1:400; eBioscience, San Diego, USA). The cells were then analyzed by flow cytometry (Gallios, Beckman Coulter) and quantified by FlowJo software.
Immunocytochemical staining
Immunocytochemical staining was performed as previously described [42]. Briefly, cells were fixed with 4% PFA, permeabilized with 0.4% Triton X-100 (Sigma-Aldrich), and blocked in 10% goat serum (Vector Laboratories, Burlingame, CA, USA). The primary antibodies used were as follows: OCT4 antibody (1:200, Abcam); SSEA4 antibody (1:200, Millipore, CA, USA); SOX2 antibody (1:200, Abcam); Nkx2.5 antibody (1:200, Santa Cruz Biotechnology, Dallas, TX, USA); ISL1 antibody (1:100; Developmental Studies Hybridoma Bank, Iowa City, IA, USA); CTNT antibody (1:200, Abcam); MLC2V antibody (1:100, Abcam) and ɑ-ACTININ antibody (1:400, Sigma-Aldrich). Alexa 488- or 569-conjugated secondary antibody (Invitrogen) was used for detection. Nuclei were stained with DAPI (Sigma-Aldrich). Images were captured by a Zeiss LSM 710 confocal microscope.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted with a RNeasy Mini kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions and then reverse-transcribed by using ReverTra Ace reverse transcriptase (Toyobo, Osaka, Japan). qRT-PCR was performed using the ViiA 7 Real-Time PCR System (Life Technologies) with SYBR Green qPCR Master Mix (Roche, Mannheim, Germany). The results are presented as fold changes normalized to GAPDH. The qRT-PCR primers are listed in Table S1.
Western blot analysis
The cells were harvested and lysed in lysis buffer containing 8 mol/L urea, 2 mol/L thiourea, 3% sodium lauryl sulfate, 75 mmol/L 1,4-dithiothreitol, 50 mmol/L TRIS, and 0.03% bromophenol blue (pH adjusted to 6.8) (Sigma-Aldrich). The antibodies against IP3R1 and IP3R2 were generated as previously reported [46]. The membranes were incubated with primary antibodies against IP3R1 (1:1000), IP3R2 (1:1000), IP3R3 (1:1000, BD Biosciences, San Jose, CA, USA), GAPDH (1:20000, Proteintech, Rosemont, IL, USA), and β-actin (1:8000; Sigma-Aldrich) in 3% BSA. IRDye 680LT donkey anti-rabbit IgG or IRDye 800LT donkey anti-mouse IgG (1:8000; Li-COR Biosciences, Lincoln, NE, USA) was used for detection. Images were captured using an Odyssey Infrared Imager (Li-COR Biosciences).
Karyotype analysis
Karyotype analysis was conducted as previously reported [47]. Briefly, hESCs were treated with colchicine (100 μg/mL) for 3 h at 37 °C and then harvested as single cells. The cells were hypotonic in 75 mmol/L KCl at 37 °C for 30 min and then fixed by freshly prepared fixative (glacial acetic acid:methanol = 1:3). After dropping the solution onto clean slides, the samples were stained with Giemsa staining solution. Twenty metaphase cells were counted, of which five cells were analyzed and karyotyped in each cell line.
Recording of Ca2+ transients
Cardiomyocytes derived from hESCs at differentiation day 90 were digested using 0.05% trypsin and plated in glass bottom cell culture dishes (Wuxi NEST Biotechnology, Wuxi, China). After 24 to 48 h of plating, the cells were loaded with 2 μmol/L Fluo4-AM (Life Technologies) dissolved in Tyrode’s buffer for 15 min at room temperature (RT). Tyrode’s buffer contained (in mmol/L) NaCl, 135; KCl, 5.4; CaCl2, 1.8; MgCl2, 1.0; glucose, 10; and HEPES, 10 (pH adjusted to 7.4). Then, the Ca2+ indicator was washed off 3 times, and the cells were incubated at RT for 15 min before use. The Ca2+ transients were captured in the line scan model using a Zeiss LSM 710 confocal microscope. A total of 5000 lines with an interval time of 10 ms were scanned. During the recording, the cells were maintained at 35 °C in a heated chamber. Phenylephrine (PE) (an ɑ1-adrenergic receptor agonist) was used at 10 μmol/L. The Ca2+ transients were analyzed with IDL software (ITT Corporation, White Plains, NY, USA).
Recording of action potentials (APs)
APs were recorded as previously described [48]. Briefly, cardiomyocytes derived from hESCs at differentiation day 90 were digested and replated onto coverslips. The cells were then recorded for APs within 24–48 h after plating. The temperature was maintained at 33 °C by perfusion with warm Tyrode’s buffer by a peristaltic pump (Cole-Parmer, IL, USA). The internal solution contained (in mmol/L): K+-aspartate,110; KCl, 20; MgCl2, 1; NaGTP, 0.1; MgATP, 5; Na2-phospocreatine, 5; EGTA, 1; and HEPES, 10; pH adjusted to 7.3 with KOH. APs were recorded using the EPC-10 amplifier (Heka Electronics, Bellmore, NY, USA) in current-clamp mode. PE was used at 10 μmol/L. The characterization of the cardiomyocyte subtypes was based on previously reported criteria [44]. Briefly, for ventricular-like cardiomyocytes, AP amplitude > 90 mV, AP duration at 90% repolarization/AP duration at 50% repolarization (APD)90/APD50 < 1.4, a rapid AP upstroke, a long plateau phase, and a negative maximum diastolic potential (MDP) (<−48 mV); for atrial-like cardiomyocytes, an absence of a prominent plateau phase, a negative diastolic membrane potential (<−48 mV) and APD90/APD50 > 1.7; and for Nodal-like cardiomyocytes, a more positive MDP, a slower AP upstroke, a prominent phase 4 depolarization and APD90/APD50 between 1.4 and 1.7.
Statistical analysis
The data are presented as the mean ± SEM. One-way analysis of variance (ANOVA) followed by Bonferroni’s multiple analysis was used for the qRT-PCR assay of ITPRs, cell cycle distribution and flow cytometry analysis of CTNT. Two-way ANOVA with Bonferroni’s multiple comparison test was used to analyze the qRT-PCR data for the expression of different genes during cardiomyocyte differentiation and the quantitative properties of APs and Ca2+ transients of the three cell lines. A paired t test was used to analyze the quantitative properties of APs and Ca2+ transients with or without PE. All of the statistical analyses were conducted using GraphPad Prism8. P < 0.05 was considered statistically significant.
Results
Differentiation of hESCs to dominant ventricular-like cardiomyocytes
To determine the role of IP3R2 in the fate of human ventricular cardiomyocytes and the function of hESC-derived cardiomyocytes, we differentiated hESCs into cardiomyocytes following a protocol reported previously [43, 44] with a small modification (Fig. S1a). Spontaneously beating cardiomyocytes appeared on differentiation day 7, and robust beating cardiomyocytes were observed on differentiation day 8 (Movie S1). Flow cytometry analysis confirmed that over 90% of cells were positive for the cardiac-specific marker CTNT at differentiation day 90 (Fig. S1b). The differentiated cardiomyocytes showed a well-arranged sarcomere structure (Fig. S1c). To determine the subtypes of differentiated cardiomyocytes, we recorded the APs of the cells at differentiation day 90. The examined cardiomyocytes derived from wild-type (wt) hESCs showed typical ventricular-like APs (Fig. S1d) with an APD90/APD50 ratio of 1.17 ± 0.01 (Fig. S1e) based on previously reported criteria [44]. These data demonstrate that cardiomyocytes differentiated from hESCs around day 90 are mainly ventricular-like cardiomyocytes.
IP3R2 is highly expressed in undifferentiated hESCs and downregulated in differentiated cardiomyocytes
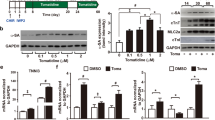
Next, we examined the expression profile of IP3R2 during cardiomyocyte differentiation. qRT-PCR analysis showed that the ITPR2 gene was highly expressed in undifferentiated hESCs and was quickly downregulated during the first 2 days of differentiation. Then, it was upregulated at differentiation days 3 and 4 and gradually downregulated in the following days, reaching a relatively stable level from differentiation day 6 to day 90 (Fig. 1a). A similar pattern of IP3R2 protein was observed during cardiac differentiation of hESCs by Western blot analysis (Fig. 1b).

a qRT-PCR analysis of the expression of the ITPR2 gene. n = 3. b Western blot analysis of IP3R2 protein levels. GAPDH was used as the loading control. n = 3.
Deficiency in IP3R2 does not significantly affect the self-renewal of hESCs
To determine the role of IP3R2 in cardiac differentiation, we used two IP3R2KO hESC lines (IP3R2KO-6 and IP3R2KO-12) generated by using transcription activator-like effector nuclease (TALEN) technology as previously reported [37]. All of the wt, IP3R2KO-6 and IP3R2KO-12 hESCs showed the normal karyotype (Fig. S2). The deficiency in IP3R2 was confirmed in IP3R2KO-6 and IP3R2KO-12 lines by Western blot, while the protein levels of other subtypes of IP3Rs, i.e., IP3R1 and IP3R3 (encoded by the ITPR1 and ITPR3 gene, respectively), were not affected (Fig. 2a), which was consistent with the qRT-PCR analysis (Fig. 2b).

a Western blot analysis of IP3R1, IP3R2 and IP3R3 proteins. β-actin was used as the loading control. b qRT-PCR analysis of ITPR1 and ITPR3 gene expression. n = 3. c The alkaline phosphatase staining and immunocytochemical staining of OCT4, SSEA4 and SOX2. Scale bar = 100 μm (ALP). Scale bar = 10 μm (immunocytochemical staining). d qRT-PCR analysis of POU5F1, NANOG and SOX2 gene expression. e The representative flow cytometry plots of cell cycle analysis. f The cell cycle analysis data. n = 3.
We previously found that IP3R2 is a dominantly functional Ca2+ channel for mediating Ca2+ release from the ER in both mESCs [49] and hESCs [22, 37]. To test whether IP3R2 deficiency affects the self-renewal of hESCs, we conducted alkaline phosphatase (ALP) staining, immunocytochemical staining and qRT-PCR. All wt, IP3R2KO-6 and IP3R2KO-12 hESCs were positive for ALP, OCT4, SSEA4 and SOX2 (Fig. 2c). The mRNA levels of POU5F1 (encodes OCT4), NANOG and SOX2 were also comparable between these three hESC lines (Fig. 2d). Furthermore, flow cytometry analysis did not detect a difference in the cell cycle distribution among the wt and two IP3R2KO cell lines (Fig. 2e, f). Therefore, IP3R2 seems to be dispensable for the maintenance of hESC self-renewal.
IP3R2 is dispensable for the differentiation of hESCs into CVPCs and cardiomyocytes
The upregulated expression of IP3R2 from differentiation day 3 to day 4 suggests a possible contribution of IP3R2 in mediating cardiac progenitor formation, as this is a critical stage for the transition of cardiac mesoderm to cardiac progenitors [44]. To determine the effect of IP3R2 on the generation of CVPCs from hESCs, we induced wt and IP3R2KO cells to differentiate into CVPCs by using a protocol established by our group [41, 45]. Immunocytochemical staining showed that all of the differentiating cells from the wt and IP3R2KO cells uniformly expressed the primitive CVPC markers NKX2-5 and ISL1 in the nuclei (Fig. 3a). Next, to determine whether IP3R2 affects cardiomyocyte generation, wt and IP3R2KO hESCs were further induced to differentiate into cardiomyocytes. We found that the percentages of CTNT-positive cells were comparable between the wt and the two IP3R2KO cell lines at differentiation day 90 (Fig. 3b). The clear and organized sarcomeric structure of induced cardiomyocytes was similar among the wt and IP3R2KO cells (Fig. 3c). In addition, the cells were double positive for MLC2V and α-ACTININ (Fig. 3d). Notably, the fiber-like structures observed in the green and red channels were well colocalized (Fig. 3d), indicating that these cells were ventricular-like myocytes. These data are consistent with the observations of the typical ventricular-like myocyte APs shown in Fig. S1d and S1e. qRT-PCR analysis further confirmed that the genes were sequentially expressed following a tandem transition from pluripotency (POU5F1), mesoderm (TBXT), cardiac mesoderm (MESP1), cardiac progenitor (ISL1 and NKX2-5) to cardiomyocytes (TNNT2) in the wt and two IP3R2KO cell lines (Fig. 3e). These data suggest that IP3R2 deficiency does not significantly affect the induction of CVPCs and cardiomyocytes from hESCs.

a Immunocytochemical staining analysis of CVPC marker NKX2-5 and ISL1 in induced CVPCs. Scale bar = 40 μm. b Flow cytometry analysis of cardiomyocytes induced from wt, IP3R2KO-6 and IP3R2KO-12 hESCs with cardiomyocyte marker CTNT. n = 3. c Immunocytochemical staining analysis of cardiomyocyte marker CTNT and ɑ-ACTININ in cardiomyocytes at differentiation day 90. Scale bar = 10 μm. d Co-immunocytochemical staining analysis of cardiomyocyte marker ɑ-ACTININ and ventricular myocyte marker MLC2V at cardiac differentiation day 90. Scale bar = 20 μm. e The mRNA expression of genes during cardiac differentiation from hESCs. POU5F1, pluripotency marker; TBXT, early mesoderm marker; MESP1, cardiac mesoderm marker; ISL1 and NKX2-5, cardiac progenitor markers; TNNT2, cardiomyocyte marker. n = 3.
IP3R2KO mildly alters APs by stimulating ɑ1-adrenergic receptors in hESC-derived cardiomyocytes
As the functional contribution of IP3R2 remains controversial in adult working cardiomyocytes [24, 33] and is unknown in early differentiated human cardiomyocytes, we examined whether IP3R2 affects APs, a crucial feature of beating cardiomyocytes [50]. The spontaneously beating cardiomyocytes at differentiation day 90 showed typical ventricular-like APs (Fig. 4a), with small variations in the MDP and amplitude but large variations in the beats per minute (Bpm), maximum rate of rise (Vmax) and depolarization (Dmax), APD50 and APD90 in the examined cardiomyocytes (Fig. 4b–h). The features of APs in cardiomyocytes derived from IP3R2KO hESCs were similar to those in the wt hESCs (Fig. 4a). To confirm this, we quantitatively analyzed the parameters of APs in these cells (Fig. 4b–h). The Bpm, amplitude, MDP, Vmax, Dmax, APD50, and APD90 in cardiomyocytes derived from IP3R2KO hESCs were comparable to those in the wt cardiomyocytes. Next, we examined whether IP3R2 deficiency affects APs under the stimulation of PE, an ɑ1-adrenergic receptor agonist that activates IP3Rs through the Gq protein [51, 52]. Spontaneous APs from the same cardiomyocyte with and without PE treatment were recorded. PE stimulation significantly accelerated the beating frequency (Fig. 4a, b), accompanied by increased MDP (Fig. 4d) and decreased amplitude (Fig. 4c), Vmax (Fig. 4e), APD50 (Fig. 4g), and APD90 (Fig. 4h), while Dmax was unchanged (Fig. 4f) compared with the corresponding values in wt hESC-derived cardiomyocytes without PE treatment. In the IP3R2KO hESC-derived cardiomyocytes, the Bpm, MDP, Vmax, APD50, and APD90 were comparable with those in the wt cardiomyocytes (Fig. 4b, d, e, g, h). However, it is notable that the amplitude in IP3R2KO-12 cardiomyocytes was decreased by 4.5% compared with that of wt cardiomyocytes, and a similar tendency was observed in IP3R2KO-6 cardiomyocytes without statistical significance (Fig. 4b). In addition, the Dmax in IP3R2KO-6 cardiomyocytes was approximately 17% slower than that in wt cardiomyocytes, and the same tendency was observed in IP3R2KO-12 cardiomyocytes (Fig. 4f). Overall, IP3R2KO does not alter spontaneous APs under baseline conditions, but it appears to contribute to the amplitude and Dmax of APs in hESC-derived cardiomyocytes under the stimulation of α1-adrenergic receptors.

a Representative AP recordings. b–h Parameters of APs. Bpm, beats per minute (b); Amplitude (c); MDP, maximum diastolic potential (d); Vmax, the maximum rate of rise of the AP (e); Dmax, the maximum rate of depolarization (f); APD50, AP duration at 50% repolarization (g); APD90, AP duration at 90% repolarization (h). PE, phenylephrine at 10 μmol/L concentration. n = 19 for wt cardiomyocytes; n = 24 for IP3R2KO-6 cardiomyocytes; n = 18 for IP3R2KO-12 cardiomyocytes. *P < 0.05, **P < 0.01, ***P < 0.001. n.s., no significant difference.
IP3R2KO does not significantly affect Ca2+ transients in hESC-derived cardiomyocytes
We next examined the contributions of IP3R2 to Ca2+ transients in hESC-derived cardiomyocytes. The rhythmic Ca2+ transients were observed in spontaneously beating cardiomyocytes (Fig. 5a). The parameters of Ca2+ transients among wt cardiomyocytes, such as the Bpm (Fig. 5b), amplitude (Fig. 5c), time to peak (Fig. 5d), time of decay to 90% (Fig. 5e) and 63% peak (T-90% decay and T-63% decay) (Fig. 5f), varied over a wide range as observed in the APs. The various parameters of Ca2+ transients in the cardiomyocytes derived from IP3R2KO hESCs were similar to those in wt cells (Fig. 5a–f). With PE treatment, the Bpm was significantly increased in wt cardiomyocytes, accompanied by decreases in time to peak, T-90% decay, and T-63% decay, while the amplitude was unchanged (Fig. 5a–f). In the cardiomyocytes derived from the two IP3R2KO hESC lines, the Bpm, amplitude, time to peak, T-90% decay and T-63% decay were comparable with those in the wt cell lines (Fig. 5b–f). Collectively, these data indicate that the stimulation of ɑ1-adrenergic receptors significantly increases the speed rising to the peak and recovery of Ca2+ transients in hESC-derived cardiomyocytes, although a large variation exists among cardiomyocytes. Moreover, IP3R2 deficiency does not significantly affect Ca2+ transients in hESC-derived cardiomyocytes with or without α1-adrenergic stimulation.

a Representative images of spontaneous intracellular Ca2+. b–e Parameters of spontaneous Ca2+ transients. The Bpm (b), amplitude (ΔF/F0, c), time to peak (d), time to 90% decay (e), and time to 63% decay (f) of the maximum amplitude. n = 30 for wt cardiomyocytes; n = 27 for IP3R2KO-6 cardiomyocytes; n = 38 for IP3R2KO-12 cardiomyocytes. ***P < 0.001 compared with the corresponding control group cells.
Discussion
In this study, using the in vitro hESC cardiac differentiation model, combined with IP3R2 knockout, Ca2+ imaging, and AP recording, we determined (i) the expression pattern of IP3R2 during cardiomyocyte differentiation of hESCs; (ii) IP3R2 deficiency does not affect the self-renewal of hESCs; (iii) IP3R2 deficiency does not significantly affect CVPC and cardiomyocyte differentiation from hESCs; (iv) hESC-derived cardiomyocytes have ventricular-like APs but with large variations in the parameters of APs and Ca2+ transients under both baseline conditions and the stimulation of α1-adrenergic receptors; (v) IP3R2 deficiency decreases the amplitude and the Dmax of APs in hESC-derived cardiomyocytes under the stimulation of α1-adrenergic receptors but not under baseline conditions; and (vi) no obvious changes are observed in the Ca2+ transients in IP3R2KO cardiomyocytes with or without the stimulation of α1-adrenergic receptors. Our results suggest the minimal contribution of IP3R2 in cardiac differentiation and derived ventricular cardiomyocytes from hESCs.
One of the findings here is the determination of the expression patterns of IP3R2 during the process of cardiomyocyte generation from hESCs. This observation is consistent with previous findings of the presence of IP3R2 in undifferentiated hESCs, hESC-derived CVPCs [22, 37] and human adult cardiomyocytes [33]. Our findings support previous observations and precisely describe the specific expression patterns of IP3R2 during the differentiation process of hESCs into cardiomyocytes, especially for ventricular-like myocytes, by revealing the following characteristics. First, the expression of IP3R2 is downregulated during the formation and maturation of cardiomyocytes from hESCs. This pattern is similar to those previously observed in the generation of CVPCs [37] and cardiomyocytes [53] from hESCs. However, notably, at the early differentiation stage (differentiation day 0 to day 1, representing the transition stage of undifferentiated hESCs to mesoderm cells), the expression of IP3R2 was sharply downregulated, followed by upregulation at differentiation days 3 and 4 (representing the transition stage from mesoderm to cardiac mesoderm)(Fig. 1). This correlation between the expression of IP3R2 and cell fate transition suggests the possible role of IP3R2 in human lineage fate determination. Although it is unknown whether this transient down- and upregulation is involved in the tissue-specific linage fate determination in humans, our previous study showed that the abundance of IP3R3 is transiently downregulated in the early differentiation stage of mESCs and that knockdown of IP3R3 in mESCs significantly suppresses the differentiation into mesoderm and cardiomyocytes by specifically increasing the apoptosis of mesodermal cells due to the alternation of Ca2+ oscillation [23]. Considering the evidence for the existence of IP3Rs in both hESCs [22, 37] and mESCs [23, 26, 54] and the functional overlap or negative feedback among these subtypes [1, 23, 25, 26, 55,56,57], the significance of the transient downregulation of IP3R2 to the specific cell fate decision during early differentiation of hESCs needs to be elucidated in the future. Moreover, in this study, we demonstrate that the IP3R2 protein exists in hESC-derived ventricular-like myocytes. We confirmed that the efficiently generated cardiomyocytes from hESCs have ventricular myocyte-like APs (Fig. S1d) and are positive for the ventricular marker MLC2V (Fig. 3d). Therefore, IP3R2 exists in both differentiated human ventricular-like myocytes (Fig. 1) and human adult left ventricular myocytes [33].
Interestingly, although IP3R2 is highly expressed in undifferentiated hESCs, it has little contribution to the maintenance of hESC self-renewal. However, the percentage of cells responding to MRS2365, a Gq protein-coupled P2Y1 receptor agonist, is significantly decreased in IP3R2KO hESCs [37], indicating the important role of IP3R2 in mediating Ca2+ release from intracellular stores in hESCs. A possible interpretation for these observations might be that IP3R2 deficiency could be compensated by other subtypes of IP3Rs or Ca2+ modulators. Although the robust Ca2+ waves are mediated by IP3R2, the intracellular Ca2+ concentration is stable with minor fluctuations in undifferentiated hESCs [22]. Thus, the small fluctuations might be enough for the maintenance of hESCs in the steady state. Moreover, in IP3R2KO hESCs, there are still some cells responding to MRS2365, despite a low percentage (<20%) [37], suggesting that IP3R3 might compensate for the loss of IP3R2. In addition, the intracellular Ca2+ channels and Ca2+ modulators located in the plasma membrane might also participate in the Ca2+ homeostats maintained after IP3R2 deficiency. Therefore, the self-renewal property of IP3R2KO hESCs is maintained. Accordingly, the functional redundancy of IP3R subtypes is also detected in mice. The Itpr1, Itpr2, or Itpr3 single mutants were indistinguishable from the control mice. No contribution of IP3R2 to CVPC and cardiomyocyte generation could also be caused by the redundancy of other IP3R subtypes and Ca2+ modulators. Indeed, cardiac progenitor and cardiomyocyte differentiation is enhanced, but the hematopoietic mesoderm is reduced in mESCs with triple knockout of Itpr1, Itpr2, and Itpr3 [26]. Although it has been reported that ET-1, which can activate IP3Rs through endothelin receptors [39, 40], is crucial in promoting the expansion of ISL1+ cardiac progenitors from hESCs [38], the underlying mechanisms are not elucidated. Moreover, a recent report [58] argues that the heart defect is caused by double knockout of Itpr1 and Itpr2 [24]. Yang and colleagues [58] found that specific deletion of Itpr1 and Itpr2 in cardiomyocytes, endothelial/hematopoietic cells or early cardiovascular lineage progenitor cells of mice do not have phenotypes, while the embryonic lethality caused by double knockout of Itpr1 and Itpr2 may be due to allantoic-placental defects. Thus, the vascular abnormalities seen in Itpr1 and Itpr2 double knockout mice might not be due to the indirect influence but are a secondary effect on allantoic/placental defects. Overall, IP3R2 deficiency alone does not affect cardiomyocyte differentiation in mice or humans.
Another interesting finding here is that the positive chronotropic effect of PE is not affected by IP3R2 deficiency, and only mild changes in the amplitude and Dmax of the APs in IP3R2KO hESC-derived cardiomyocytes under PE stimulation are observed compared with those in wt cardiomyocytes (Fig. 4c, f). The positive chronotropic effect of PE in immature cardiomyocytes is mediated by α1-adrenergic receptors [59]; however, the underlying mechanism remains unknown. The positive chronotropic effect induced by β-adrenergic receptor activation is related to the regulation of Ca2+ influx through the L-type Ca2+ channel [60,61,62,63]. This channel is also regulated by the activation of α1-adrenergic receptors [64]. Thus, the chronotropic effect of PE in immature cardiomyocytes might also be mediated through L-type Ca2+ channels, although we could not exclude the participation of other channels, such as sodium channels or potassium channels. In addition, given that IP3R2 can be located near the plasma membrane [28, 30, 53, 65], IP3R2 might affect the voltage-gated ion channels on the plasma membrane by regulating the surrounding electrical environment, as hypothesized [66, 67], and this possibility needs to be tested.
Despite the possible role of IP3R2 in the regulation of ion channels, the contribution of IP3R2 to the functional properties of hESC-derived cardiomyocytes is weak. Under basal conditions, the APs and Ca2+ transients of the cardiomyocytes derived from wt and IP3R2KO hESCs are similar. It has been shown that IP3Rs contribute to the regulation of Ca2+ transients and/or contraction in mESC- [49] or hESC-derived cardiomyocytes [53, 65], isolated rabbit ventricular myocytes [28, 29], and adult human ventricular cardiomyocytes [33]. However, these conclusions are mainly based on the usage of agonists and antagonists of IP3Rs, lacking confirmation by using direct gene interference. Therefore, the nonspecific effects of these reagents could not be excluded. For example, the commonly used pan-IP3R antagonist 2-APB can also inhibit Ca2+ release-activated Ca2+ channels [68]. Thus, the results collected from studies without direct gene interference should be carefully interpreted. In the present study, the significant chronotropic response to the activation of α1-adrenergic receptors with PE in wt cardiomyocytes is consistent with a previous report [59, 69], while the amplitude of Ca2+ transients remained unchanged under the stimulation of α1-adrenergic receptors. However, activation of other Gq-coupled receptors in cardiomyocytes, such as ET-1 receptors and purinergic receptors, correlates with enhanced Ca2+ transients and increased contractility [33, 70]. Such differences may be due to the diversity of downstream cascades of different receptors. These possibilities need to be tested further. Notably, the PE-induced chronotropic response remained unchanged in IP3R2KO cardiomyocytes. This may have several interpretations. (i) IP3R2 minimally contributes to the function of hESC-derived cardiomyocytes. This is supported by the findings that heart function is normal in IP3R2-deficient mice [24, 31]. (ii) The functional redundancy of other IP3R subtypes may compensate for the loss of function of IP3R2. (iii) The large variations in the different parameters of APs and Ca2+ transients indicate the heterogeneous maturation of hESC-derived cardiomyocytes, which may conceal the mild involvement of IP3R2 in the function of early cardiomyocytes. Accordingly, the amplitude and recovery of APs under the activation of α1-adrenergic receptors were altered in IP3R2KO cardiomyocytes. (iv) Lastly, IP3R2 might be involved in the regulation of cardiomyocyte function under long-term stimulation. In the current study, APs were recorded immediately after PE addition, reflecting the acute response of cardiomyocytes. In a mouse model, the overexpression of α1A-adrenergic receptor [71] or IP3R2 [35] results in cardiac hypertrophy, suggesting that the chronic activation of IP3R2 might act as a pathological stimulus to the heart. In this circumstance, the relationship of the mild changes of the APs with the pathological outcomes under the chronic activation of IP3R2 needs to be studied in the future.
In conclusion, we have determined the precise expression pattern of IP3R2 during cardiomyocyte differentiation of hESCs and demonstrated that IP3R2 is dispensable in the self-renewal and cardiac differentiation of hESCs. IP3R2 has minimal contribution to the function of hESC-derived ventricular-like cardiomyocytes. These findings provide new insights into the role of IP3Rs in human early cardiac development and in human immature ventricular myocytes. This knowledge would also provide references for the use of hESC-derived cardiomyocytes as disease models and drug development.
References
Clapham DE. Calcium signaling. Cell. 2007;131:1047–58.
Carafoli E, Krebs J. Why Calcium? How calcium became the best communicator. J Biol Chem. 2016;291:20849–57.
Slusarski DC, Pelegri F. Calcium signaling in vertebrate embryonic patterning and morphogenesis. Dev Biol. 2007;307:1–13.
Webb SE, Miller AL. Calcium signalling during embryonic development. Nat Rev Mol Cell Biol. 2003;4:539–51.
Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol. 2011;3:a004242.
Marks AR. Calcium and the heart: a question of life and death. J Clin Invest. 2003;111:597–600.
Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–13.
Spater D, Hansson EM, Zangi L, Chien KR. How to make a cardiomyocyte. Development. 2014;141:4418–31.
He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: action potential characterization. Circ Res. 2003;93:32–9.
Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–14.
Ma Z, Wang J, Loskill P, Huebsch N, Koo S, Svedlund FL, et al. Self-organizing human cardiac microchambers mediated by geometric confinement. Nat Commun. 2015;6:7413.
Lee J, Shao NY, Paik DT, Wu H, Guo H, Termglinchan V, et al. SETD7 drives cardiac lineage commitment through stage-specific transcriptional activation. Cell Stem Cell. 2018;22:428–44.e5.
Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–91.
Wang J, Liu M, Wu Q, Li Q, Gao L, Jiang Y, et al. Human embryonic stem cell-derived cardiovascular progenitors repair infarcted hearts through modulation of macrophages via activation of signal transducer and activator of transcription 6. Antioxid Redox Signal. 2019;31:369–86.
Zhu K, Wu Q, Ni C, Zhang P, Zhong Z, Wu Y, et al. Lack of remuscularization following transplantation of human embryonic stem cell-derived cardiovascular progenitor cells in infarcted nonhuman primates. Circ Res. 2018;122:958–69.
Fu JD, Li J, Tweedie D, Yu HM, Chen L, Wang R, et al. Crucial role of the sarcoplasmic reticulum in the developmental regulation of Ca2+ transients and contraction in cardiomyocytes derived from embryonic stem cells. FASEB J. 2006;20:181–3.
Fu JD, Yang HT. Developmental regulation of intracellular calcium homeostasis in early cardiac myocytes. Sheng Li Xue Bao. 2006;58:95–103.
Guo A, Yang HT. Ca2+ removal mechanisms in mouse embryonic stem cell-derived cardiomyocytes. Am J Physiol Cell Physiol. 2009;297:C732–41.
Lam AK, Galione A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim Biophys Acta. 2013;1833:2542–59.
Yang HT, Tweedie D, Wang S, Guia A, Vinogradova T, Bogdanov K, et al. The ryanodine receptor modulates the spontaneous beating rate of cardiomyocytes during development. Proc Natl Acad Sci USA. 2002;99:9225–30.
Yanagida E, Shoji S, Hirayama Y, Yoshikawa F, Otsu K, Uematsu H, et al. Functional expression of Ca2+ signaling pathways in mouse embryonic stem cells. Cell Calcium. 2004;36:135–46.
Huang JJ, Wang YJ, Zhang M, Zhang P, Liang H, Bai HJ, et al. Functional expression of the Ca(2+) signaling machinery in human embryonic stem cells. Acta Pharmacol Sin. 2017;38:1663–72.
Liang J, Wang YJ, Tang Y, Cao N, Wang J, Yang HT. Type 3 inositol 1,4,5-trisphosphate receptor negatively regulates apoptosis during mouse embryonic stem cell differentiation. Cell Death Differ. 2010;17:1141–54.
Uchida K, Aramaki M, Nakazawa M, Yamagishi C, Makino S, Fukuda K, et al. Gene knock-outs of inositol 1,4,5-trisphosphate receptors types 1 and 2 result in perturbation of cardiogenesis. PLoS One. 2010;5:e12500.
Mikoshiba K. Role of IP receptor in development. Cell Calcium. 2011;49:331–40.
Wang YJ, Huang J, Liu W, Kou X, Tang H, Wang H, et al. IP3R-mediated Ca2+ signals govern hematopoietic and cardiac divergence of Flk1+ cells via the calcineurin-NFATc3-Etv2 pathway. J Mol Cell Biol. 2017;9:274–88.
Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. 1997;272:23961–9.
Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, et al. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol. 2000;10:939–42.
Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H596–604.
Ju YK, Liu J, Lee BH, Lai D, Woodcock EA, Lei M, et al. Distribution and functional role of inositol 1,4,5-trisphosphate receptors in mouse sinoatrial node. Circ Res. 2011;109:848–57.
Cooley N, Ouyang K, McMullen JR, Kiriazis H, Sheikh F, Wu W, et al. No contribution of IP3R2 to disease phenotype in models of dilated cardiomyopathy or pressure overload hypertrophy. Circ Heart Fail. 2013;6:318–25.
Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005;96:1274–81.
Signore S, Sorrentino A, Ferreira-Martins J, Kannappan R, Shafaie M, Del Ben F, et al. Inositol 1, 4, 5-trisphosphate receptors and human left ventricular myocytes. Circulation. 2013;128:1286–97.
Higazi DR, Fearnley CJ, Drawnel FM, Talasila A, Corps EM, Ritter O, et al. Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol Cell. 2009;33:472–82.
Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, et al. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res. 2010;107:659–66.
Tompkins JD, Jung M, Chen CY, Lin Z, Ye J, Godatha S, et al. Mapping human pluripotent-to-cardiomyocyte differentiation: methylomes, transcriptomes, and exon DNA methylation “memories”. EBioMedicine. 2016;4:74–85.
Huang J, Zhang M, Zhang P, Liang H, Ouyang K, Yang HT. Coupling switch of P2Y-IP3 receptors mediates differential Ca2+ signaling in human embryonic stem cells and derived cardiovascular progenitor cells. Purinergic Signal. 2016;12:465–78.
Soh BS, Ng SY, Wu H, Buac K, Park JH, Lian X, et al. Endothelin-1 supports clonal derivation and expansion of cardiovascular progenitors derived from human embryonic stem cells. Nat Commun. 2016;7:10774.
Mazzuca MQ, Khalil RA. Vascular endothelin receptor type B: structure, function and dysregulation in vascular disease. Biochem Pharmacol. 2012;84:147–62.
Deliu E, Brailoiu GC, Mallilankaraman K, Wang H, Madesh M, Undieh AS, et al. Intracellular endothelin type B receptor-driven Ca2+ signal elicits nitric oxide production in endothelial cells. J Biol Chem. 2012;287:41023–31.
Cao N, Liang H, Yang HT. Generation, expansion, and differentiation of cardiovascular progenitor cells from human pluripotent stem cells. Methods Mol Biol. 2015;1212:113–25.
Bai HJ, Zhang P, Ma L, Liang H, Wei G, Yang HT. SMYD2 drives mesendodermal differentiation of human embryonic stem cells through mediating the transcriptional activation of key mesendodermal genes. Stem Cells. 2019;37:1401–15.
Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. 2012;109:E1848–57.
Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–60.
Cao N, Liang H, Huang J, Wang J, Chen Y, Chen Z, et al. Highly efficient induction and long-term maintenance of multipotent cardiovascular progenitors from human pluripotent stem cells under defined conditions. Cell Res. 2013;23:1119–32.
Ouyang K, Leandro Gomez-Amaro R, Stachura DL, Tang H, Peng X, Fang X, et al. Loss of IP3R-dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat Commun. 2014;5:4814.
Fu X, Toh WS, Liu H, Lu K, Li M, Cao T. Establishment of clinically compliant human embryonic stem cells in an autologous feeder-free system. Tissue Eng Part C Methods. 2011;17:927–37.
Poon EN, Luo XL, Webb SE, Yan B, Zhao R, Wu SCM, et al. The cell surface marker CD36 selectively identifies matured, mitochondria-rich hPSC-cardiomyocytes. Cell Res. 2020;30:626–9.
Fu JD, Yu HM, Wang R, Liang J, Yang HT. Developmental regulation of intracellular calcium transients during cardiomyocyte differentiation of mouse embryonic stem cells. Acta Pharmacol Sin. 2006;27:901–10.
Monfredi O, Maltsev VA, Lakatta EG. Modern concepts concerning the origin of the heartbeat. Physiology (Bethesda). 2013;28:74–92.
O’Connell TD, Jensen BC, Baker AJ, Simpson PC. Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev. 2014;66:308–33.
Kohi M, Yang HT, Endoh M. Myocardial alpha 1A-adrenoceptor subtypes in rabbit: differentiation by a selective antagonist, HV723. Eur J Pharmacol. 1993;250:95–101.
Satin J, Itzhaki I, Rapoport S, Schroder EA, Izu L, Arbel G, et al. Calcium handling in human embryonic stem cell-derived cardiomyocytes. Stem Cells. 2008;26:1961–72.
Resende RR, Adhikari A, da Costa JL, Lorencon E, Ladeira MS, Guatimosim S, et al. Influence of spontaneous calcium events on cell-cycle progression in embryonal carcinoma and adult stem cells. Biochim Biophys Acta. 2010;1803:246–60.
Garcia MI, Boehning D. Cardiac inositol 1,4,5-trisphosphate receptors. Biochim Biophys Acta Mol Cell Res. 2017;1864:907–14.
Berridge MJ, Bootman MD, Lipp P. Calcium−a life and death signal. Nature. 1998;395:645–8.
Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res. 2013;113:690–708.
Yang F, Huang L, Tso A, Wang H, Cui L, Lin L, et al. Inositol 1,4,5-trisphosphate receptors are essential for fetal-maternal connection and embryo viability. PLoS Genet. 2020;16:e1008739.
Wobus AM, Wallukat G, Hescheler J. Pluripotent mouse embryonic stem cells are able to differentiate into cardiomyocytes expressing chronotropic responses to adrenergic and cholinergic agents and Ca2+ channel blockers. Differentiation. 1991;48:173–82.
Lorenzen-Schmidt I, Schmid-Schonbein GW, Giles WR, McCulloch AD, Chien S, Omens JH. Chronotropic response of cultured neonatal rat ventricular myocytes to short-term fluid shear. Cell Biochem Biophys. 2006;46:113–22.
Xiang Y, Rybin VO, Steinberg SF, Kobilka B. Caveolar localization dictates physiologic signaling of beta 2-adrenoceptors in neonatal cardiac myocytes. J Biol Chem. 2002;277:34280–6.
Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–102.
van der Heyden MA, Wijnhoven TJ, Opthof T. Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc Res. 2005;65:28–39.
Liu QY, Karpinski E, Pang PK. Changes in alpha 1-adrenoceptor coupling to Ca2+ channels during development in rat heart. FEBS Lett. 1994;338:234–8.
Itzhaki I, Rapoport S, Huber I, Mizrahi I, Zwi-Dantsis L, Arbel G, et al. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS One. 2011;6:e18037.
Hund TJ, Ziman AP, Lederer WJ, Mohler PJ. The cardiac IP3 receptor: uncovering the role of “the other” calcium-release channel. J Mol Cell Cardiol. 2008;45:159–61.
Hirose M, Stuyvers B, Dun W, Ter Keurs H, Boyden PA. Wide long lasting perinuclear Ca2+ release events generated by an interaction between ryanodine and IP3 receptors in canine Purkinje cells. J Mol Cell Cardiol. 2008;45:176–84.
Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol. 2001;536:3–19.
Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–40.
Goldberg AT, Bond BR, Mukherjee R, New RB, Zellner JL, Crawford FA Jr, et al. Endothelin receptor pathway in human left ventricular myocytes: relation to contractility. Ann Thorac Surg. 2000;69:711–5. discussion 716.
Milano CA, Dolber PC, Rockman HA, Bond RA, Venable ME, Allen LF, et al. Myocardial expression of a constitutively active alpha 1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc Natl Acad Sci USA. 1994;91:10109–13.
Acknowledgements
This work was supported by grants from National Key R&D Program of China (2017YFA0103700 and 2016YFC1301204 to HTY), National Natural Science Foundation of China (81520108004, 81470422 to HTY), the Strategic Priority Research Program of the CAS (No. XDA16010201 to HTY), Shanghai Natural Science Foundation (17ZR1435500 to JJH), and the Shenzhen Basic Research Foundation (KQJSCX20170330155020267). The authors thank WiCell Research Institute for providing the H7 hESCs, Dr. Hua-jun Bai (Shanghai Institute of Nutrition and Health) for the assistance in hESC culture, and Prof. Ping Liang from Zhejiang University for the constructive discussion and suggestions.
Author information
Authors and Affiliations
Contributions
HTY and PZ contributed to the concept and experiment design; PZ, JJH, HL and YJW contributed to the data collection; PZ, HTY, KFOY and MLL contributed to the data analysis; HTY and PZ contributed to the data interpretation and manuscript writing; HTY approved the manuscript and provided the funding support.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Zhang, P., Huang, Jj., Ou-yang, Kf. et al. Minimal contribution of IP3R2 in cardiac differentiation and derived ventricular-like myocytes from human embryonic stem cells. Acta Pharmacol Sin 41, 1576–1586 (2020). https://doi.org/10.1038/s41401-020-00528-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00528-w
Keywords
This article is cited by
-
Myosin light chain 2 marks differentiating ventricular cardiomyocytes derived from human embryonic stem cells
Pflügers Archiv - European Journal of Physiology (2021)
