
Abstract
The immune system plays an essential and central role in tumor cell differentiation, proliferation, angiogenesis, apoptosis, invasion, and metastasis. Over the past decade, cancer therapy has rapidly evolved from traditional approaches, such as surgery, chemotherapy, and radiotherapy, to revolutionary new treatment options with immunotherapy. This new era of cancer treatment options has now been clinically tested and applied to many forms of human malignancies, often with quite dramatic results. As we develop more effective combinations of cancer treatment, several agents have been recently investigated, putatively identified as anticancer agents, or immunostimulatory molecules. One such agent is metformin, originally developed as a fairly standard first-line therapy for patients with type-2 diabetes mellitus (T2DM). Given the underlying mechanisms of action, researchers began to examine the alternative functions and possible utility of metformin, finding that the cancer risk in patients with T2DM was reduced. It appears that metformin, at least in part, has an antitumor effect through activation of the 5’ adenosine monophosphate-activated protein kinase (AMPK) signaling pathway. Moreover, numerous studies have demonstrated that metformin interferes with key immunopathological mechanisms that are involved in the pathological processes or associated with malignant progression. Such insights may shed light on further analyzing whether metformin enhances the effectiveness of the immunotherapy and overcomes the immunotherapy resistance in the patients. Herein, we provide a comprehensive review of the literature examining the impact of metformin upon the host immune system and cancer immunity.
Similar content being viewed by others

Introduction
Worldwide, there are 18.1 million new cancer cases and 9.6 million cancer deaths every year [1]. In the United States alone, in 2020, the incidence of new cases for all types of cancer is 1,806,590, with 606,520 deaths [2]. Cancers of all types are ranked as either the first or second most common cause of premature death in almost 100 countries worldwide. We have begun to understand the critical role of the human immune system in protecting us not only from foreign pathogens but also from cancer in terms of cancer prevention, development, and metastasis [3, 4]. The pathophysiology of a neoplasm begins with the establishment of a protumor and immunosuppressive microenvironment that supports growth, metastatic potential, and immune escape mechanisms.
In general, tumor cells can give rise to nondividing cells that form part of the tumor microenvironment and secrete proteins and growth factors that support tumor cell growth and progression. The tumor immune microenvironment (TIME) represents a complex network of cellular mechanisms mediated by both tumor cells and host immune cells, with the capacity to predict overall immunologic responsiveness to cancer. This phenomenon occurs not only at the primary tumor site but also in regional and distant sites of metastatic disease. The latest advances in molecular and cellular tumor immunology have demonstrated that cancer cells can actively recruit and alter immune cell phenotypes, as well as cellular functions, promoting either immune suppression or tolerance of tumor-associated antigens [5].
Recently, we have experienced nothing short of a revolution in regards to the development of efficacious cancer treatments, especially in the area of new cancer immunotherapy with immune checkpoint inhibitors (ICIs) [6]. Derived from ICI research, some agents have also been demonstrated to modulate cancer immunity and enhance ICI immunotherapy. One of these agents, metformin, also known as 1,1-dimethyl biguanide hydrochloride, is an FDA-approved drug to treat type-2 diabetes mellitus (T2DM) in the clinic. With a well-established mechanism of action via the activation of 5’ adenosine monophosphate-activated protein kinase (AMPK), metformin regulates cellular energy metabolism, surprisingly resulting in a reduction in incidence and mortality for several types of cancer [7, 8]. For instance, in breast cancer patients, treatment with neoadjuvant chemotherapy in combination with metformin appears to increase the rate of complete pathological response to therapy [9]. Intriguingly, metformin was also found to boost the immune system and increase the potency of cancer treatment, although the molecular mechanisms involved in this effect have not been fully understood [10, 11]. In this review, we outline the immune features of metformin and its potential effects on immune systems, including its defining role as an anticancer agent and in preventing the development of cancer.
Mechanism of action of the antidiabetic and anticancer activities of metformin
Metformin is one of the first-line treatments for T2DM and exerts its antidiabetic effects by suppressing hepatic gluconeogenesis. The widely accepted mechanism of action for metformin is the stimulation of AMPK. Inhibiting mitochondrial respiratory chain complex I by metformin can activate AMPK by reducing ATP production, leading to an increase in the AMP:ATP ratio [12, 13]. AMPK activation can not only suppress the transcription of gluconeogenic genes but also inhibit lipogenesis and improve insulin sensitivity [12, 13]. Deactivating the mammalian target of rapamycin (mTOR) signaling is one of the dominant outcomes of the AMPK-dependent action of metformin, and mTOR is often regarded as an essential regulatory mechanism to control cell growth and proliferation in both diabetes and cancer [14,15,16,17]. Recently, Kalender et al. reported that mTOR complex 1 (mTORC1) could also be targeted by metformin in an AMPK-independent mechanism involving the inhibition of Rag GTPases in two distinct preclinical models of cancer and diabetes [18].
Reduced reactive oxygen species (ROS) is another AMPK-independent mechanism accounting for the activity of metformin. Algire et al. reported that metformin could reduce ROS production to protect cells from DNA damage and mutagenesis [19]. Nguyen et al. further demonstrated that metformin could diminish lithocholic acid (LCA)-stimulated ROS production, which in turn blocked the nuclear factor kappa B (NF-κB) signaling that was critical for upregulating interleukin (IL)-8 in human colorectal cancer (CRC) HCT116 cells [20]. Furthermore, metformin can target hexokinase (HK) in breast cancer cells [21], which is an essential enzyme for glycolysis that catalyzes the phosphorylation of glucose by ATP to glucose-6-phosphate (G6P). In the breast cancer MDA-MB-231 model, metformin was found to inhibit HK activity, which in turn, partially impaired glucose metabolism and tumor growth [21]. Recently, DeWaal et al. reported that metformin treatment in hepatocellular carcinoma (HCC) cells with HK-2 deletion could effectively inhibit mTORC1 through an AMPK-independent mechanism involving a gene called regulated in development and DNA damage responses 1 (REDD1) [22]. Ben Sahra et al. reported that REDD1 was a negative regulator of mTOR and a molecular target of metformin involved in cell cycle arrest [23]. They also found that cyclin D1 was an alternative target of metformin in the regulation of the cell cycle [24]. Transforming growth factor-β (TGF-β) has been shown to be involved in the pathogenesis of numerous human diseases, including a variety of cancers. Xiao et al. demonstrated that metformin could directly influence the dimerization of type II TGF-β1 receptor and block the binding of TGF-β1, effectively reducing TGF-β oncogenic signal transduction [25]. These mechanisms provide novel insights into the AMPK-independent antidiabetic and anticancer activities of metformin.
Mitochondrial respiratory chain complex 1, known as NADH:ubiquinone oxidoreductase, was also reported as a vital target of metformin [12, 26]. Metformin can suppress NADH oxidation, the electron transport chain (ETC), and ATP production, leading to an increase in the AMP:ATP ratio. In turn, AMPK is activated, resulting in the inhibition of fructose-1,6-bisphosphatase (FBP1), an essential gluconeogenic enzyme involved in the process of gluconeogenesis [13]. Metformin can target mitochondrial glycerophosphate dehydrogenase (mGPDH) and impair the production of dihydroxyacetone phosphate (DHAP), leading to a decrease in gluconeogenesis from glycerol [27]. Furthermore, Madiraju et al. reported that the cytosolic redox state promoted by mGPDH inhibition could suppress the activity of the redox-dependent enzyme lactate dehydrogenase (LDH), resulting in the reduction of pyruvate production and gluconeogenesis from lactate [27]. They also demonstrated that metformin could suppress hepatic gluconeogenesis in vivo in a redox-dependent manner, independent of reductions in citrate synthase flux, hepatic nucleotide concentrations, acetyl-CoA carboxylase activity, and gluconeogenic enzyme protein expression [28]. Alshawi et al. reported that a low dose of metformin could cause a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibit gluconeogenesis by a redox-independent mechanism. They found that a compromised malate-aspartate shuttle and altered allosteric effectors of phosphofructokinase-1 (PFK1) and FBP1 could attenuate trans-mitochondrial electrogenic transport mechanisms, leading to changes in the lactate/pyruvate ratio and glucose production [29].
Overall, both AMPK-dependent and AMPK-independent pathways contribute to the mechanism of action of the antidiabetic and anticancer activities of metformin.
Tumor microenvironment, immune cells, and cancer progression
The tumor microenvironment (TME) is defined as a complex with the tumor mass at the center surrounded by fibroblasts, blood vessels, immune and inflammatory cells, adipose cells, neuroendocrine cells, and the extracellular matrix (ECM) [30]. In the TME, immune cells have been recognized as having a dominant influence and control over tumorigenesis, immune tolerance, and escape. Multiple immune cell types, including neutrophils, macrophages, dendritic cells (DCs), natural killer (NK) cells, and T- and B-lymphocytes, have been shown to infiltrate the tumor and actively participate in the modulation of the TME.
The infiltration of immune cells, such as T-lymphocytes, has been shown to predict patient outcomes for many solid tumor types following treatment with different immunotherapy approaches [31]. In many instances, clinically detectable cancer has likely evaded the host immune system, continuing to grow unabated over time [32]. The tumor escape phenomena fall into two broad categories, one based on cellular characteristics and the other based on molecular characteristics of the TME [32]. The growth of a cancer cell is quite complicated and comprises an interdependency upon cells, adjacent stromal tissue, and other growth and inhibitory factors that ultimately determine the outcome of tumor growth, progression, or metastasis [33].
Although neutrophils were initially thought to play a critical role in immunosurveillance, recent evidence suggests that a population of neutrophils, called tumor-associated neutrophils (TANs), has tumor-supportive functions [34]. Under certain conditions within the TME, immature and mature neutrophils undergo proliferative expansion, inducing the suppression of T-cell proliferation [35]. These suppressing cells are known as myeloid-derived suppressor cells (MDSCs), which consist of two major subpopulations: monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs). The discovery of MDSCs is quite important, along with the recognition of their crucial role in determining the ultimate outcome of tumors, either growth or regression, within the TME [36]. One of the main features of MDSCs is their potent immunosuppressive activity. Initially generated within the bone marrow of tumor-bearing hosts, MDSCs eventually leave the bone marrow and migrate to the peripheral lymphoid organs, as well as the established tumor, suppressing the T-cell response even with immunotherapy.
Tumor-associated macrophages (TAMs) represent a class of robust regulators of the complex interplay between the immune system and cancer. Macrophages are one of the most common immune cells within the TME of solid tumors, and their presence correlates with the survival of cancer patients [37]. TAMs can be found at all stages of tumor progression and can stimulate angiogenesis and tumor cell invasion/migration from the primary site. TAMs and monocytes can facilitate tumor cell motility and survival by suppressing immune surveillance and support the formation of a premetastatic niche at distant metastatic sites where TAMs can promote the growth of disseminated tumor cells [37]. The development of agents that are capable of inhibiting the recruitment of macrophages is regarded as a promising anticancer strategy in the future.
The identification of a “cancer-immunity cycle” was first hypothesized and developed by Chen and Mellman [38]. In this seminal article, the authors referred to a cycle consisting of seven major steps, starting with neoantigen release, presentation, priming and activation, T-lymphocyte trafficking, infiltration, recognition of cancer cells, and ending with the killing of cancer cells by cytotoxic T-lymphocytes (CTLs). Therefore, cancer-directed immunotherapy has focused on a specific antitumor CTL response, recognizing the potent cytotoxicity of CD8+ T-lymphocytes on established tumors [39].
Immunity to cancer is a cyclic process that can be self-propagating, leading to an accumulation of immune-stimulatory factors that amplify and broaden T-cell responses. The most clinically effective immunotherapies to date, in the form of ICIs, target the programmed cell death protein 1 (PD-1) receptor or its ligand, PD-L1 [40,41,42,43]. However, only select tumor types and cancer patients have benefited from ICI therapies. Therefore, it is of utmost importance to understand the interactions of cancer cells with different immune cells and the mechasnim how they form a complement system as an essential component of innate immunity to regulate cancer immunity [44]. For instance, the complement factors, C1Q, anaphylatoxins C3a and C5a, and their associated receptors, C3aR and C5aR1, have been reported to be involved in tolerogenic cell death and inhibition of antitumor T-cell responses functionally via the recruitment of several immunosuppressive cell populations, such as MDSCs, regulatory T-cells (T-regs), and TAMs, as previously discussed [44].
The initial identification of NK cells showed that these lymphocytes were readily capable of killing tumor cells without specific immunization or activation [45]. Subsequently, it was shown that NK cells could also kill cells that were infected with certain viruses, as well as preferentially, and nonselectively, attacking cells that lack the expression of major histocompatibility complex (MHC) class I antigens [46]. The cytotoxic ability of NK cells can be enhanced by cytokines, specifically IL-2 and IL-12 [47, 48]. After activation, NK cells are able to release cytokines and chemokines to induce inflammatory responses and modulate the growth and differentiation of monocytes, DCs, and granulocytes, which further influence subsequent adaptive immune responses [46,47,48].
While T-cells remain a critical component of an effective antitumor response, NK cells are one of the “first responders” to the scene, even before other T-cell subsets arrive. It is NK cells that possess the innate ability to detect transformed cells, and thus, are the key to cancer immunosurveillance and antitumor immunity, particularly in hematological cancers, to potentially minimize the chance for metastatic dissemination of tumor cells [49]. Therefore, NK cells display a rapid and potent antitumor immunity, with major studies currently being undertaken to fully exploit NK cell antitumor properties in the clinical setting [50].
The clinical utility of metformin as an immune modulator to enhance immunotherapy
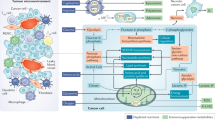
Cancer immunotherapy enhances the immune system’s ability to recognize, target, and eliminate tumor cells, wherever they may reside in the body. It has been demonstrated that metformin has antitumor effects and can inhibit tumor growth and development in many types of cancer, including colon, prostate, liver, and breast cancer [7, 51,52,53,54,55]. The underlying mechanism of action has been extensively examined, with the activation of the AMPK-dependent and AMPK-independent signaling pathways resulting in a preventive effect on tumor cells, which also gives them the necessary time to reverse the aberrant AMP:ATP ratio for protecting cells against early cell death. These pathways represent both upstream and downstream networks that govern cell growth in response to the microenvironments in diabetes and cancer. It is likely that the real anticancer effects of metformin may be related to its involvement in host immunity and inflammation, as shown in Fig. 1. Thus, we hope to describe the role of metformin in antitumor immunity and its potential as a component of combination immunotherapy for the treatment of a variety of human cancers [11, 56,57,58,59]. Table 1 lists the studies that have utilized a combination of metformin and anti-PD-1/PD-L1/CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) in both clinical and preclinical settings.

AMPK: 5’ adenosine monophosphate-activated protein kinase; BECN1: beclin1; COX2: cyclooxygenase 2; CTLs: cytotoxic T-lymphocytes; CXCL1: C-X-C motif chemokine ligand 1; DACH1: dachshund family transcription factor 1; ERAD: ER-associated protein degradation; Eomes: eomesodermin; MDSCs: myeloid-derived suppressor cells; MICA: major histocompatibility complex class I related chain A; mTOR: mammalian target of rapamycin; NF-κB: nuclear factor kappa B; NK cells: natural killer cells; NLRP3: NOD-like receptor protein 3; PD-1: programmed cell death protein 1; PD-L1: programmed cell death protein 1 ligand 1; PGE2: prostaglandin E2; ROS: reactive oxygen species; TAMs: tumor-associated macrophages.
Metformin enhances the antitumor effect of CTLs
Solid tumors are able to establish, sustain, and grow within the TME, creating an immunosuppressive milieu that favors immune escape and is not recognized by the host immune system [60]. The effectiveness of ICIs has changed the treatment landscape for many cancers, with researchers worldwide now examining in more detail the pathobiology and immunology of such dramatic responses to treatment. One mechanism, referred to as immune exhaustion, describes the stepwise and progressive loss of functions and proliferative capacity of CTLs [61]. One hallmark of exhausted CTLs is the increased surface expression of checkpoint proteins, such as PD-1 and CTLA-4, that can bind with PD-L1, which is dominantly expressed on the surface of tumor cells, leading to suppression of the CTL response and tumor immune resistance [62].
Metformin has been reported to possess both antitumor activities and the ability to help maintain a threshold of immunosurveillance CTLs [63]. Metformin interacts with CD8+ tumor-infiltrating lymphocytes (TILs), preventing them from progressing to apoptotic cell death. Cha et al. elegantly showed that metformin interacts with CTL activation as a critical player in cancer immunity, partly resulting in a reduction in PD-L1 stability and membrane localization [57]. Of significance, they demonstrated that AMPK, after activation by metformin, could directly result in the phosphorylation of PD-L1, which in turn led to the accumulation of PD-L1 within the endoplasmic reticulum (ER) with ER-associated protein degradation (ERAD), and then PD-L1 was found to be abnormally glycosylated and degraded. Therefore, blocking the inhibitory signal of PD-L1 with metformin can enhance CTL activity against cancer cells [57]. In addition, in the syngeneic animal models of breast cancer (4T1), melanoma (B16F10), and CRC (CT26), the combination of metformin and anti-CTLA-4 therapy displayed robust improvement in tumor burden, survival rate, and CTL activity, but no significant toxicity was detected [57]. A recent study also found that metformin could facilitate the antitumor activity of CD8+ T-lymphocytes in lung cancer [64]. The molecular mechanism involves AMPK activation, which decreases miR-107 expression, thus enhancing the expression of eomesodermin and suppressing the transcription of the PD-1 gene in metformin-treated CD8+ T-lymphocytes [64].
Metformin affects macrophage polarization
The activation state of macrophages can be categorized into two main subsets, classically-activated M1 and alternatively-activated M2 [65]. For most cancers, TAMs resemble the M2-like subset. As they tend to enhance tumor growth through the production of specific cytokines that subsequently downregulate the antitumor immune responses, TAMs are associated with the development and poor prognosis in several types of cancers, such as colon, breast, lung, ovarian, lymphoma, glioblastoma, liver, and kidney [66,67,68,69,70,71,72]. Ding et al. revealed that the treatment of lung cancer cells with metformin in vitro increases the expression of M1-related cytokines and attenuates M2-related cytokine expression [73]. Furthermore, others have found that the AMPK-NF-κB signaling is involved with the regulation of M1- and M2-related cytokine expression-inducing genes for macrophage polarization towards an antitumor phenotype [74]. Wang et al. reported that, by skewing TAM polarization from an M2- to an M1-like phenotype, metformin inhibited both tumor growth and angiogenesis in vivo [75]. Given its importance in cancer immunity, TAM polarization could offer a novel therapeutic opportunity for the addition of metformin as part of an immunotherapy regimen.
Metformin has also been shown to improve wound healing and enhance angiogenesis, providing an additional anti-inflammatory effect through the regulation of the AMPK/mTOR/NLRP3 inflammasome signaling axis in which NLRP3 (NOD-like receptor protein 3) can boost M2 macrophage polarization to accelerate wound healing [76]. Therefore, by regulating the AMPK/mTOR signaling pathway to inhibit NLRP3 inflammasome activation, metformin is able to inhibit cancer development potentially [76]. In addition, metformin was also demonstrated to induce a significant reduction in ROS levels in CD11+ MDSCs and TAMs in tumors [77], and inhibit the progression of prostate cancer by blocking the infiltration of TAMs through the suppression of the COX2/PGE2 axis, suggesting that combination therapy with metformin could be a more efficient method of treatment [78].
Metformin suppresses MDSCs activity
MDSCs induce an immunosuppressive TME that, in essence, protects tumor cells from immune recognition and elimination by the host immune system. Studies have shown that MDSCs suppress cell immunity through the enzymatic activity of CD39/CD73 in mouse models [79, 80]. It was reported that metformin was involved in the regulation of human MDSC-mediated immunosuppression, both in vitro and in vivo, mainly through AMPK activation with subsequent induction of MDSC activation. Metformin treatment was demonstrated to inhibit C-X-C motif chemokine ligand 1 (CXCL1) secretion in esophageal squamous cell carcinoma (ESCC) cells and tumor xenografts via AMPK phosphorylation to upregulate dachshund homolog 1 (DACH1), resulting in a reduction of MDSC migration [81]. These data, as well as other related research, show that metformin plays an integral role as an antitumor agent by reducing PMN-MDSC accumulation in the TME via the AMPK/DACH1/CXCL1 axis [81]. Additionally, metformin is capable of downregulating the gene expression of CD39 and CD73, thereby inhibiting their activity in MDSCs through the activation of AMPK-α and downregulation of gene expression in the hypoxia-inducible factor 1-alpha (HIF-1α) pathway, ultimately resulting in an overall decrease in MDSC suppressive activity [82].
Metformin facilitates NK-cell activation
NK cells are critical for immunosurveillance and are especially sensitive to metastatic cells or certain hematological cancers [49]. The latest study reported that metformin in combination with nelfinavir, an antiviral drug for treating HIV infection, could induce NAD-dependent deacetylase sirtuin-3 (SIRT3) and mitochondrial ROS (mROS)-dependent autophagy and sensitization to NK cell-mediated lysis in cervical cancer cells by upregulating major histocompatibility complex class I related chain A (MICA) and downregulating beclin-1 (BECN1) [83]. MICA is a natural ligand of NK cells, and BECN1 is a documented marker of autophagy. Recently, Mgrditchian et al. demonstrated that targeting BECN1 could inhibit melanoma tumor growth by inducing a massive infiltration of functional NK cells into the TME of melanoma [84].
In addition to affecting immune cells, metformin was reported to inhibit angiogenesis by downregulating the expression of vascular endothelial growth factor (VEGF) [75] and suppressing HIF-1α-induced expression of angiogenesis-associated factors (AAFs) [85]. These findings are complemented by others that demonstrated the ability of metformin to inhibit HER2-induced breast tumor angiogenesis by triggering the HIF-1α/VEGF signaling axis [86]. Cancer- or tumor-associated fibroblasts (CAFs or TAFs) are the most abundant and critical components of stromal cells in the TME [87]. In ovarian cancer, metformin was found to reduce the stromal fibroblast-derived IL-6, which was relevant to chemoresistance to cisplatin, and the mechanism of action involved metformin blocking the NF-κB signaling that regulated IL-6 at the transcriptional level [88].
In gastric cancer (GC) cells, metformin was reported to selectively target calmodulin‑like protein 3 (Calml3) that was secreted by TAFs, leading to suppression of GC progression [89]. Metformin was also reported to trigger ECM in the TME. Shen et al. found that metformin could inhibit Ang II-induced ECM production in renal fibroblasts [90]. In addition, Incio et al. demonstrated that in prostate cancer, metformin could reduce desmoplasia in preclinical animal models, with the mechanism of action involving a reduction of ECM components, specifically hyaluronan (HA) [91]. Matrix metalloproteinases (MMPs) play a dominant role in the proteolysis of ECM components, with Hwang et al. finding that the inhibitory effect of metformin on MMP-9 could address the antimetastatic activity of metformin in human fibrosarcoma cells [92].
Conclusion and future perspectives
The proper activation of the human immune system in response to the transformation, establishment, and growth of cancer is both complex and not well understood. We continue to explore and learn more about the interactions between specific immune cell subsets, the TME, and the responses of both to combination immunotherapy [44]. We have many examples of the robust efficacy of various monotherapy and combination immunotherapy regimens that have proven clinical efficacy for a variety of cancers, such as non-small-cell lung cancer, bladder cancer, brain cancer, breast cancer, renal cancer, colorectal cancer, lymphoma, ovarian cancer, prostate cancer, and gastric cancer [93,94,95,96,97,98].
During this time of intense research into optimizing our treatment approaches for cancer patients, metformin has been identified as a potentially efficacious antitumor agent in its own right. Metformin likely acts in a more collaborative manner with other immunotherapeutic agents involved in tumor elimination [11, 56,57,58,59]. It clearly works through many cellular and molecular mechanisms, with one of the main antitumor effects elicited through activation of AMPK-dependent and AMPK-independent pathways (Fig. 1). There are very likely to be many other mechanisms by which metformin impacts the tumor response of host immune cells. These unrecognized and undefined interactions of metformin will require a more in-depth understanding of the tumor milieu and microenvironment as it relates to cancer immunity. Last, it appears that nontraditional treatments and approaches to cancer will play a more significant role in the future of cancer immunity.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Candeias SM, Gaipl US. The immune system in cancer prevention, development and therapy. Anticancer Agents Med Chem. 2016;16:101–7.
Janssen LME, Ramsay EE, Logsdon CD, Overwijk WW. The immune system in cancer metastasis: friend or foe? J Immunother Cancer. 2017;5:79. https://doi.org/10.1186/s40425-017-0283-9.
Gun SY, Lee SWL, Sieow JL, Wong SC. Targeting immune cells for cancer therapy. Redox Biol. 2019;25:101174. https://doi.org/10.1016/j.redox.2019.101174.
Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. 2018;9:1739.
Lee MS, Hsu CC, Wahlqvist ML, Tsai HN, Chang YH, Huang YC. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: a representative population prospective cohort study of 800,000 individuals. BMC Cancer. 2011;11:20. https://doi.org/10.1186/1471-2407-11-20.
Zhang P, Li H, Tan X, Chen L, Wang S. Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol. 2013;37:207–18.
Martin-Castillo B, Pernas S, Dorca J, Alvarez I, Martinez S, Perez-Garcia JM, et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: the METTEN study. Oncotarget. 2018;9:35687–704.
Pereira FV, Melo ACL, Low JS, de Castro IA, Braga TT, Almeida DC, et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget. 2018;9:25808–25.
Afzal MZ, Mercado RR, Shirai K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J Immunother Cancer. 2018;6:64. https://doi.org/10.1186/s40425-018-0375-1.
Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its antidiabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–14.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62.
Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–23.
Zachariah Tom R, Garcia-Roves PM, Sjogren RJ, Jiang LQ, Holmstrom MH, Deshmukh AS, et al. Effects of AMPK activation on insulin sensitivity and metabolism in leptin-deficient ob/ob mice. Diabetes. 2014;63:1560–71.
Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf). 2009;196:65–80.
Xu J, Ji J, Yan XH. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr. 2012;52:373–81.
Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401.
Algire C, Moiseeva O, Deschenes-Simard X, Amrein L, Petruccelli L, Birman E, et al. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev Res (Philos). 2012;5:536–43.
Nguyen TT, Ung TT, Li S, Lian S, Xia Y, Park SY, et al. Metformin inhibits lithocholic acid-induced interleukin 8 upregulation in colorectal cancer cells by suppressing ROS production and NF-kB activity. Sci Rep. 2019;9:2003.
Marini C, Salani B, Massollo M, Amaro A, Esposito AI, Orengo AM, et al. Direct inhibition of hexokinase activity by metformin at least partially impairs glucose metabolism and tumor growth in experimental breast cancer. Cell Cycle. 2013;12:3490–9.
DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9:446. https://doi.org/10.1038/s41467-017-02733-4.
Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–72.
Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86.
Xiao H, Zhang J, Xu Z, Feng Y, Zhang M, Liu J, et al. Metformin is a novel suppressor for transforming growth factor (TGF)-beta1. Sci Rep. 2016;6:28597.
El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–8.
Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–6.
Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang XM, Zhang D, et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat Med. 2018;24:1384–94.
Alshawi A, Agius L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J Biol Chem. 2019;294:2839–53.
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8:761–73.
Liu M, Guo F. Recent updates on cancer immunotherapy. Precis Clin Med. 2018;1:65–74.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Mizuno R, Kawada K, Itatani Y, Ogawa R, Kiyasu Y, Sakai Y. The role of tumor-associated neutrophils in colorectal cancer. Int J Mol Sci. 2019;20:529.
Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16:601–20.
Tesi RJ. MDSC; the most important cell you have never heard of. Trends Pharmacol Sci. 2019;40:4–7.
Nielsen SR, Schmid MC. Macrophages as key drivers of cancer progression and metastasis. Mediators Inflamm. 2017;2017:9624760. https://doi.org/10.1155/2017/9624760.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–8.
Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. https://doi.org/10.3389/fphar.2017.00561.
Asaoka Y, Ijichi H, Koike K. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;373:1979. https://doi.org/10.1056/NEJMc1510353.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71.
Pio R, Ajona D, Ortiz-Espinosa S, Mantovani A, Lambris JD. Complementing the cancer-immunity cycle. Front Immunol. 2019;10:774.
Wu J, Lanier LL. Natural killer cells and cancer. Adv Cancer Res. 2003;90:127–56.
Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. 2017;8:1124.
Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol. 2017;8:930. https://doi.org/10.3389/fimmu.2017.00930.
Lehmann C, Zeis M, Uharek L. Activation of natural killer cells with interleukin 2 (IL-2) and IL-12 increases perforin binding and subsequent lysis of tumour cells. Br J Haematol. 2001;114:660–5.
Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. 2019;40:142–58.
Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–36.
Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70:2465–75.
Lee JH, Kim TI, Jeon SM, Hong SP, Cheon JH, Kim WH. The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. Int J Cancer. 2012;131:752–9.
Zhang ZJ, Zheng ZJ, Shi R, Su Q, Jiang Q, Kip KE. Metformin for liver cancer prevention in patients with type 2 diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97:2347–53.
Tang GH, Satkunam M, Pond GR, Steinberg GR, Blandino G, Schunemann HJ, et al. Association of metformin with breast cancer incidence and mortality in patients with type II diabetes: a GRADE-Assessed systematic review and meta-analysis. Cancer Epidemiol Biomark Prev. 2018;27:627–35.
Zhang ZJ, Yuan J, Bi Y, Wang C, Liu Y. The effect of metformin on biomarkers and survivals for breast cancer- a systematic review and meta-analysis of randomized clinical trials. Pharmacol Res. 2019;141:551–5.
Afzal MZ, Dragnev K, Sarwar T, Shirai K. Clinical outcomes in non-small-cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung Cancer Manag. 2019;8:LMT11.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–20. e7.
Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. 2017;5:9–16.
Shen X, Zhao Y, Liu G, Zhou HL, Fan J, Zhang L, et al. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci. 2020;256:117923.
Noman MZ, Berchem G, Janji B. Targeting autophagy blocks melanoma growth by bringing natural killer cells to the tumor battlefield. Autophagy. 2018;14:730–2.
Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. 2010;129:474–81.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99.
Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015;112:1809–14.
Zhang Z, Li F, Tian Y, Cao L, Gao Q, Zhang C, et al. Metformin enhances the antitumor activity of CD8+ T lymphocytes via the AMPK-miR-107-Eomes-PD-1 pathway. J Immunol. 2020;204:2575–88.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96.
Xu L, Zhu Y, Chen L, An H, Zhang W, Wang G, et al. Prognostic value of diametrically polarized tumor-associated macrophages in renal cell carcinoma. Ann Surg Oncol. 2014;21:3142–50.
Lan C, Huang X, Lin S, Huang H, Cai Q, Wan T, et al. Expression of M2-polarized macrophages is associated with poor prognosis for advanced epithelial ovarian cancer. Technol Cancer Res Treat. 2013;12:259–67.
Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013;15:1079–87.
Niino D, Komohara Y, Murayama T, Aoki R, Kimura Y, Hashikawa K, et al. Ratio of M2 macrophage expression is closely associated with poor prognosis for Angioimmunoblastic T-cell lymphoma (AITL). Pathol Int. 2010;60:278–83.
Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T, et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today. 2012;42:1–7.
Tang X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013;332:3–10.
Zhang BC, Gao J, Wang J, Rao ZG, Wang BC, Gao JF. Tumor-associated macrophages infiltration is associated with peritumoral lymphangiogenesis and poor prognosis in lung adenocarcinoma. Med Oncol. 2011;28:1447–52.
Ding L, Liang G, Yao Z, Zhang J, Liu R, Chen H, et al. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget. 2015;6:36441–55.
Chiang CF, Chao TT, Su YF, Hsu CC, Chien CY, Chiu KC, et al. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-kappaB signaling. Oncotarget. 2017;8:20706–18.
Wang JC, Sun X, Ma Q, Fu GF, Cong LL, Zhang H, et al. Metformin’s antitumour and anti-angiogenic activities are mediated by skewing macrophage polarization. J Cell Mol Med. 2018;22:3825–36.
Qing L, Fu J, Wu P, Zhou Z, Yu F, Tang J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am J Transl Res. 2019;11:655–68.
Uehara T, Eikawa S, Nishida M, Kunisada Y, Yoshida A, Fujiwara T, et al. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int Immunol. 2019;31:187–98.
Liu Q, Tong D, Liu G, Gao J, Wang LA, Xu J, et al. Metformin inhibits prostate cancer progression by targeting tumor-associated inflammatory infiltration. Clin Cancer Res. 2018;24:5622–34.
Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. 2011;187:6120–9.
Ye C, Geng Z, Dominguez D, Chen S, Fan J, Qin L, et al. Targeting ornithine decarboxylase by alpha-difluoromethylornithine inhibits tumor growth by impairing myeloid-derived suppressor cells. J Immunol. 2016;196:915–23.
Qin G, Lian J, Huang L, Zhao Q, Liu S, Zhang Z, et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology. 2018;7:e1442167.
Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018;78:1779–91.
Xia C, He Z, Liang S, Chen R, Xu W, Yang J, et al. Metformin combined with nelfinavir induces SIRT3/mROS-dependent autophagy in human cervical cancer cells and xenograft in nude mice. Eur J Pharmacol. 2019;848:62–9.
Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A. 2017;114:E9271–9.
Wang JC, Li GY, Li PP, Sun X, Li WM, Li Y, et al. Suppression of hypoxia-induced excessive angiogenesis by metformin via elevating tumor blood perfusion. Oncotarget. 2017;8:73892–904.
Wang J, Li G, Wang Y, Tang S, Sun X, Feng X, et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget. 2015;6:44579–92.
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anticancer immunotherapy. J Hematol Oncol. 2019;12:86.
Xu S, Yang Z, Jin P, Yang X, Li X, Wei X, et al. Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol Cancer Ther. 2018;17:1291–302.
Chen G, Yu C, Tang Z, Liu S, An F, Zhu J, et al. Metformin suppresses gastric cancer progression through calmodulinlike protein 3 secreted from tumorassociated fibroblasts. Oncol Rep. 2019;41:405–14.
Shen Y, Miao N, Xu J, Gan X, Xu D, Zhou L, et al. Metformin prevents renal fibrosis in mice with unilateral ureteral obstruction and inhibits Ang II-induced ECM production in renal fibroblasts. Int J Mol Sci. 2016;17:146.
Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T, et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS ONE. 2015;10:e0141392.
Hwang YP, Jeong HG. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase Calpha-dependent pathway: phorbol-12-myristate-13-acetate-induced/extracellular signal-regulated kinase/activator protein-1. Br J Pharmacol. 2010;160:1195–211.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28.
Jackson CM, Lim M, Drake CG. Immunotherapy for brain cancer: recent progress and future promise. Clin Cancer Res. 2014;20:3651–9.
Konala VM, Adapa S, Aronow WS. Immunotherapy in bladder cancer. Am J Ther. 2019; online ahead of print.
Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–50.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Acknowledgements
The authors sincerely wish to apologize to the many colleagues who have contributed significantly to this field of research, but whose publications were not cited due to the space limitations imposed upon this review.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Ma, R., Yi, B., Riker, A.I. et al. Metformin and cancer immunity. Acta Pharmacol Sin 41, 1403–1409 (2020). https://doi.org/10.1038/s41401-020-00508-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00508-0
Keywords
This article is cited by
-
Stroma AReactive Invasion Front Areas (SARIFA) proves prognostic relevance in gastric carcinoma and is based on a tumor–adipocyte interaction indicating an altered immune response
Gastric Cancer (2024)
-
A new approach of nano-metformin as a protector against radiation-induced cardiac fibrosis and inflammation via CXCL1/TGF-Β pathway
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Radiation-induced senescence: therapeutic opportunities
Radiation Oncology (2023)
-
Hyperglycemia induces PFKFB3 overexpression and promotes malignant phenotype of breast cancer through RAS/MAPK activation
World Journal of Surgical Oncology (2023)
-
Metformin may improve the outcome of patients with colorectal cancer and type 2 diabetes mellitus partly through effects on neutrophil extracellular traps
BJC Reports (2023)
