
Abstract
Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disease caused by a mutation in the gene encoding the dystrophin protein. Catalpol is an iridoid glycoside found in Chinese herbs with anti-inflammatory, anti-oxidant, anti-apoptotic, and hypoglycemic activities that can protect against muscle wasting. In the present study we investigated the effects of catalpol on DMD. Aged Dystrophin-deficient (mdx) mice (12 months old) were treated with catalpol (100, 200 mg·kg−1·d−1, ig) for 6 weeks. At the end of the experiment, the mice were sacrificed, and gastrocnemius (GAS), tibialis anterior (TA), extensor digitorum longus (EDL), soleus (SOL) muscles were collected. We found that catalpol administration dose-dependently increased stride length and decreased stride width in Gait test. Wire grip test showed that the time of wire grip and grip strength were increased. We found that catalpol administration dose-dependently alleviated skeletal muscle damage, evidenced by reduced plasma CK and LDH activity as well as increased the weight of skeletal muscles. Catalpol administration had no effect on dystrophin expression, but exerted anti-inflammatory effects. Furthermore, catalpol administration dose-dependently decreased tibialis anterior (TA) muscle fibrosis, and inhibited the expression of TGF-β1, TAK1 and α-SMA. In primary myoblasts from mdx mice, knockdown of TAK1 abolished the inhibitory effects of catalpol on the expression levels of TGF-β1 and α-SMA. In conclusion, catalpol can restore skeletal muscle strength and alleviate skeletal muscle damage in aged mdx mice, thus may provide a novel therapy for DMD. Catalpol attenuates muscle fibrosis by inhibiting the TGF-β1/TAK1 signaling pathway.
Similar content being viewed by others

Introduction
Duchenne muscular dystrophy (DMD) is a severe progressive muscular dystrophy that is caused by mutations in the X-linked gene encoding the cytoskeletal protein dystrophin and affects approximately 1 in 3,500–5,000 newborn males worldwide [1]. Dystrophin, a large cytoskeletal protein localized in the sarcolemma of skeletal muscle, connects the intracellular sarcolemmal cytoskeleton to the extracellular matrix, maintaining the stability, strength, and flexibility of muscle fibers during movement and protecting muscle fibers from damage during contraction [2]. Loss of dystrophin damages the sarcolemma, leading to cycles of myofiber degeneration and regeneration, chronic inflammation, and accumulation of fibrous tissues [3]. Symptoms in subjects with DMD first appear at the age of 3–5 years [4]. Due to the progression of muscle wasting and severe impairment of muscle function, patients require wheelchair support at approximately 12 years of age and eventually die prematurely from cardiac and respiratory failure at 20–30 years of age [5]. Severe oxidative stress, inflammation, fibrosis, and necrosis of myofibers can be observed in DMD patients and mdx mice (a mouse model of DMD) [6]. Although improvements in patient care and disease management have slowed down disease progression, there are no effective drug therapies for DMD. The lack of functional dystrophin is the fundamental cause of pathology, and restoration of functional dystrophin expression is considered a logical therapeutic approach. Currently, research on dystrophin-targeted gene therapy is increasing [7]. A lack of dystrophin also triggers many different pathological pathways. In addition to therapies aimed at restoring dystrophin to correct the primary pathology, small-molecule compounds targeting secondary pathology are also potential treatments.
The transforming growth factor-β (TGF-β) family is a family of multifunctional cytokines. The TGF-β cytokine family member TGF-β1 plays an important role in regulating various biological processes, including cell growth, differentiation, apoptosis, tissue development, and inflammation [8]. TGF-β1 promotes the synthesis of extracellular matrix and inhibits matrix degradation, thereby inducing skeletal muscle fibrosis and modulating the repair of damaged skeletal muscle [3]. In a previous study, the TGF-β1 pathway was reported to be activated in the pathological process of DMD [9]. In addition, inhibition of the TGF-β1 pathway has been confirmed to improve pathology in mdx mice [10]. Transforming growth factor-β-activated kinase 1 (TAK1), a member of the MEK kinase family, is an important signaling molecule and a central mediator that regulates different downstream signaling pathways in response to multiple cytokines, such as TGF-β1, Toll-like receptors, interleukin-1, and tumor necrosis factor-α (TNF-α) [11]. TAK1 is essential for myogenesis and plays an important role in the regulation of skeletal muscle mass and function [12]. In a previous study, curcumin was shown to protect the peritoneal membrane from fibrosis via the TGF-β1/TAK1 signaling pathway [13]. Based on the above findings, we hypothesized that TGF-β1/TAK1 may be involved in skeletal muscle fibrosis in DMD.
Catalpol is an iridoid glucoside widely found in Chinese herbs and has been confirmed to exert anti-inflammatory, antioxidant, antiapoptotic, and hypoglycemic activities [14, 15]. Catalpol can reportedly reverse neuromuscular damage and was shown in our previous study to enhance myogenic differentiation through activation of MyoD/MyoG-mediated myogenesis [16]. However, whether catalpol can alleviate DMD skeletal muscle injury remains unclear. Pathological features appear in mdx mice at ~3–4 weeks of age and are accompanied by massive degeneration of a large number of myofibers [17]. Then, degeneration and regeneration cycles of new fibers occur at ~6–8 weeks of age [18]. Severe inflammation, oxidative stress, and fibrosis develop in the diaphragm and limb muscles of mdx mice during the pathological process. However, unlike the progressive and deteriorating pathological status in DMD patients, the pathology of the limb muscles of mdx mice is gradually ameliorated due to the compensatory repair of body damage [19]. Cardiac and skeletal muscles become dysfunctional due to fibrosis and severe infiltration of inflammatory cells in aged mdx mice [20]. In addition, aged mdx mice undergo fewer degeneration and regeneration cycles than young mdx mice [21], and muscle mass and function are more severely impaired in aged mdx mice than in young mdx mice [22]. Therefore, how catalpol improves disease phenotype in aged (approximately 12-month-old) mdx mice, when the pathology is more severe and similar to what is observed in DMD patients, was explored in the present study.
Materials and methods
Materials
Catalpol (CAS number 2415-24-9, purity >98%) was purchased from Nanjing Jingzhu Biological Engineering Co., Ltd. (Nanjing, China). Other reagents were purchased from Millipore Sigma (St. Louis, MO, USA) unless otherwise stated.
Animal studies
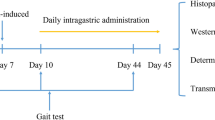
All experimental procedures were conducted in accordance with the guidelines of the Animal Ethics Committee of China Pharmaceutical University for the Care and Use of Laboratory Animals and were approved by the Laboratory Animal Management Committee of China (Nanjing, China; NCT number: 2019-11-006). Dystrophin-deficient C57BL/10ScSnJNju-Dmdem3Cd4/Gpt (mdx) mice and C57BL/10ScSn/J mice (male, aged 12 months) were acquired from Nanjing University Animal Model Research Center (Nanjing, China). Mice were housed in an environmentally controlled room on a standard 12:12 h light/dark cycle and provided free access to food and water. Twenty-four mdx mice were randomly assigned to the DMD group, Cat 100 group (catalpol 100 mg·kg−1·d−1), or Cat 200 group (catalpol 200 mg·kg−1·d−1; n = 8 per group), and wild-type mice (n = 8) were used as the control group. Catalpol was administered by gavage for 6 consecutive weeks, while the control group and DMD group were treated with saline. At the end of the experiment, the mice were sacrificed, and the gastrocnemius (GAS), tibialis anterior (TA), extensor digitorum longus (EDL), and soleus (SOL) muscles were collected.
Gait experiment
The forelimbs of each mouse were stained with ink, and the mice were placed on a track covered with white paper and motivated to move forward. The width of the runway was 3 cm, and the length was 2 m. A desk lamp was placed at the starting point of the runway, and a small black box was placed at the end of the runway. The rest of the runway was kept in relative darkness. The affinity of mice for darkness motivated them to move forward. The four groups of mice were tested sequentially. The forelimbs of the mice were wiped, and the same experiment was performed for the hindlimbs. After the experiment, six prints on the paper near the end point were selected to measure the step length and step width.
Wire grip test
The wire test is a standard method for assessing whole-body strength. The mice were allowed to grasp a 2-mm metal wire, and the length of time until the mice fell from the wire was recorded. The experiment was performed two days before the mice were sacrificed. This experiment was repeated at least three times. When the mice fell, they were allowed to recover for 3 min before the next trial. The test score was determined by calculating the average of at least three trial.
Grip strength test
The test was performed as described previously [15]. The forelimb grip strength test was used to determine the forelimb grip strength by using a calibrated grip strength tester (YLS-13A, Yiyan Bio, Jinan, China). At least three trials of the grip strength test were performed.
Serum analysis and tissue collection
Before the mice were euthanized, blood was taken from the inferior vena cava. Serum was obtained after centrifuging blood samples at 3500 rpm for 15 min at 4 °C. Creatine kinase (CK) and lactate dehydrogenase (LDH) activity in the serum was measured by using a HITACHI7080 Automatic Clinical Analyzer (Tokyo, Japan). Serum inflammatory cytokine levels were determined using enzyme-linked immunosorbent assay (ELISA) kits (ExCell Biotech, Shanghai, China). The TA, GAS, EDL, and SOL muscles were dissected and weighed.
Histological analysis
The TA, GAS, EDL, and SOL muscles from the left leg were embedded in tragacanth gum, frozen in liquid nitrogen-cooled isopentane, and stored at −80 °C until use. Cryosections of the TA muscle were mounted on a slide and then horizontally sectioned into 5-μm slices. The slices of the TA muscle were stained with hematoxylin and eosin (H&E) according to standard protocols. TA paraffin sections (8 μm) were stained with Masson’s trichrome. All slides were viewed, and photomicrographs were captured under a light microscope (BX53, Olympus, Tokyo, Japan).
Isolated myoblasts
Primary myoblasts were isolated from the TA muscles of mdx mice as described previously [23]. The cells were isolated by digesting the muscles with 1.2 U/mL dispase type II (D4693, Millipore Sigma) and 5 mg/mL collagenase type D (35799223; Roche Diagnostics, Ltd., Shanghai, China) in DMEM at 37 °C for 1 h. Myoblasts were cultured in DMEM containing 20% fetal bovine serum (Gibco, Waltham, MA, USA).
Immunofluorescence
Mounted TA sections were blocked in blocking buffer (10% goat serum and phosphate-buffered saline) for 1 h at room temperature. Then, the slides were incubated overnight at 4 °C with primary antibodies against dystrophin (1:50; 7A10, DSHB, Iowa City, IA, USA); utrophin (1:50; 8A4, DSHB); laminin (1:200; L9393, Sigma, St. Louis); F4/80 (1:100; 123140, BioLegend, San Diego, CA, USA); CD68 (1:100; 141710, BioLegend); and fibronectin (1:200; ab32419, Abcam, Cambridge, MA, USA). The slides were incubated with secondary antibody (Alexa Fluor 633-conjugated, Alexa Fluor 488-conjugated; Thermo Fisher, Waltham, MA, USA) for 1 h at room temperature, and 4’,6-diamidino-2-phenylindole (DAPI, Thermo Fisher) was used to stain the nuclei. The sections were viewed, and photomicrographs were captured under a fluorescence microscope (FV1000, Olympus, Tokyo, Japan).
Western blot analysis
The TA muscles from the right leg were frozen in liquid nitrogen, shredded and placed in total protein extraction buffer containing phosphatase inhibitor and protease inhibitor. Then, the muscles were further homogenized with a tissue grinder. The samples were lysed on ice, and the supernatant was obtained by centrifugation at 12,000 × g for 10 min at 4 °C. The concentration of the protein was detected by the BCA assay. In short, the protein was fractionated on 6% to 12% sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA) by electroblotting using a miniature transfer apparatus (BioRad Laboratories, Hercules, CA, USA). At room temperature, the membranes were blocked in Tris buffered saline containing 5% bovine serum albumin (BSA) for 90 min. Next, the membranes were incubated with primary antibody at 4 °C overnight. The information for all antibodies is provided in Supplementary Table S1. After washing in TBST three times (7 min each), the membranes were incubated with secondary antibody for 1 h. Then, the membranes were washed in TBST three times, and the blots were imaged using the Bio-Rad ECL system (Hercules, CA, USA). The bands were subjected to optical density analysis with Image-Pro Plus software (Media Cybernetics, Rockville, MD, USA).
Statistical analysis
All data provided are expressed as the mean ± SD and were analyzed by one-way or two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison posttest using GraphPad Prism software (ver. 8.0; GraphPad Software Inc., La Jolla, CA, USA). Differences with P values < 0.05 were considered statistically significant.
Results
Catalpol increased skeletal muscle function in aged mdx mice
DMD is a degenerative disease, and skeletal muscle injury is more severe in aged mdx mice than in young mdx mice [24]. In the present study, 12-month-old mdx mice were treated with different doses of catalpol to examine its therapeutic effects on DMD. To explore the effects of catalpol on skeletal muscle function, the stride length of the forelimbs and hindlimbs was measured (Fig. 1a). Compared with that of the control mice, the stride length of aged mdx mice was decreased. After 6 weeks treatment with catalpol, the stride length of aged mdx mice increased; 200 mg/kg catalpol resulted in a greater increase in stride length than 100 mg/kg catalpol (Fig. 1b, d). In addition, the stride width of the forelimbs and hindlimbs was measured; the stride width of aged mdx mice was increased compared with that of control mice. After treatment with catalpol, the stride width of aged mdx mice decreased (Fig. 1c, e). To further show that catalpol can improve skeletal muscle function, the ending position and duration that the mice grasped the wire in the wire test were recorded. Ending positions (Fig. 1f) revealed skeletal muscle weakness in the limbs of aged mdx mice, and the wire test time showed decreased skeletal muscle function in the limbs of aged mdx compared to those of control mice. Catalpol improved skeletal muscle function, as evidenced by increased duration in the wire test (Fig. 1g, h).

The gait and wire tests were used to assess the behavioral function of skeletal muscle. a Diagrams of the gait of the forelimbs and hindlimbs of wild-type mice (CON), Duchenne muscular dystrophy (DMD) mice, mdx mice treated with 100 mg·kg−1·d−1 catalpol for 6 weeks (Cat 100), and mdx mice treated with 200 mg·kg−1·d−1 catalpol for 6 weeks (Cat 200). b–e Statistical analysis of step length and width of the forelimbs and hindlimbs. f Different views of CON, DMD, Cat 100, and Cat 200 mice during the wire test. g, h Suspension time and physical impulse. All data are shown as the means ± standard deviations (n = 8 animals), **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. DMD.
Catalpol ameliorated muscle damage in aged mdx mice
Grip strength was measured to further show the ameliorating effects of catalpol on skeletal muscle injury. Compared with that of control mice, the grip strength of aged mdx mice was decreased, and grip strength generally returned to normal after catalpol treatment (Fig. 2b). In previous research, serum CK and LDH levels were increased in mdx mice compared to control mice. In the present study, catalpol significantly reduced serum CK and LDH levels, and 200 mg·kg−1·d−1 catalpol significantly reduced serum levels (Fig. 2c, d). In addition, the mouse SOL, TA, EDL, and GAS muscles were weighed. The weight of the SOL, TA, and EDL muscles were significantly decreased in aged mdx mice compared with control mice. After treatment with catalpol, the weight of the SOL, TA, and EDL muscles was increased. The weight of the GAS muscle was slightly decreased in aged mdx mice compared to control mice, suggesting that catalpol had minimal effects (Fig. 2e–h). In addition, hematoxylin and eosin (H&E) staining of mouse skeletal muscles was performed. H&E staining of the TA muscle showed a large amount of inflammatory cell infiltration in the skeletal muscle of aged mdx mice and an increase in the gap between muscle cells compared to that in control mice. After treatment with catalpol, the number of inflammatory cells in skeletal muscle was significantly reduced, and the tightly packed arrangement of the muscle cells was restored, indicating that catalpol had anti-inflammatory effects (Fig. 3a). To further evaluate the anti-inflammatory effects of catalpol, F4/80 immunofluorescence, and CD68 immunofluorescence were evaluated. Macrophages appeared in the skeletal muscles of aged mdx mice, but the number of macrophages was reduced in catalpol-treated mice compared with control mice. In addition, catalpol repaired damaged skeletal muscle in aged mdx mice based on laminin immunofluorescence (Fig. 3b). Next, we detected inflammatory cytokine (TNF-α, IL-1β, and TGF-β1) levels by ELISA. The results showed that catalpol treatment reduced inflammatory cytokine (TNF-α, IL-1β, TGF-β1) levels in aged mdx mice (Fig. 3c).

a Chemical structure of catalpol. b Grip strength of control mice (CON), Duchenne muscular dystrophy (DMD) mice, mdx mice treated with 100 mg·kg−1·d−1 catalpol (Cat 100), and mdx mice treated with 200 mg·kg−1·d−1 catalpol for 6 weeks (Cat 200). c, d Serum creatine kinase (CK) and lactic dehydrogenase (LDH) levels. Blood was collected from CON, DMD, Cat 100, and Cat 200 mice. The serum was obtained by centrifugation. e–h The weight of the soleus (SOL), tibialis anterior (TA), extensor digitorum longus (EDL), and gastrocnemius (GAS) muscles of CON, DMD, Cat 100, and Cat 200 mice. All data are shown as the means ± standard deviations (n = 8 animals), *P < 0.05, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. DMD.

a Hematoxylin and eosin (H&E)-stained tibialis anterior (TA) muscles of control mice, Duchenne muscular dystrophy (DMD) mice, mdx mice treated with 100 mg·kg−1·d−1 catalpol (Cat 100), and mdx mice treated with 200 mg·kg−1·d−1 catalpol for 6 weeks (Cat 200). Scale bar, 100 μm. b The TA muscle was stained with laminin (green), F4/80 (red), and CD68 (arrowheads), and DAPI was used to stain the nuclei (blue). Scale bar, 100 μm. c Serum IL-1β, TNF-α, and TGF-β1 levels were measured by using enzyme-linked immunosorbent assay kits. All data are shown as the means ± standard deviations (n = 8 animals), *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. DMD.
Catalpol did not restore the expression of dystrophin in aged mdx mice
Based on current research, mutations in the dystrophin gene and consequent loss of the dystrophin protein cause DMD. To explore whether catalpol can restore dystrophin protein expression in aged mdx mice, dystrophin protein immunofluorescence was evaluated. Immunofluorescence analysis of skeletal muscles showed that the dystrophin protein was not expressed in aged mdx mice and that catalpol did not restore dystrophin expression (Fig. 4a). These results were validated by Western blot analysis (Fig. 4b). Utrophin (also known as dystrophin-related protein) is a member of the dystrophin superfamily and has a wide range of sequence similarities and functional motifs with dystrophin, including the ability to bind to dystrophin-associated glycoprotein compounds [25]. Utrophin is expressed in the sarcolemma during early embryonic development and myofiber regeneration [26]. Utrophin can reportedly ameliorate the phenotype of mdx mice [27]. Therefore, utrophin protein immunofluorescence was examined in the present study. Significant upregulation of utrophin protein was observed in aged mdx mice compared to control mice. In addition, the utrophin protein level was higher after treatment with catalpol than after control treatment (Fig. 4c). However, increased utrophin expression did not improve DMD.

a The tibialis anterior (TA) muscle was stained with an anti-dystrophin (green) antibody, and DAPI was used to stain the nuclei (blue). Scale bar, 100 μm. b Western blot analysis of dystrophin protein levels in the TA muscle and the pooled values of each group. c The TA muscle was stained with an anti-utrophin (red) antibody. Scale bar, 100 μm. All data are shown as the means ± standard deviations (n = 8 animals), ***P < 0.001 vs. control.
Catalpol attenuated skeletal muscle fibrosis by inhibiting the TGF-β1/TAK1 signaling pathway
Skeletal muscle fibrosis is a symptom of DMD [28]. Therefore, fibrosis in the TA muscle was evaluated using Masson’s trichrome staining, which was increased in aged mdx mice compared to control mice and reduced in catalpol-treated mdx mice compared to control-treated mdx mice. The improvement in skeletal muscle fibrosis was greater following treatment with 200 mg·kg−1·d−1 catalpol than following treatment with 100 mg·kg−1·d−1 catalpol (Fig. 5a, b). To further demonstrate the antifibrotic effects of catalpol, fibronectin immunofluorescence was evaluated. The results indicated that skeletal muscle fibrosis was increased in aged mdx mice compared with control mice and that skeletal muscle fibrosis was reduced after treatment with catalpol. The normalized results showed that the antifibrotic effects of 200 mg·kg−1·d−1 catalpol were more apparent than those of 100 mg·kg−1·d−1 catalpol (Fig. 5c, d).

a Representative Masson’s trichrome-stained images of tibialis anterior (TA) muscle sections and b analysis of collagen fiber area percentage in each group. c, d Representative images showing connective tissue levels in the TA muscle based on fibronectin (green) immunostaining. Scale bar, 100 μm. All data are shown as the means ± standard deviations (n = 8 animals), ***P < 0.001 vs. control; #P < 0.05, ###P < 0.001 vs. DMD.
To confirm the fibrosis-inhibiting mechanism of catalpol, the TGF-β1/TAK1 signaling pathway in skeletal muscle was analyzed. Based on Western blotting, the protein ratios of p-TAK1 to t-TAK1 and p-Smad 2/3 to t-Smad 2/3 were increased in aged mdx mice compared with control mice (Fig. 6a–d). Furthermore, TGF-β1 and α-smooth muscle actin (α-SMA) protein expression was increased in aged mdx mice compared with control mice (Fig. 6e, f). After treatment with catalpol, the p-TAK1/t-TAK1 protein ratio was decreased, and α-SMA protein and TGF-β1 expression were downregulated (Fig. 6a–f). Therefore, we hypothesized that catalpol inhibited skeletal muscle fibrosis by inhibiting the TGF-β1/TAK1 signaling pathway.

a Western blot of p-TAK1 and t-TAK1 protein levels in TA muscle and b the pooled values of each group. c Western blot analysis of p-Smad 2/3 and t-Smad 2/3 protein levels in the TA muscle and (d) the pooled values of each group. e TGF-β1 and α-smooth muscle actin (α-SMA) protein levels in the tibialis anterior (TA) muscle and (f) the pooled values of each group. All data are shown as the means ± standard deviations (n = 8 animals), *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. DMD.
To confirm that TGF-β1/TAK1 signaling mediates the effect of catalpol on the attenuation of muscle fibrosis, we blocked TAK1 activity by shRNA-TAK1 in primary myoblasts from mdx mice. We found that the catalpol-induced decreases in TAK1 protein levels were abolished after TAK1 inhibition. Moreover, there were no changes in α-SMA and TGF-β1 protein levels between the TGF-β1 + shRNA-TAK1 group and TGF-β1 + shRNA-TAK1 + catalpol group (Fig. 7a–c). These results indicate that TGF-β1/TAK1 signaling mediates the effect of catalpol on the attenuation of muscle fibrosis.

a p-TAK1 and t-TAK1 protein levels and b TGF-β1 and α-SMA protein levels in primary myoblasts from mdx mice. c Pooled data for p-TAK1/t-TAK1, TGF-β1 and α-SMA are shown. d Schematic diagram depicting the possible mechanism by which catalpol inhibits the TGF-β1/TAK1 signaling pathway in DMD. All data are shown as the means ± standard deviations (n = 3), **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. TGF-β1.
Discussion
Catalpol is isolated from the roots of Rehmannia glutinosa [15], which has antioxidative, anti-inflammatory [8, 29], antifibrotic [30], and hypoglycemic properties [30]. Catalpol has been shown to significantly suppress the expression of proinflammatory factors such as TNF-α and inducible NO synthase and reduce reactive oxygen species levels [31]. Xu et al. suggested that catalpol improves skeletal muscle mitochondrial function by activating adenosine 5′-monophosphate-activated protein kinase-mediated mitochondrial biogenesis [32]. In the present study, only 200 mg·kg−1·d−1 catalpol had a mild anti-inflammatory effect in skeletal muscle (Fig. 3). Catalpol has been shown to increase skeletal muscle grip strength and weight by upregulating the expression of MyoD, MyoG, and myosin heavy chain [16]. In the present study, catalpol improved skeletal muscle function, including enhancing muscle grip strength, prolonging suspension time, and improving walking posture, and this effect was accompanied by decreased serum CK and LDH levels (Figs. 1 and 2). Catalpol was beneficial in increasing TA muscle weight in mdx mice (Fig. 2), which could potentially inhibit skeletal muscle atrophy, as previously reported [33]. In a previous study, catalpol was shown to ameliorate liver fibrosis and exert anti-inflammatory activities [30]. In the present study, the TA muscles of mdx mice exhibited obvious fibrosis, inflammatory infiltration, centralization of the nucleus, and increased intercellular space. In addition, catalpol attenuated skeletal muscle fibrosis (Fig. 5). When fibronectin and α-SMA, which are markers of fibrosis, were investigated, it was found that α-SMA was downregulated and fibronectin immunofluorescence was decreased by catalpol treatment, confirming that catalpol inhibits skeletal muscle fibrosis (Figs. 5 and 6).
DMD, a muscular dystrophy disease, is caused by deletion of dystrophin. DMD mainly affects young males, and subjects usually die of heart or respiratory failure. Corticosteroids remain the main drugs for DMD treatment in China. Steroid therapy can improve muscle function and prolong walking ability. However, long-term daily steroid regimens are associated with side effects, such as obesity, bone fractures, and behavioral changes [34]. Currently, there is no effective drug treatment for DMD; however, gene therapy is used to improve dystrophin gene function [35]. Clinical trials have been performed using exon-skipping approaches targeting exons 45 and 53 using 2′-O-methyl phosphorothioate antisense oligonucleotides [36]. Eteplirsen, an antisense oligonucleotide, was the first drug approved for DMD in the United States [37] and is used for the treatment of DMD caused by a mutation in exon 51. In addition, other antisense oligonucleotide therapies are in clinical trials, such as casimersen (exon 45-skipping), NS-065 (exon 53-skipping), and suvodirsen (exon 51-skipping) [36]. These gene therapies each target a single exon mutation in dystrophin; however, many mutations of the gene encoding dystrophin cause DMD. Therefore, gene therapy is not suitable for the majority of DMD patients. In the present study, dystrophin expression in the TA muscle was analyzed in mdx mice, and it was found that catalpol did not affect dystrophin expression (Fig. 4a). Coenzyme Q10, an electron acceptor for NADH and succinate dehydrogenase, inhibits inflammation and has antioxidative capabilities [38]. Furthermore, coenzyme Q10 is currently being evaluated in clinical trials as an antifibrotic drug for DMD. Tamoxifen, an estrogen receptor modulator, was previously reported to improve leg structure, diminish cardiac and diaphragm fibrosis, and improve respiratory function in mdx mice [39]. Tamoxifen is in a phase III clinical trial [40] and is expected to be an effective treatment for DMD. The results of the present study indicate that catalpol can significantly inhibit skeletal muscle fibrosis and that catalpol is a potential agent for DMD treatment.
In a recent study, TAK1 was shown to be highly expressed and activated in the developing skeletal muscle of mice [41]. TAK1 deficiency in satellite cells results in increased oxidative stress and necrosis [42]. In a previous study, it was shown that TGF-β1a activates inflammation and that overexpression of TGF-β1a upregulates liver fibrosis during early tumorigenesis and downregulates liver fibrosis during late tumorigenesis [43]. In addition, TGF-β plays a major role in peritoneal fibrosis, and Zhao et al. [13] suggested that the TGF-β1/TAK1 signaling pathway may be involved in peritoneal fibrosis suppression caused by curcumin. Based on these results, TAK1 and TGF-β1 expression were analyzed in the present study. Catalpol inhibited TAK1 phosphorylation and reduced the total TAK1 level. In addition, catalpol inhibited the overexpression of the fibrosis biomarkers TGF-β1 and α-SMA (Fig. 6). In the TA muscle, catalpol relieved fibrosis. Overall, our findings indicate that catalpol may attenuate fibrosis by inhibiting the TGF-β1/TAK1 signaling pathway (Fig. 7d).
As reported previously, aged mdx mice display more severe muscle damage than young mdx mice, especially in the diaphragm muscle [21]. The pathology of aged mdx mice is more severe than that of young mice and more similar to what is observed in DMD patients. In the current study, catalpol attenuated muscle fibrosis by possibly blocking the TGF-β1/TAK1 signaling pathway. However, the inhibition of the TGF-β1/TAK1 signaling pathway should be investigated in detail in future studies. The TGF-β1/TAK1 signaling pathway is a potential target for the treatment of DMD.
In conclusion, catalpol not only improves muscle strength and function but also protects muscles from fibrosis in aged mdx mice. Furthermore, catalpol-mediated attenuation of muscle fibrosis appears to via inhibiting the TGF-β1/TAK1 signaling pathway.
References
Govoni A, Magri F, Brajkovic S, Zanetta C, Faravelli I, Corti S, et al. Ongoing therapeutic trials and outcome measures for Duchenne muscular dystrophy. Cell Mol Life Sci. 2013;70:4585–602.
Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7:762–73.
Delaney K, Kasprzycka P, Ciemerych MA, Zimowska M. The role of TGF-β1 during skeletal muscle regeneration. Cell Biol Int. 2017;41:707–15.
Ponnusamy S, Sullivan RD, You D, Zafar N, Yang CH, Thiyagarajan T, et al. Androgen receptor agonists increase lean mass, improve cardiopulmonary functions and extend survival in preclinical models of Duchenne muscular dystrophy. Hum Mol Genet. 2017;26:2526–40.
Emery AE. The muscular dystrophies. Lancet. 2002;359:687–95.
Nguyen LN, Ferry A, Schnell FJ, Hanson GJ, Popplewell L, Dickson G, et al. Functional muscle recovery following dystrophin and myostatin exon splice modulation in aged mdx mice. Hum Mol Genet. 2019;28:3091–100.
Guiraud S, Chen H, Burns DT, Davies KE. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp Physiol. 2015;100:1458–67.
Bi J, Wang XB, Chen L, Hao S, An LJ, Jiang BO, et al. Catalpol protects mesencephalic neurons against MPTP-induced neurotoxicity via attenuation of mitochondrial dysfunction and MAO-B activity. Toxicol Vitr. 2008;136:1883–9.
Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65:826–34.
Nelson CA, Hunter RB, Quigley LA, Girgenrath S, Weber WD, McCullough JA, et al. Inhibiting TGF-β activity improves respiratory function in mdx mice. Am J Pathol. 2011;178:2611–21.
Mihaly SR, Ninomiya-Tsuji J, Morioka S, Differentiation. TAK1 control of cell death. Cell Death Differ. 2014;21:1667–76.
Hindi SM, Sato S, Xiong G, Bohnert KR, Gibb AA, Gallot YS, et al. TAK1 regulates skeletal muscle mass and mitochondrial function. JCI Insight. 2018;3:e98441.
Zhao JL, Zhang T, Shao X, Zhu JJ, Guo MZ. Curcumin ameliorates peritoneal fibrosis via inhibition of transforming growth factor-activated kinase 1 (TAK1) pathway in a rat model of peritoneal dialysis. BMC Complement Alter Med. 2019;19:280.
Huang WJ, Niu HS, Lin MH, Cheng JT, Hsu FL. Antihyperglycemic effect of catalpol in streptozotocin-induced diabetic rats. J Nat Prod. 2010;73:1170–2.
Liu JY, Zheng CZ, Hao XP, Zhang DJ, Mao AW, Yuan P. Catalpol ameliorates diabetic atherosclerosis in diabetic rabbits. Am J Transl Res. 2016;8:4278–88.
Xu DQ, Wang L, Jiang ZZ, Zhao GL, Hassan HM, Sun LX, et al. A new hypoglycemic mechanism of catalpol revealed by enhancing MyoD/MyoG-mediated myogenesis. Life Sci. 2018;209:313–23.
Mcdonald AA, Hebert SL, Kunz MD, Ralles SJ, Mcloon LK. Disease course in mdx:utrophin+/− mice: comparison of three mouse models of Duchenne muscular dystrophy. Physiol Rep. 2015;3:e12391.
Haddix SG, Lee YI, Kornegay JN, Thompson WJ. Cycles of myofiber degeneration and regeneration lead to remodeling of the neuromuscular junction in two mammalian models of Duchenne muscular dystrophy. PLoS One. 2018;13:e0205926.
Carnwath JW, Shotton DM. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J Neurol Sci. 1987;80:39–54.
Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;129:97–105.
Roig M, Roma J, Fargas A, Munell F. Longitudinal pathologic study of the gastrocnemius muscle group in mdx mice. Acta Neuropathol. 2004;107:27–34.
Koopman R, van Loon LJ. Aging, exercise, and muscle protein metabolism. J Appl Physiol. 2009;106:2040–8.
Xu DQ, Jiang ZZ, Sun ZR, Wang L, Zhao GL, Hassan HM, et al. Mitochondrial dysfunction and inhibition of myoblast differentiation in mice with high-fat-diet-induced pre-diabetes. J Cell Physiol. 2019;234:7510–23.
Harris JB. Myotoxic phospholipases A2 and the regeneration of skeletal muscles. Toxicon 2003;42:933–45.
Loro E, Sengupta K, Bogdanovich S, Whig K, Schultz DC, Huryn DM, et al. High-throughput identification of post-transcriptional utrophin up-regulators for Duchenne muscle dystrophy (DMD) therapy. Sci Rep. 2020;10:2132.
Duan D. Micro-utrophin therapy for duchenne muscular dystrophy. Mol Ther. 2019;27:1872–4.
Gilbert R, Nalbantoglu J, Petrof BJ, Ebihara S, Guibinga GH, Tinsley JM, et al. Adenovirus-mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum Gene Ther. 1999;10:1299–310.
Desguerre I, Arnold L, Vignaud A, Cuvellier S, Yacoub-Youssef H, Gherardi RK, et al. A new model of experimental fibrosis in hindlimb skeletal muscle of adult mdx mouse mimicking muscular dystrophy. Muscle Nerve. 2012;45:803–14.
Liu JY, Zhang DJ. Amelioration by catalpol of atherosclerotic lesions in hypercholesterolemic rabbits. Planta Med. 2015;81:175–84.
Liu Z, Zhu P, Zhang L, Xiong B, Tao J, Guan W, et al. Autophagy inhibition attenuates the induction of anti-inflammatory effect of catalpol in liver fibrosis. Biomed Pharmacother. 2018;103:1262–71.
Choi HJ, Jang HJ, Chung TW, Jeong SI, Cha J, Choi JY, et al. Catalpol suppresses advanced glycation end-products-induced inflammatory responses through inhibition of reactive oxygen species in human monocytic THP-1 cells. Fitoterapia. 2013;86:19–28.
Xu DQ, Li CJ, Jiang ZZ, Wang L, Huang HF, Li ZJ, et al. The hypoglycemic mechanism of catalpol involves increased AMPK-mediated mitochondrial biogenesis. Acta Pharmacol Sin. 2020;41:791–9.
Dowling JJ. Eteplirsen therapy for Duchenne muscular dystrophy: skipping to the front of the line. Nat Rev Neurol. 2016;12:675–6.
Crone M, Mah JK. Current and emerging therapies for duchenne muscular dystrophy. Curr Treat Options Neurol. 2018;20:17–31.
Stein CA. Eteplirsen approved for duchenne muscular dystrophy: the FDA faces a difficult choice. Mol Ther. 2016;24:1884–5.
Verhaart IE, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019;15:373–86.
Aartsma-Rus A, Krieg AM. FDA approves eteplirsen for duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017;27:1–3.
Spurney CF, Rocha CT, Henricson E, Florence J, Mayhew J, Gorni K, et al. CINRG pilot trial of coenzyme Q10 in steroid-treated Duchenne muscular dystrophy. Muscle Nerve. 2011;44:174–8.
Dorchies OM, Reutenauer-Patte J, Dahmane E, Ismail HM, Petermann O, Patthey- Vuadens O, et al. The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am J Pathol. 2013;182:485–504.
Nagy S, Hafner P, Schmidt S, Rubino-Nacht D, Schadelin S, Bieri O, et al. Tamoxifen in Duchenne muscular dystrophy (TAMDMD): study protocol for a multicenter, randomized, placebo-controlled, double-blind phase 3 trial. Trials. 2019;20:637.
Ogura Y, Hindi SM, Sato S, Xiong G, Akira S, Kumar A. TAK1 modulates satellite stem cell homeostasis and skeletal muscle repair. Nat Commun. 2015;6:10123.
Xu D, Zhao L, Jiang J, Li S, Sun Z, Huang X. A potential therapeutic effect of catalpol in Duchenne muscular dystrophy revealed by binding with TAK1. J Cachexia Sarcopenia Muscle. 2020. https://doi.org/10.1002/jcsm.12581.
Yan C, Yang Q, Gong Z. Transgenic expression of tgfb1a induces hepatic inflammation, fibrosis and metastasis in zebrafish. Biochem Biophys Res Commun. 2019;509:175–81.
Acknowledgements
This work was supported by grants from the Scholar of the 14th Batch of “Six Talents Peak” High-level Talent Selection Program (SWYY-094); the Postgraduate Research Practice Innovation Program of Jiangsu Province (KYCX19-0763); the “Double First-Class” University Project (CPU2018GY33); and the National Natural Science Foundation of China (Nos. 81773827 and 81573514 to ZZJ; No. 81773995 to LYZ).
Author information
Authors and Affiliations
Contributions
DQX, SJL, and LZ, did the research work, discussed the results, and wrote the manuscript. XFH and CJL collected and analyzed the data. LXS, and XHL, reviewed the manuscript. ZZJ, conceived the experiments, researched the data, and edited/reviewed the manuscript. LYZ, is the guarantor of this work, has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
About this article

Cite this article
Xu, Dq., Zhao, L., Li, Sj. et al. Catalpol counteracts the pathology in a mouse model of Duchenne muscular dystrophy by inhibiting the TGF-β1/TAK1 signaling pathway. Acta Pharmacol Sin 42, 1080–1089 (2021). https://doi.org/10.1038/s41401-020-00515-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00515-1
