
Abstract
Ion channels are the third largest class of targets for therapeutic drugs. The pharmacology of ion channels is an important research area for identifying new treatment options for human diseases. The past decade or so has seen increasing interest in an ion channel protein belonging to the transient receptor potential (TRP) family, namely the melastatin subfamily member 7 (TRPM7), as an emerging drug target. TRPM7 is a bifunctional protein with a magnesium and calcium-conducting divalent ion channel fused with an active kinase domain. TRPM7 is ubiquitously expressed in human tissues, including the brain, and regulates various cell biology processes such as magnesium and calcium homeostasis, cell growth and proliferation, and embryonic development. TRPM7 provides a link between cellular metabolic status and intracellular calcium homeostasis in neurons due to TRPM7’s unique sensitivity to fluctuating intracellular Mg·ATP levels. Thus, the protein plays a key role in ischemic and hypoxic neuronal cell death and brain injury, and is one of the key nonglutamate mechanisms in cerebral ischemia and stroke. Currently, the most potent and specific TRPM7 inhibitor is waixenicin A, a xenicane diterpenoid from the Hawaiian soft coral Sarcothelia edmondsoni. Using waixenicin A as a pharmacological tool, we demonstrated that TRPM7 is involved in promoting neurite outgrowth in vitro. Most recently, we found that waixenicin A reduced hypoxic–ischemic brain injury and preserved long-term behavioral outcomes in mouse neonates. We here suggest that TRPM7 is an emerging drug target for CNS diseases and disorders, and waixenicin A is a viable drug lead for these disorders.
Similar content being viewed by others

Introduction
In drug discovery and development, ion channels are the third largest class of drug targets [1], behind only receptors and enzymes. The drugs that modulate these proteins act as (1) agonists or antagonists of receptors, (2) activators or inhibitors of enzymes, or (3) openers or blockers of ion channels. Hence, ion channels are important drug targets [2,3,4,5,6], comprising 19% of all human proteins with corresponding drugs approvals [7], largely for the treatment of diseases of the nervous, cardiovascular, and endocrine systems. In the pursuit of new classes of drugs for human diseases, ion channels represent a promising group of proteins in the discovery and development of new therapeutics.
Functionally, ion channels are membrane proteins forming pores that selectively allow ions to cross organellar and cell plasma membranes. Ion channels conduct different ionic species and thus are classified as sodium (Na+), potassium (K+), calcium (Ca2+), nonselective cation, or chloride (Cl−) anion channels. Ion channels are expressed in both excitable and nonexcitable cells and transport their respective ion species relying on an open channel state and the electrochemical gradient across the cellular membrane. Ion channels are gated either by changes in membrane potential voltages (voltage-operated ion channels), by second messengers, or receptor ligands [8]. Ion channels can be activated depending on the replenishment of intracellular calcium stores and are involved in the sensing of physical stimuli such as cold, heat, pH, mechanical stress, and ultimately pain [9]. Intracellular calcium, ATP, pH, and many other factors may regulate basic ion channel activity, while mutations of ion channels may cause the ion channel dysfunction underlying many known channelopathies [10, 11]. Thus, ion channels play important roles in both physiology and pathophysiology.
An important pathophysiology is brain ischemia. According to the World Health Organization [12, 13], stroke [14, 15] is a leading cause of mortality and immobility in adults worldwide. Stroke prevalence has consistently increased in recent years, accompanied by high mortality rates. Except for tissue plasminogen activator, which has a limited therapeutic window [16], there are no other effective therapeutic measures against stroke, resulting in high social and economic impacts worldwide [17].
Neonates can undergo hypoxic–ischemic brain injury, and hypoxic–ischemic encephalopathy (HIE) is a subsequent related early-onset brain and neuropsychological disorder in children [18, 19]. HIE manifests as a group of brain impairments, such as neurodevelopmental delay, cognitive and motor dysfunctions, and permanent neuropsychological handicaps in the form of cerebral palsy with or without epilepsy, learning and intellectual disabilities. Neonatal hypoxic–ischemic brain injury and its related brain disorders, HIE and cerebral palsy, cause significant lifetime health care costs, and lifelong disability causes a noticeable burden worldwide. For years, studies examined the mechanism of HI brain injury and the related HIE, but these research efforts have not yet yielded positive results for therapeutic treatment. Currently, the limited clinical management for HI brain injury is using ventilation and inducing mild hypothermia [20]. In both adults and children, hypoxia-ischemia injury causes a substantial health and economic burden.
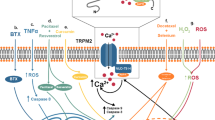
Mechanistically, cerebral ischemia triggers calcium overload and intracellular ionic imbalance and eventually leads to neuronal cell death [14, 15]. During cerebral ischemia, glutamate, an excitatory neurotransmitter, is released from neurons to act on glutamate receptor channels and trigger calcium overload and neuronal cell death [14, 15]. Mediated by glutamate NMDA and AMPA receptor channels, glutamate excitotoxicity has been the traditionally accepted mechanism [21, 22] and has been in the spotlight for stroke research for decades. Theoretically, inhibition of calcium overload is deemed to be neuroprotective. Consistent with this idea, numerous in vitro and in vivo studies have demonstrated that blocking calcium-mediated glutamate receptor channels during oxygen and glucose deprivation and cerebral ischemia reduces intracellular calcium overload and subsequently ischemic brain damage. Despite overwhelming positive experimental results, subsequent human clinical trials of anti-excitotoxic therapies (AET) did not show the expected therapeutic outcomes [23]. As a result, stroke researchers started probing for alternative pathways instead of the traditional excitotoxicity glutamate mechanism. This effort led to many new studies showing that a nonglutamate mechanism of cerebral ischemia can trigger intracellular ionic imbalance and ischemic neuronal cell death [21, 22]. Therefore, neuronal cell death and ischemic brain damage during cerebral ischemia have been recognized as the result of both glutamate-mediated excitotoxicity mechanisms and the newly identified nonglutamate mechanisms of cerebral ischemia and stroke [21, 22]. The nonglutamate mechanisms of cerebral ischemia are thought to partially explain the unsatisfactory outcomes of previous AET clinical trials. It is therefore critical to investigate these new mechanisms and their therapeutic potential for stroke. As with drug development for the traditional excitotoxicity pathway, new therapies must be developed strictly based on the guidelines set by the Stroke Therapy Academic Industry Roundtable Protocol [24] for testing potential stroke therapies using in vivo models of stroke with multiple species. It is also essential to consider the effects of a potential drug on both the glutamate and nonglutamate mechanisms of stroke. The nonglutamate mechanisms of cerebral ischemia that have emerged in recent years involve ATP-sensitive potassium (KATP) channels [25,26,27,28], transient receptor potential (TRP) channels [29,30,31], volume-regulated anion channels [32], acid-sensing ion channels [33], hemichannels [34], sodium-calcium exchangers [35], or other nonselective cation channels [36]. The sequence of activation of nonglutamate mechanisms must be considered. For instance, KATP channels cause anoxic depolarization at the early stage of ischemic events, followed by traditional glutamate excitotoxicity and calcium overload; after glutamate excitotoxicity, TRPM7 activity can be triggered, and TRPM2 ion channels can be prompted in later phase of cerebral ischemia and initiated during ischemia-induced neuroinflammation, accelerating the devastating ischemia cascade. Thus, in the treatment of stroke, the nonglutamate mechanisms of cerebral ischemia are attractive targets for neuroprotection and bring attention to the related ion channels.
Screening of chemical libraries for new drug targets, including ion channels, is a major strategy in drug discovery. Two important sources of drug leads as screening targets include natural products and synthetic libraries [37], the former of which has been a focus of our group’s chemists. Naturally occurring secondary metabolites produced by organisms have evolved to interact with biomolecules and are well established leads for drug development as potent ion channel blockers [37]. For instance, the development of Prialt® (ziconotide approved by the FDA in 2004), a peptide from the toxin of the cone snail Conus magus, for the management of chronic pain stands as a representative example of a therapeutically important calcium ion channel blocker. Here, we discuss waixenicin A, a potent ion channel blocker targeting TRPM7 that was isolated from the marine soft coral Sarcothelia edmondsoni. The effective in vitro and in vivo activity of waixenicin A gives it great potential as a lead for the development of treatments against brain disorders related to the overactivity of the TRPM7 protein.
TRP channels
TRP channels were initially described in Drosophila photoreceptors [38], as the mutant trp gene initiates a transient response to light [38] and is involved in phototransduction [39, 40]. Located on the cell membrane of various cell types, TRP channels are a superfamily of cation channels [41,42,43]. The family of TRP channels has nearly 30 mammalian homologs [43]. The classification of TRP channels has been based on their homologous sequences, and they have been separated into six subfamilies: (1) TRPC (canonical), (2) TRPV (vanilloid), (3) TRPM (melastatin), (4) TRPA (ankyrin), (5) TRPML (mucolipin), and (6) TRPP (polycystin) channels. Different physical and chemical stimuli can activate different TRP channels, which in turn play various physiological and pathological roles in different cells and tissues [41,42,43].
The melastatin subfamily TRPM channels have eight members, TRPM1 to TRPM8. TRPM7 [41, 44] is broadly expressed in many cell types and tissues, including neurons and the brain. TRPM7 is a bifunctional protein with a channel domain fused to an active kinase domain [44, 45]. The TRPM7 channel pore is selective for divalent cations, such as magnesium and calcium, and trace metal ions with a selectivity profile of Zn2+ ≈ Ni2+ » Ba2+ > Co2+ > Mg2+ ≥ Mn2+ ≥ Sr2+ ≥ Cd2+ ≥ Ca2+ ions [46].
Associated with important functions in physiology and pathophysiology, TRPM7 channel activity can be modulated by distinct intracellular and extracellular factors [47], such as Mg2+- and Mg2+-complexed nucleotides (such as Mg·ATP and Mg·GTP) [48, 49], extracellular pH [50, 51], osmolarity [52], and halides [53]. TRPM7 participates in a diverse range of cell biology, such as cellular and systemic magnesium homeostasis [54, 55] and cell growth, proliferation and adhesion [44, 56]. Overexpression of TRPM7 reduces cell viability [44, 57, 58]. TRPM7 is crucial for embryonic development [59] and skeleton formation [60]. Since TRPM7 channel activity is inversely correlated with the amount of cellular Mg·ATP [48], it was hypothesized [44] and subsequently shown that ischemia activates TRPM7 channels [61]. During ischemic conditions, low concentrations of Mg2+-nucleotides [48] and acidic conditions [51, 62] may thus contribute to TRPM7 activity and lead to ischemic cell death and brain damage.
Under physiological conditions, TRPM7 plays an important role in cell growth and neurite outgrowth [44, 56, 63]. We have recently shown that a specific TRPM7 inhibitor, waixenicin A (Fig. 1), suppressed TRPM7 currents in vitro and enhanced neurite outgrowth and maturation in primary cultures of mouse hippocampal neurons [63].

Structures of various inhibitors of TRPM7 including natural products and compounds from screens: waixenicin A is a selective and potent TRPM7 inhibitor; other non-selective and less potent TRPM7 inhibitors include carvacrol, xyloketal B, spermine, FTY720, sphingosine, AA861, MK886, NS8593 and NDGA. (IC50 = half maximal inhibitory concentration toward TRPM7; ND = not determined).
Under pathophysiological conditions, TRPM7 also plays key roles in ischemia- and/or hypoxia-induced neuronal cell death [29, 30, 61]. Studies on the role of TRPM7 in anoxia and cerebral ischemia were initially conducted using virally mediated shRNA when a specific TRPM7 inhibitor was not available [29, 61]. As a “proof-of-principle” study, we were able to show that suppression of TRPM7 using virally mediated shRNA reduced ischemic neuronal cell death and preserved behavioral outcomes in vivo using a rat model of global ischemia [29]. Using the nonselective TRPM7 inhibitor carvacrol (Fig. 1), we were later able to show that pharmacological inhibition of TRPM7 also reduced neonatal hypoxic–ischemic brain injury in mice and preserved related behavioral outcomes [30]. We also tested another marine compound, xyloketal B [64, 65] (Fig. 1), as an inhibitor of TRPM7-mediated currents and attenuator of mouse neonatal hypoxic–ischemic brain injury and adult brain damage in a focal stroke model. Recently, we were able to demonstrate that waixenicin A, a specific TRPM7 ion channel inhibitor, attenuated hypoxic–ischemic brain injury and preserved both short-term and long-term behavioral outcomes and affected the related TRPM7 signaling in hypoxic–ischemic brain injury [66]. While our data implicated a clear impact of the TRPM7 channel domain in ischemia, recent data confirmed a critical role of the TRPM7 kinase domain in arterial thrombosis and ischemic stroke in vivo [67]. It has been shown that the TRPM7 kinase domain regulates store-operated calcium entry (SOCE) in DT40 B cells in vitro [68]. In a follow-up study, a single point mutation rendering the TRPM7 kinase domain inactive in vivo led to SOCE suppression in mouse platelets through impaired phosphatidylinositol-4,5-bisphosphate (PIP2) signaling, ultimately impairing platelet aggregation [67]. Such genetic impairment of the TRPM7 kinase domain has also been shown to negatively impact SOCE in mouse T lymphocytes in vivo [69]. Notably, it is currently unknown whether waixenicin A interferes with the activity of the TRPM7 kinase in addition to its effect on channel conductance; this is a very real possibility that, if found to be true, would show a broadened impact of this compound as a potential drug in brain injury, including SOCE-mediated inflammatory processes.
Finally, and in relation to TRPM7 and its impact on brain cancer, we were able to show that carvacrol and xyloketal B inhibited TRPM7 currents along with cell survival in glioma (GBM) cell lines in vitro, albeit at quite high concentrations [70, 71]. Furthermore, our most recent data demonstrate that waixenicin A inhibited GBM cell proliferation, migration, and invasion in vitro and inhibited GBM cell functions in vivo, as well as the related signaling (unpublished results).
Thus, we have now demonstrated the potential of the specific TRPM7 inhibitor waixenicin A in three in vitro [63] and three in vivo studies [66]. Therefore, we offer additional details on the promise that waixenicin A may hold in drug development by targeting TRPM7 for brain disorders and diseases.
Development of the TRPM7 inhibitor waixenicin A
Several inhibitors of TRPM7 are now known, including the natural products carvacrol and xyloketal B, which exhibit in vivo activity [30, 64, 65]. Other reported inhibitors of TRPM7 with in vitro potency of <10 μM include spermine, FTY720, sphingosine, docebenone (AA861), MK886, NS8593, and nordihydroguaiaretic acid (NDGA) [72]. Although all of these small molecules are reported to be inhibitors of TRPM7 (Fig. 1), inhibitory concentration curves (IC50) were not determined for some of them, with inhibition reported only at one or two concentrations for xyloketal B, AA861, MK886, and NDGA. Of these TRPM7 inhibitors, FTY720 and sphingosine were effective at IC50 values of 0.7 and 0.6 µM, respectively. The focus here is on the natural product waixenicin A, which has shown the greatest potency in vitro (with an IC50 value as low as 16 nM [73]) and in vivo activity against TRPM7.
Waixenicin A [63, 73] is a marine compound isolated from the Hawaiian soft coral S. edmondsoni. Using our groups’ established TRPM7 medium-throughput cell-based assay measuring TRPM7-mediated Mn2+ quenching of the Fura-2 fluorophore [73], the Horgen and Fleig groups identified an extract of S. edmondsoni as a dose-dependent inhibitor of TRPM7. Following bioassay-directed isolation, they showed that waixenicin A, a xenicane diterpene, inhibits cell proliferation through an Mg2+-dependent blockade of TRPM7 [73]. Subsequent in vivo studies proved that administration of waixenicin A in a cancer mouse model induced hypomagnesemia via insufficient magnesium absorption in the colon [74], showing proof-of-principle of Waixenicin A effectiveness in suppressing TRPM7 activity on a systemic level.
Waixenicin A has been extensively tested on various heterologously overexpressed TRPM channels in HEK293 cells [63, 73] and in primary neurons [63] for its selectivity for TRPM7 inhibition. This natural product demonstrates exceptional selectivity, failing to inhibit a number of related TRPM channels, including its closest homolog, TRPM6 [69], and in fact, it has no effect against zebrafish TRPM7 [75]. Furthermore, waixenicin A has been extensively tested in vitro as an inhibitor of TRPM7 currents and calcium signaling with electrophysiology and calcium imaging, and through IC50 and in vivo dose determinations [63, 66, 73]. Notably, in numerous laboratories around the world, waixenicin A has demonstrated in vitro effects that are consistent with the TRPM7 inhibition of biological effects in broad applications, such as the inhibition of malaria parasite invasion into erythrocytes [76] and a range of anticancer activities [77, 78], reduced hyperglycemia-induced neuronal damage [79], and inhibition of store-operated calcium entry in lymphocytes [68] and neuronal outgrowth [63].
In addition to waixenicin A demonstrating high selectivity against TRPM7 and consistent in vitro efficacy, it shows exceptional in vivo potency for reducing brain injury in mice. The effective in vivo testing dose was 37 µg/kg body weight by intraperitoneal injection using an hypoxic–ischemic brain injury model [66]. This outcome aligns with the high index of waixenicin A for blood brain barrier (BBB) permeation, as determined by a central nervous system multiparameter optimization (CNS MPO) analysis [80], leading to a brain/plasma equilibration rate of −3.1 for this compound, which predicts sufficient BBB penetration for CNS activity. Aside from the excellent predicted BBB properties of waixenicin A, hypoxic and ischemic conditions damage and open the BBB to enable drugs to more easily enter into brain tissue and cells. Finally, the neonatal hypoxic–ischemic brain injury model is based on p7 mouse pups, which are in a developmental stage with an incomplete BBB [81]. Since waixenicin A shows potent and specific inhibitory effects on TRPM7, it is important to determine the structure–activity relationship of waixenicin A and congeners including simplified derivatives as a next step and evaluate active candidates in critical in vivo models, including a neonatal hypoxic–ischemic brain injury model [66], adult middle cerebral artery occlusion model [28], and GBM xenograft models.
Conclusions and future direction
Waixenicin A, a potent and specific TRPM7 inhibitor, holds great promise as a drug lead for further development in therapeutics for brain diseases and disorders. Our group has demonstrated both in vitro and in vivo models showing that waixenicin A promotes neurite outgrowth, reduces brain damage in ischemia and hypoxia, and suppresses GBM cell functions. Thus, TRPM7 is an excellent drug development lead compound, and waixenicin A is an excellent candidate for further biological studies and directed medicinal chemistry research.
References
Dabrowski MA, Dekermendjian K, Lund PE, Krupp JJ, Sinclair J, Larsson O. Ion channel screening technology. Cns Neurol Disord Drug Targets. 2008;7:122–8.
Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, et al. Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem. 2013;56:593–624.
Waszkielewicz AM, Gunia A, Szkaradek N, Sloczynska K, Krupinska S, Marona H. Ion channels as drug targets in central nervous system disorders. Curr Med Chem. 2013;20:1241–85.
Yu HB, Li M, Wang WP, Wang XL. High throughput screening technologies for ion channels. Acta Pharmacol Sin. 2016;37:34–43.
Kaczorowski GJ, McManus OB, Priest BT, Garcia ML. Ion channels as drug targets: the next GPCRs,. J Gen Physiol. 2008;131:399–405.
Carnero A. High throughput screening in drug discovery. Clin Transl Oncol. 2006;8:482–90.
Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19–34.
Penner R, Fasolato C, Hoth M. Calcium influx and its control by calcium release. Curr Opin Neurobiol. 1993;3:368–74.
De LF, Geppetti P. Ion channel pharmacology for pain modulation. Handb Exp Pharmacol. 2019;260:161–86.
Ashcroft FM. The Walter B. Cannon Physiology in Perspective Lecture, 2007. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab. 2007;293:E880–E889.
Zaydman MA, Silva JR, Cui J. Ion channel associated diseases: overview of molecular mechanisms. Chem Rev. 2012;112:6319–33.
WHO. WHO: the top 10 causes of death. WHO; 2016. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e60.
Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–7.
Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–568.
Zivin JA. Acute stroke therapy with tissue plasminogen activator (tPA) since it was approved by the U.S. Food and Drug Administration (FDA). Ann Neurol. 2009;66:6–10.
Stroke. 2000. http://www.strokeassociation.org/STROKEORG/AboutStroke/About-Stroke_UCM_308529_SubHomePage.jsp.
Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–8.
Vannucci RC. Hypoxic-ischemic encephalopathy. Am J Perinatol. 2000;17:113–20.
Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574–84.
Tymianski M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci. 2011;14:1369–73.
Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–75.
Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, et al. Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke. 2000;31:347–54.
Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–8.
Sun HS, Xu B, Chen W, Xiao A, Turlova E, Alibraham A, et al. Neuronal KATP channels mediate hypoxic preconditioning and reduce subsequent neonatal hypoxic-ischemic brain injury. Exp Neurol. 2015;263:161–71.
Sun HS, Feng ZP, Miki T, Seino S, French RJ. Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol. 2006;95:2590–601.
Sun HS, Feng ZP, Barber PA, Buchan AM, French RJ. Kir6.2-containing ATP-sensitive potassium channels protect cortical neurons from ischemic/anoxic injury in vitro and in vivo. Neuroscience. 2007;144:1509–15.
Liu R, Wang H, Xu B, Chen W, Turlova E, Dong N, et al. Cerebrovascular safety of sulfonylureas: the role of KATP channels in neuroprotection and the risk of stroke in patients with type 2 diabetes. Diabetes. 2016;65:2795–809.
Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci. 2009;12:1300–7.
Chen W, Xu B, Xiao A, Liu L, Fang X, Liu R, et al. TRPM7 inhibitor carvacrol protects brain from neonatal hypoxic-ischemic injury. Mol Brain. 2015;8:11.
Alim I, Teves L, Li R, Mori Y, Tymianski M. Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci. 2013;33:17264–77.
Alibrahim A, Zhao LY, Bae CY, Barszczyk A, Sun CLF, Wang GL, et al. Neuroprotective effects of volume-regulated anion channel blocker DCPIB on neonatal hypoxic-ischemic injury. Acta Pharmacol Sin. 2013;34:113–8.
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98.
Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–7.
Pignataro G, Tortiglione A, Scorziello A, Giaccio L, Secondo A, Severino B, et al. Evidence for a protective role played by the Na+/Ca2+ exchanger in cerebral ischemia induced by middle cerebral artery occlusion in male rats. Neuropharmacology. 2004;46:439–48.
Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, et al. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433–40.
Otvos RA, Still KBM, Somsen GW, Smit AB, Kool J. Drug discovery on natural products: from ion channels to nAChRs, from nature to libraries, from analytics to assays. SLAS Discov. 2019;24:362–85.
Minke B, Wu C, Pak WL. Induction of photoreceptor voltage noise in the dark in Drosophila mutant. Nature. 1975;258:84–7.
Montell C, Rubin GM. The Drosophila ninaC locus encodes two photoreceptor cell specific proteins with domains homologous to protein kinases and the myosin heavy chain head. Cell. 1988;52:757–72.
Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–23.
Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–24.
Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–52.
Wu LJ, Sweet TB, Clapham DE. International union of basic and clinical pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010;62:381–404.
Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–5.
Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–7.
Monteilh-Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol. 2003;121:49–60.
Penner R, Fleig A. The Mg2+ and Mg2+-nucleotide-regulated channel-kinase TRPM7. Handb Exp Pharmacol. 2007;179:313–28.
Demeuse P, Penner R, Fleig A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol. 2006;127:421–34.
Takezawa R, Schmitz C, Demeuse P, Scharenberg AM, Penner R, Fleig A. Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain. Proc Natl Acad Sci USA. 2004;101:6009–14.
Jiang J, Li M, Yue L. Potentiation of TRPM7 inward currents by protons. J Gen Physiol. 2005;126:137–50.
Li M, Du J, Jiang J, Ratzan W, Su LT, Runnels LW, et al. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J Biol Chem. 2007;282:25817–30.
Bessac BF, Fleig A. TRPM7 channel is sensitive to osmotic gradients in human kidney cells. J Physiol. 2007;582:1073–86.
Yu H, Zhang Z, Lis A, Penner R, Fleig A. TRPM7 is regulated by halides through its kinase domain. Cell Mol Life Sci. 2013;70:2757–71.
Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, et al. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200.
Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, et al. TRPM7 is essential for Mg2+ homeostasis in mammals. Nat Commun. 2010;1:109.
Inoue K, Xiong ZG. Silencing TRPM7 promotes growth/proliferation and nitric oxide production of vascular endothelial cells via the ERK pathway. Cardiovasc Res. 2009;83:547–57.
Chen HC, Xie J, Zhang Z, Su LT, Yue L, Runnels LW. Blockade of TRPM7 channel activity and cell death by inhibitors of 5-lipoxygenase. PLoS ONE. 2010;5:e11161.
Su LT, Agapito MA, Li M, Simonson WT, Huttenlocher A, Habas R, et al. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J Biol Chem. 2006;281:11260–70.
Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–60.
Elizondo MR, Arduini BL, Paulsen J, MacDonald EL, Sabel JL, Henion PD, et al. Defective skeletogenesis with kidney stone formation in dwarf zebrafish mutant for trpm7. Curr Biol. 2005;15:667–71.
Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–77.
Rehncrona S. Brain acidosis. Ann Emerg Med. 1985;14:770–6.
Turlova E, Bae CY, Deurloo M, Chen W, Barszczyk A, Horgen FD, et al. TRPM7 regulates axonal outgrowth and maturation of primary hippocampal neurons. Mol Neurobiol. 2016;53:595–610.
Xiao AJ, Chen W, Xu B, Liu R, Turlova E, Barszczyk A, et al. Marine compound xyloketal B reduces neonatal hypoxic-ischemic brain injury. Mar Drugs. 2015;13:29–47.
Pan N, Lu LY, Li M, Wang GH, Sun FY, Sun HS, et al. Xyloketal B alleviates cerebral infarction and neurologic deficits in a mouse stroke model by suppressing the ROS/TLR4/NF-kappaB inflammatory signaling pathway. Acta Pharmacol Sin 2017;38:1236–47.
Turlova E, Wong R, Xu B, Li F, Du L, Habbous S, et al. TRPM7 mediates neuronal cell death upstream of calcium/calmodulin-dependent protein kinase II and calcineurin mechanism in neonatal hypoxic-ischemic brain injury. Transl Stroke Res. 2020. https://doi.org/10.1007/s12975-020-00810-3.
Gotru SK, Chen W, Kraft P, Becker IC, Wolf K, Stritt S, et al. TRPM7 kinase controls calcium responses in arterial thrombosis and stroke in mice. Arterioscler Thromb Vasc Biol. 2018;38:344–52.
Faouzi M, Kilch T, Horgen FD, Fleig A, Penner R. The TRPM7 channel kinase regulates store-operated calcium entry. J Physiol. 2017;595:3165–80.
Beesetty P, Wieczerzak KB, Gibson JN, Kaitsuka T, Luu CT, Matsushita M, et al. Inactivation of TRPM7 kinase in mice results in enlarged spleens, reduced T-cell proliferation and diminished store-operated calcium entry. Sci Rep. 2018;8:3023.
Chen WL, Barszczyk A, Turlova E, Deurloo M, Liu B, Yang BB, et al. Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget. 2015;6:16321–40.
Chen WL, Turlova E, Sun CL, Kim JS, Huang S, Zhong X, et al. Xyloketal B suppresses glioblastoma cell proliferation and migration in vitro through inhibiting TRPM7-regulated PI3K/Akt and MEK/ERK signaling pathways. Mar Drugs. 2015;13:2505–25.
Chubanov V, Schafer S, Ferioli S, Gudermann T. Natural and synthetic modulators of the TRPM7 channel. Cells. 2014;3:1089–101.
Zierler S, Yao G, Zhang Z, Kuo WC, Porzgen P, Penner R, et al. Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J Biol Chem. 2011;286:39328–35.
Huang J, Furuya H, Faouzi M, Zhang Z, Monteilh-Zoller M, Kawabata KG, et al. Inhibition of TRPM7 suppresses cell proliferation of colon adenocarcinoma in vitro and induces hypomagnesemia in vivo without affecting azoxymethane-induced early colon cancer in mice. Cell Commun Signal. 2017;15:30.
Jansen C, Sahni J, Suzuki S, Horgen FD, Penner R, Fleig A. The coiled-coil domain of zebrafish TRPM7 regulates Mg nucleotide sensitivity. Sci Rep. 2016;6:33459.
Sisquella X, Nebl T, Thompson JK, Whitehead L, Malpede BM, Salinas ND, et al. Plasmodium falciparum ligand binding to erythrocytes induce alterations in deformability essential for invasion. Elife. 2017;6:e21083.
Liu H, Dilger JP, Lin J. The role of transient receptor potential melastatin 7 (TRPM7) in cell viability: a potential target to suppress breast cancer cell cycle. Cancers. 2020;12:131.
Visser D, Langeslag M, Kedziora KM, Klarenbeek J, Kamermans A, Horgen FD, et al. TRPM7 triggers Ca2+ sparks and invadosome formation in neuroblastoma cells. Cell Calcium. 2013;54:404–15.
Huang Y, Leng TD, Inoue K, Yang T, Liu M, Horgen FD, et al. TRPM7 channels play a role in high glucose-induced endoplasmic reticulum stress and neuronal cell apoptosis. J Biol Chem. 2018;293:14393–406.
Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1:435–49.
Ek CJ, D’Angelo B, Baburamani AA, Lehner C, Leverin AL, Smith PL, et al. Brain barrier properties and cerebral blood flow in neonatal mice exposed to cerebral hypoxia-ischemia. J Cereb Blood Flow Metab. 2015;35:818–27.
Acknowledgements
This paper was supported by the following grants: NIH NIGMS P20 (GM103466) to FDH; DR acknowledges support by Baylor University for start-up funds to initiate this project and partial support by NIH NIGMS MERIT (GM052964) and MIRA (GM134910); Hamamatsu/Queen’s PET Imaging, LLC to AF; Canadian Institutes of Health Research (CIHR PJT-153155) to ZPF; Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants (RGPIN-2016-04574) to HSS.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Sun, HS., Horgen, F.D., Romo, D. et al. Waixenicin A, a marine-derived TRPM7 inhibitor: a promising CNS drug lead. Acta Pharmacol Sin 41, 1519–1524 (2020). https://doi.org/10.1038/s41401-020-00512-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00512-4
