
Abstract
Immune system-mediated tumor killing has revolutionized anti-tumor therapies, providing long-term and durable responses in some patients. The phosphoinositide 3-kinase (PI3K) pathway controls multiple biological processes and is frequently dysregulated in malignancies. Enormous efforts have been made to develop inhibitors against class I PI3K. Notably, with the increasing understanding of PI3K, it has been widely accepted that PI3K inhibition not only restrains tumor progression, but also reshapes the immunosuppressive tumor microenvironment. In this review, we focus on the pivotal roles of class I PI3Ks in adaptive and innate immune cells, as well as other stromal components. We discuss the modulation by PI3K inhibitors of the tumor-supportive microenvironment, including eliminating the regulatory immune cells, restoring cytotoxic cells or regulating angiogenesis. The potential combinations of PI3K inhibitors with other therapies to enhance the anti-tumor immunity are also described.
Similar content being viewed by others

Introduction
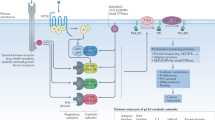
Phosphatidylinositol 3-kinases (PI3Ks) are lipid kinases capable of phosphorylating the 3’-hydroxyl group of the inositol ring of phosphatidylinositol. PI3Ks integrate signals from growth factors, cytokines and other extracellular stimuli into intracellular signals to regulate cell growth, proliferation, survival, motility and metabolism [1, 2]. Based on substrate preference and sequence homology, PI3Ks can be divided into three classes. Class I PI3Ks are further grouped into class IA and IB PI3Ks according to their distinct regulatory modes. Class IA PI3Ks are heterodimers consisting of a p110 catalytic subunit (p110α, p110β, or p110δ) and a p85 regulatory subunit (p85α, p55α, p50α, p85β or p85γ). Class IB PI3K is composed of a p110γ catalytic subunit and a p101 or p87 regulatory subunit [3]. Class IA PI3Ks can be activated by receptor tyrosine kinases (RTKs), G protein-coupled receptors (GPCRs), RAS and other adapter proteins, while class IB PI3K is activated exclusively by GPCRs [4]. In this review, PI3Ks refer to class I PI3Ks unless noted otherwise. In response to activating signals, PI3Ks are recruited to the plasma membrane. The catalytic subunit p110 is released from inhibition by the regulatory subunit p85 and phosphorylates PtdIns(4,5)P2 to generate PtdIns(3,4,5)P3, which acts as a second messenger to recruit pleckstrin homology (PH) domain-containing proteins, such as AKT and PDK1 (Fig. 1). Phosphorylation of AKT by PDK1 or mTORC2 further regulates multiple downstream signaling pathways. For instance, AKT relieves the tuberous sclerosis protein 2 (TSC2)-mediated inhibition of mTORC1, which then phosphorylates ribosomal S6 kinase (p70S6K) and eIF4E-binding proteins (4EBPs) to regulate proliferation and metabolism [5] (Fig. 1). PtdIns(3,4,5)P3 induced by growth factor stimulation is rapidly removed by the lipid phosphatase PTEN to terminate PI3K signaling under physiological conditions.

Class I PI3K isoforms are heterodimers consisting of p110 and p85 or p87/p101 subunits. Class IA PI3Ks can be activated by RTKs, GPCRs, RAS and other adapter proteins, while class IB PI3K is exclusively activated by GPCRs. When PI3K is activated by upstream signals, PtdIns(3,4,5)P3 (PIP3) is generated from PtdIns(4,5)P2 (PIP2) and activates downstream signaling pathways, such as the AKT/mTOR pathway. The activated PI3K pathway ultimately contributes to cell growth, proliferation, survival, motility and migration.
The PI3K/AKT/mTOR pathway is one of the most frequently dysregulated signaling pathways in human cancer due to the hyperactivation of upstream RTKs, RAS mutations, the amplification and mutation of PIK3CA, and functional loss of PTEN or INPP4B [6]. PI3K has been validated as a promising target for cancer therapy based on its significant roles in cell growth and proliferation. A few PI3K inhibitors have been approved for the treatment of cancer originating from different tissue types [7]. For example, BYL719 (alpelisib), the first PI3Kα-selective inhibitor approved by the U.S. Food and Drug Administration, is used in combination with endocrine therapy for the treatment of postmenopausal women and men with hormone receptor (HR)-positive, human epidermal growth factor-2 (HER2)-negative, PIK3CA-mutated advanced or metastatic breast cancer. The PI3Kδ inhibitor CAL-101 (idelalisib) has been approved for the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma, while the dual PI3Kγ/δ inhibitor IPI-145 (duvelisib) was approved in 2018 for the treatment of adult relapsed or refractory (RR) CLL or small lymphocytic lymphoma (SLL) and RR follicular lymphoma (FL).
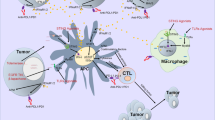
As PI3Ks are ubiquitously expressed and involved in many biological processes, they also play important roles in shaping the tumor microenvironment. The tumor microenvironment contains cancer-associated fibroblasts (CAFs); blood vessels; and immune cells, including T and B lymphocytes, macrophages, neutrophils, natural killer (NK) cells, dendritic cells (DCs) and myeloid-derived suppressor cells (MDSCs) [8]. CD8+ T effector cells and CD4+ T helper cells mediate the immune response to eliminate cancer cells. Meanwhile, tumor-associated macrophages (TAMs), regulatory T cells (Tregs), MDSCs, and CAFs contribute to a milieu favoring tumor growth and immune escape by the production of suppressive cytokines and depletion of nutrients. Immune escape may also result from the loss of tumor antigens, expression of checkpoint receptor ligands, and generation of physical barriers [9]. Abnormal activation of the PI3K pathway is comprehensively involved in these processes and promotes a suppressive phenotype in immune cells (Fig. 2). PI3K activation could decrease the infiltration of CD8+ T cells, impair their cytotoxic function, upregulate suppressive cytokines, and enhance immunosuppression mediated by protumoral immune cells. Hyperactivation of PI3K signaling is also correlated with tumor angiogenesis and CAF-promoted tumor progression.

Within the tumor microenvironment, accumulated regulatory cells and suppressed cytotoxic T cells constitute a microenvironment that favors tumor progression. PI3K inhibitors have been shown to enhance the infiltration of immune cells, restrain suppressive immune cells, and improve the function of CD8+ T cells. Notably, isoform-specific PI3K inhibitors display selective modulation of the tumor-immune interaction. The black lines indicate the regulatory effects of stromal components on malignant cells or CD8+ T cells, while the red lines indicate the counteraction of tumor cells on immune cells. The impact of PI3K inhibitors is indicated by the blue lines.
In the last few years, a plethora of therapies aimed at boosting host immunity to detect and kill tumor cells termed immunotherapy have achieved remarkable progress. However, a large proportion of patients display intrinsic resistance to this therapy due to variable mechanisms. The emerging roles of PI3K in shaping the tumor microenvironment provide new insights into how kinase inhibitors can contribute to enhancing antitumor immunity and their application in combination therapy to improve the outcome of immunotherapy. In this review, we have summarized the roles of PI3K in the immune system and the modulation of PI3K inhibitors in the suppressive tumor microenvironment. The combined use of PI3K inhibitors with other antitumor therapies to boost host immunity and execute potential synergy has also been discussed.
Functions of PI3Ks in the tumor microenvironment
Functions of PI3Ks in B and T lymphocytes
B and T lymphocytes are the key components in adaptive immune systems and provide powerful antigen-specific immunity. While PI3Kα and PI3Kβ are ubiquitously expressed in most cells and tissues, PI3Kγ and PI3Kδ are expressed mainly in leukocytes and act as the dominant isoforms responsible for their function [10]. Receptors on the surface of lymphocytes, such as CD19 on B cells, CD28 on T cells, cytokine receptors, Toll-like receptors (TLR), and GPCRs, containing PI3K-binding motifs directly activate PI3K upon receiving environmental cues. Alternatively, receptors indirectly link to the PI3K pathway through mediators such as the tyrosine kinase SYK, Bruton agammaglobulinemia tyrosine kinase (BTK) or IL-2-inducible T cell kinase (ITK) [11]. The activation of PI3K leads to multiple biological processes involved in the development, activation, or migration of immune cells.
PI3K in B cell development and function
B cell development starts inside bone marrow and occurs through several discrete stages. Formation of the pre-B cell receptor (pre-BCR) and pre-BCR signaling lead to the transition from the pro-B cell to pre-B cell stage. BCR formation and signaling facilitate the formation of immature B cells, which finally differentiate into three distinguishable subsets of mature B cells, including follicular B cells, marginal-zone B cells, and B1 cells, and then migrate to the spleen [12].
PI3Ks, especially the p85α and p110δ subunits, are involved in the development of B cells at multiple stages, including the transition from the pro-B cell to pre-B cell. Mice deficient in the p85α subunit displayed partial blockade at the pro-B cell stage with reduced proliferation [13, 14]. A similar phenotype was found in p110δ-mutated or p110δ-deficient mice. Okkenhaug et al. [15]. and Jou et al. [16]. found that the differentiation of B cells was blocked at the bone marrow stage, and the cell number decreased in p110δ-mutated mice. PI3K is also involved in the maturation of B cells. The absence of either p85α or p110δ in mice led to a reduced number of mature B cells [13, 15]. Mutations in PIK3CD and PIK3R1 (the genes encoding p110δ and p85α, respectively) cause immune system abnormalities, such as activated PI3Kδ syndrome (APDS). Patients harboring mutant PI3Kδ displayed lower numbers of total B cells and the perturbed maturation of peripheral B cells [17, 18].
PI3Ks also act as important mediators of antigen receptor signals in B cells. BCR-dependent Ca2+ flux in response to anti-IgM crosslinking in B cells was attenuated in p110δD910A/D910A mice [15]. Furthermore, p110δ deficiency in mice significantly impaired the ability of B cells to respond to T cell-independent (TI) and T cell-dependent (TD) antigens [16]. Though B cells displayed a normal immune response to TD antigens in mice deficient in p85α, they failed to respond to TI type-2 antigens [13, 14].
PI3Ks in T cell development and function
The development of T cells takes place in the thymus, where double-negative (DN) thymocyte precursors undergo a four-step differentiation process (DN1-DN4) and β-selection. DN thymocytes further differentiate into CD4+CD8+ double-positive (DP) cells, which are subsequently subjected to positive selection. Finally, CD4+ or CD8+ single-positive (SP) cells undergo negative selection and maturation. Mature T cells migrate out of the thymus and circulate in the periphery [12]. Upon receiving the peptide antigen presented by activated DCs, T cells secrete IL-2, undergo rapid proliferation and differentiate into distinct subsets determined by the cytokines to which T cells are exposed [11].
Multiple studies have suggested that PI3Ks play vital roles in the development and maturation of thymocytes, including the transition from DN3 to DN4, β-selection, and pre-TCR signaling. Pre-TCR signaling was attenuated in both p85α-deficient and p110δ-knockout T cells [19, 20]. Mice with genetic deletion of p110δ/γ exhibited profound blockade at the β-selection checkpoint with impaired pre-TCR signaling [21]. In addition, deletion of p110δ/γ resulted in a significantly reduced total number of DP thymocytes and increased number of apoptotic cells, indicating their crucial roles in the survival of thymocytes. The diminished number of thymocytes further resulted in a paucity in the number of mature T cells [22].
PI3Ks are well recognized to regulate the survival and function of T cells, which integrate signaling from the TCR and CD28 or other costimulatory receptors and cytokine receptors. For example, PI3Ks are thought to be important in IL-2 receptor-mediated cell cycle progression and cell survival [23]. Similar to their roles in B cells, PI3Ks mediate antigen receptor signaling in T cells. Ca2+ flux in response to anti-CD3 crosslinking in T cells was attenuated in p110δD910A/D910A mice [15]. A reduction in TCR-induced Ca2+ flux was also observed in mature CD4+ T cells from p110δ/γ−/− mice [22]. In CD4+ T cells, PI3K signaling is essential for antigen-driven clonal expansion and differentiation into the Th1 or Th2 subset but is not required for the proliferation of CD8+ T cells [24]. Moreover, PI3K and AKT are necessary to induce and sustain the expression of cytotoxic T lymphocyte (CTL) effector molecules, such as perforin, IFN-γ and granzymes, and other biomarkers that distinguish memory and effector T cells [25]. The PI3K pathway has also been implicated in the homing and trafficking of T lymphocytes. Evidence has shown that p110γ is required for CTL chemotaxis and trafficking to the infection site of CTLs [26] and effector CD4+ T cells [27].
CD4+CD25+ Tregs represent a unique lineage of T cells that potently suppress the function of effector T cells, thus limiting the clinical outcomes of cancer treatment [28]. Studies have indicated that activation of PI3Kδ is required for the immunosuppression of Tregs [29]. Using p110δD910A mice, Khaled Ali et al. showed that p110δ inactivation impaired the maintenance and functionality of Tregs, unleashed CD8+ cytotoxic T cells, and suppressed tumor growth [30]. Likewise, genetic inactivation of p110δ in a murine CLL model significantly undermined the expansion of Tregs, and reconstitution with wild-type p110δ restored the occurrence of leukemia [31]. Consequently, PI3K is critical for the function of Tregs, which act as a barrier to cancer therapy.
Functions of PI3Ks in innate immune cells
Macrophages, monocytes, DCs, granulocytes, mast cells and NK cells are the major components in the innate immune response, acting as the first line of the host defense against infection. Chemokines produced by macrophages in the infected tissue, together with the complement fragments C3a and C5a, increase local vascular permeability and attract neutrophils. Neutrophils and macrophages then phagocytize bacteria via multiple cell surface receptors. Nearly all of these processes require the activity of class I PI3Ks [32].
PI3Ks in neutrophils
Neutrophils are terminally differentiated cells that can be attracted by cytokines and quickly migrate to the site of inflammation, where they kill microorganisms through phagocytosis, degranulation and respiratory burst [33]. It is becoming clear that PI3Ks are essential for the chemotaxis and function of neutrophils. Neutrophils from PI3Kγ-knockout mice showed impaired chemoattractant-induced migration in response to IL-8, fMLP, C5a or MIP-1α [34,35,36]. PI3K also regulates neutrophil phagocytosis. During phagocytosis, neutrophils produce reactive oxygen species (ROS) to kill microbes, referred to as respiratory burst [37]. Neutrophils in mice lacking p110γ displayed defects in respiratory burst in response to a GPCR agonist [36]. p110β-knockout neutrophils exhibited the reduced production of ROS upon interaction with immobilized immune complexes (ICs), though depletion of p110β did not affect their ability to ingest and kill complement-opsonized bacteria [38]. Hence, PI3Ks, principally p110γ and p110β, play important roles in the proper functioning of neutrophils.
PI3Ks in NK cells
NK cells play a pivotal role in the control of viral infection. The lytic function of NK cells is mediated by formation of the immunological synapse, a signaling platform that directs the secretion of specialized lysosomes containing perforin and granzymes [39]. Both p110δ and p110γ are crucial in the maturation and function of NK cells, including cytokine secretion and cytotoxicity [40]. For instance, inactivation of PI3Kδ prevented the degranulation of NK cells, impairing their function in immune surveillance [41]. Inhibition of PI3K by pharmacological inhibitors blocked the PAK1/MEK/ERK pathway and interfered with the movement of perforin and granzyme B to target cells, indicating the vital role of PI3Ks in the cytotoxicity of NK cells [42]. Furthermore, PI3K signaling, mainly mediated by p110γ and p110δ, was shown to be required for the chemotaxis of NK cells to the chemokines CCL2, CCL5, CXCL10, and SDF1α [43, 44].
PI3Ks in myeloid cells, monocytes and macrophages
Macrophages are derived from monocytes and can be divided into proinflammatory M1 and anti-inflammatory M2 subtypes [45]. The PI3K/AKT/mTOR pathway is well recognized to regulate the polarization, chemotaxis, and proliferation of macrophages. For instance, PI3K/AKT/mTOR activation in macrophages led to increased histone acetylation and the induction of a subset of genes supporting the M2 phenotype [46]. Correspondingly, the deficiency of SHIP and PTEN, which are negative regulators of the PI3K pathway, enhanced differentiation towards M2 macrophages [47,48,49]. The chemotactic response of macrophages also requires PI3K activity. PI3Kγ-null macrophages displayed reduced migration in response to chemotactic stimuli and defective accumulation in a septic peritonitis model [34]. Furthermore, PI3K is important for the survival and proliferation of macrophages [50]. TAMs are M2-like macrophages that facilitate tumor progression by eliminating M1 macrophage-mediated immune responses and impairing the activation of T cells [51]. In TAMs, p110δ and p110γ are both abundantly expressed. p110γ promotes an immunosuppressive phenotype characterized by secretion of the cytokines IL-10 and TGF-β, which is correlated with reduced survival in cancer patients [52].
In addition, the PI3K pathway can act as a negative feedback regulator in the inflammatory response of monocytes and macrophages. Various lines of evidence have implicated PI3Ks in TLR signaling and shown that PI3Ks regulate the cellular response to pathogens by limiting the production of proinflammatory cytokines (such as IL-12) and enhancing the secretion of anti-inflammatory IL-10 [53,54,55]. The PI3K/AKT/mTOR pathway was demonstrated to antagonize TLR signaling partially by promoting STAT3 activity and suppressing the NF-κB-mediated transcriptional program [9].
MDSCs, a heterogeneous population of immature myeloid cells characterized by the expression of CD11b and GR1, are thought to be the precursors of DCs, macrophages, and granulocytes [56]. The presence of MDSCs promotes tumor progression by strongly suppressing the activity of T cells, NK cells, and DCs, in which PI3Kδ has been implicated [57]. PI3Kδ inactivation has been reported to reduce the number of MDSCs and alleviate tumor burden in vivo. Unlike wild-type MDSCs, MDSCs from PI3Kδ-inactivated mice did not suppress the proliferation of T cells [30]. Thus, PI3Kδ may dominate the immune suppression of MDSCs and may be a therapeutic target for cancer therapy.
Functions of PI3Ks in the extracellular matrix
Blood vessels and cellular components of the extracellular matrix (ECM), such as CAFs, are significant constituents in the tumor microenvironment. The growth and progression of solid tumors is critically dependent on the formation of new blood vessels [58]. Notably, PI3K signaling is particularly important in tumor angiogenesis [59]. PI3Ks, especially PI3Kα, are activated downstream of VEGFR in endothelial cells and promote tumor neovascularization [60]. Moreover, angiogenic factors secreted by cancer cells and TAMs are regulated by the PI3K pathway [58].
CAFs are one of the most abundant stromal cell types in tumor tissue and contribute to various malignant phenotypes and tumor progression [61]. The roles of PI3Ks in CAFs are less well understood, but several studies have validated that the PI3K pathway is involved in the protumoral function of CAFs, which is mediated by the secretion of tumor-promoting chemokines and enzymes. Activation of PI3K due to the loss of PTEN in stromal fibroblasts accelerated the initiation, progression, and malignant transformation of mammary epithelial tumors. These effects were ascribed to the induction of genes involved in ECM remodeling and the recruitment of macrophages, including Mmp9 and Ccl3 [62]. Matrix metalloproteinases (MMPs) function in multiple biological processes at different stages of cancer development and are primarily expressed by fibroblasts [63]. PI3Kγ has been shown to regulate TNF-mediated secretion of MMPs from fibroblasts, which is crucial for the migration of tumor cells [64]. CAFs also mediate immunosuppression of the T cell response. Ziqian Li et al. revealed that CAFs were positively correlated with PD-L1 expression in melanoma and colorectal carcinoma (CRC) tumor. They found that CXCL5 derived from CAFs activated the PI3K/AKT signaling pathway and promoted the expression of PD-L1 in tumor cells [65]. These studies suggest that PI3Ks also promote malignancy by inducing the secretion of protumoral factors by CAFs.
Modulation of immune cells and the immune-tumor interaction by PI3K inhibitors
Hyperactivation of the PI3K pathway in cancer and its vital function in cell survival and proliferation have made it an ideal target for treatment. Meanwhile, the hyperactivation of PI3K is increasingly recognized as an important player in shaping the immunosuppressive environment (Fig. 2). In addition to the roles of PI3Ks in TAMs, MDSCs, and Tregs, the loss of PTEN and PI3K activation in melanoma resulted in the decreased infiltration of CD8+T cells and protected tumor cells from T cell-mediated killing [66]. Activation of PI3K also induced the expression of immunosuppressive cytokines and PD-L1 [67]. The expression of VEGF is tightly regulated by PI3K/mTOR, which not only promotes tumor vascularization but also enhances the infiltration of regulatory immune cells, including MDSCs and Tregs [68, 69]. The nonredundant roles of PI3Ks in immunosuppression suggest that PI3K inhibitors may exert their activity by modulating both sides of the tumor-immune interface.
Pan-PI3K inhibitors
Pan-PI3K inhibitors target all PI3K isoforms with similar potencies without selectivity for certain isoforms. The development of pan-PI3K inhibitors was in part driven by the common expression of multiple PI3K isoforms with nonredundant functions in malignancies and the unidentified structures of PI3K isoforms, which are difficultly exploited to design isoform-specific inhibitors [70]. The fact that PI3K isoforms may functionally compensate for one another and limit the efficacy of isoform-selective inhibitors in some tumors highlights the advantage of pan-PI3K inhibition [71]. However, due to their broad inhibition profiles, the doses of pan-PI3K inhibitors needed to fully block the four isoforms might not be well tolerated. Thus, the therapeutic windows and efficacies of pan-PI3K inhibitors are limited in some situations due to their on-target and off-target toxicities [70].
Apart from their antiproliferative activity against cancer cells, pan-PI3K inhibitors exert immunomodulatory effects by enhancing the infiltration of immune cells, especially T cells. BKM120 restrained tumor growth and increased the infiltration of CD45+ immune cells and CD3+ T cells in human VMCUB1 bladder xenografts harboring a PIK3CA mutation. BKM120 was found to upregulate several cytokines and chemokines responsible for the migration of immune cells [72]. BKM120 treatment also increased the proportion of CD4+ T cells in MMTV-PyMT mammary tumor tissue [73].
Pan-PI3K inhibitors restrain the function of immunoregulatory cells. In MMTV-PyMT mammary tumors, BKM120 significantly reduced tumor-infiltrated macrophages, which was accompanied by a shift in the differentiation of bone marrow progenitors towards a proinflammatory phenotype [73]. SF1126, a prodrug derivative of LY294002, was reported to restrain the growth and metastasis of Lewis lung carcinoma (LLC) by blocking the induction of HIF1α and HIF2α and its transcriptional target, VEGF, in macrophages [74]. Wortmannin selectively inhibited the proliferation of CD4+CD25+ Tregs over conventional CD4+CD25− T cells. The growth of immortalized TC-1 cells by expression of the HPV16 E6 and E7 genes could be enhanced by Tregs, but this effect could be abrogated by wortmannin treatment in vivo [75].
Pan-PI3K inhibitors also directly modulate T cells to reinforce their activity. LY294002 converted the program of CD33-specific CAR-T cell differentiation from a shorter-lived effector state to a less differentiated state without affecting their expansion and improved their in vivo persistence and antitumor potency [76]. As antigen-presenting cells such as DCs are critical for the activation of T cells during the immune response, ZSTK474 could reduce the production of IL-10 and TGF-β by DCs, enhance DC-based immunotherapy and augment the antitumor T cell response in a B16 melanoma model [77].
Isoform-selective PI3Kα and PI3Kβ inhibitors
Despite the antineoplastic activity and positive impact of pan-PI3K inhibitors on immune cells, blockade of all class I PI3Ks may not be well tolerated. Isoform-selective PI3K inhibitors developed and tested in clinical settings have achieved impressive results with few toxic effects.
The high frequency of PIK3CA mutations and reliance on p110α in solid tumors highlight the need to develop p110α-selective inhibitors. In recent years, PI3Kα has been found to play important roles in immune modulation in addition to its pivotal role in the proliferation of tumor cells [78]. PI3Kα inhibitors may modulate the tumor microenvironment by interfering with angiogenesis. For example, GDC-0326 blocked the growth of RIP1-Tag2 pancreatic neuroendocrine tumors by repressing angiogenesis motivated by endothelial cells [79]. Moreover, a PI3Kα inhibitor was reported to modulate the polarization of TAMs and overcome TAM-mediated resistance to radiation therapy. CYH33, a novel PI3Kα-selective inhibitor with a distinctive structure, is currently in clinical trials for advanced solid tumors (NCT03544905). CYH33 was found to enhance the activity of radiation against esophageal squamous cell carcinoma by abrogating the radiation-induced phosphorylation of AKT and infiltration of M2-like macrophages [80].
The intrinsic activation of PI3Kα in malignant cells might influence the composition of the tumor microenvironment and function of cytotoxic immune cells. For instance, PI3Kα/AKT signaling in KrasG12D/Trp53R127H-driven KPC pancreatic tumors limited the infiltration of T cells and their capability to recognize and obliterate cancer cells. Further studies revealed that suppressed expression of MHC-I and CD80 in tumor cells was dependent on PI3Kα [81]. Tumor-associated neutrophils (TANs) are a population of distinct neutrophils with protumoral functions [82]. By analyzing the mRNA-seq data from uterine corpus endometrial carcinoma (UCEC) patients, high PIK3CA expression was found to be correlated with the neutrophil-related pathway. Consistently, neutrophil-related genes and neutrophils were significantly altered by PIK3CA expression in UCEC [83]. However, little is known about the effect of PI3Kα inhibitors on the tumor microenvironment, which deserves further investigation.
PI3Kβ regulates AKT activity in tumors with PTEN loss yet is dispensable in TCR activation [84, 85]. Treatment with the PI3Kβ inhibitor GSK2636771 reinforced T cell-mediated tumor killing in PTEN-null human melanoma cells coincubated with T cells without impairing the proliferation of antigen-specific T cells. In BP mice bearing spontaneously developed PTEN-loss/BRAF-mutated melanoma, the combination of GSK2636771 and anti-PD-1 antibody or anti-CTLA-4 antibody dramatically improved the antitumor efficacy and survival of mice compared to those following monotherapy, associated with the enhanced infiltration of CD4+ T and CD8+ T cells [66]. Nevertheless, further studies to dissect the underlying mechanism of PI3Kβ inhibitors in the immune environment are warranted.
Isoform-selective PI3Kδ and PI3Kγ inhibitors
As PI3Kδ and PI3Kγ are highly expressed in leukocytes, a number of preclinical studies have suggested that the efficacy of PI3Kδ and PI3Kγ inhibitors is not limited to hematologic malignancies but that these inhibitors are also potent in suppressing solid tumor growth. Due to their regulatory roles in immune cells, targeting these isoforms is helpful to remodel the tumor microenvironment.
PI3Kγ and PI3Kδ inhibitors have been reported to reestablish the immune response against tumors by mitigating the immunosuppression of TAMs. The PI3Kγ inhibitor IPI-549 delayed the growth of multiple solid tumors by inhibiting the functions of macrophages and stimulating the immune responses of CD8+ T cells [52, 86, 87]. Similarly, pharmacologic inhibition by the PI3Kγ/δ inhibitor TG100-115 or genetic deletion of PI3Kγ led to a transcriptional switch to immune stimulation in macrophages, increased the infiltration of CD8+ T cells and attenuated tumor growth [88]. A recent study revealed that the PI3Kγ/δ inhibitor IPI-145 not only directly inhibited the proliferation of tumor cells but also blocked the extended survival of CLL cells supported by M2 macrophages [89].
PI3Kδ inhibitors may convert immune suppression by Tregs and activate effector T cells, as PI3Kδ is the predominant isoform that maintains the proliferation and function of Tregs. The PI3Kδ inhibitor IC87114 was demonstrated to selectively inhibit the proliferation of Tregs over CD4+ T effector cells [90]. Administration of the PI3Kδ inhibitor PI-3065 restrained KPC pancreatic tumor growth and prolonged survival, associated with the reduced abundance of Tregs and elevated levels of infiltrating CD8+ T cells [30]. The PI3Kδ inhibitor CAL-101 (idelalisib) was also shown to preferentially inhibit the proliferation of Tregs and attenuate their immunosuppressive effect on effector cells [91].
Meanwhile, PI3Kδ inhibitors may display an immune-activating effect by directly regulating CD8+ T cells. CAL-101, but not a PI3Kα or PI3Kβ inhibitor, delayed terminal differentiation and maintained the memory phenotype of CD8+T cells [92]. Similarly, Jacob S. Bowers et al. revealed that antigen-specific CD8+T cells differentiated to a memory phenotype upon CAL-101 treatment, which was associated with prolonged survival in mice bearing syngeneic melanoma or human M108 mesothelioma tumors [93].
Taken together, these studies demonstrate that the dependence of regulatory immune cells on the PI3K pathway can be exploited to release immune suppression and restore the cytotoxic function of CD8+ T cells by treatment with pan-PI3K and isoform-selective PI3K inhibitors. Nevertheless, the detailed mechanism has not been fully elucidated, and controversial results regarding the effect of PI3K inhibitors on the immune response have been obtained, which might reflect different experimental contexts, including cellular heterogeneity, dynamic interactions and crosstalk between different nodes of the PI3K pathway, off-target effects on immune cells, and cellular plasticity [94]. Therefore, the efficacy of PI3K inhibitors would be optimal when balance among cancer cells, regulatory immune cells, and effector immune cells in the microenvironment is achieved.
PI3K inhibitor-based drug combination to improve antitumor immunity
While PI3K inhibitors have been proven to boost antitumor immunity, their efficacy as monotherapy might be mild. The combination of PI3K inhibitors with immunotherapy, chemotherapy or molecularly targeted therapy has been shown to achieve synergy by immunomodulation.
Cancer immunotherapies have emerged as promising innovative treatments for many types of tumors and achieved remarkable performance, especially in melanoma and hematologic tumors [95]. Among these immunotherapies, immune checkpoint inhibitors targeting PD-1 or CTLA-4 have displayed an encouraging clinical benefit. However, their highly variable response and associated adverse effects severely impair therapeutic outcome [96, 97]. Because the PI3K pathway is comprehensively involved in shaping the immunosuppressive tumor microenvironment, the combination of PI3K inhibitors with immune checkpoint inhibitors has been studied in both preclinical and clinical settings (Table 1). The PI3Kβ inhibitor GSK2636771 was reported to sensitize PTEN-loss melanoma cells to T cell-mediated cytotoxicity and enhance the efficacy of anti-PD-1 or anti-CTLA-4 antibodies in vivo [66]. A phase I/II study to test the efficacy of GSK2636771 in combination with pembrolizumab (anti-PD-1) in patients with PD-1 refractory metastatic melanoma deficient in PTEN is ongoing [98]. The pan PI3K inhibitor BKM120 was found to improve the activity of anti-PD-1 against PIK3CA-mutated bladder cancer by increasing the infiltration of immune cells in the tumor tissue [72]. The PI3Kγ/δ inhibitor TG100-115 was also found to significantly improve the efficacy of anti-PD-1 antibody in a preclinical MEER HPV+ head and neck squamous cell carcinoma model [52].
Resistance to immune checkpoint blockade has been a significant obstacle in tumor therapy. PI3K inhibitors may overcome this issue by regulating immune cells. The PI3Kγ/δ inhibitor IPI-145 was reported to partially abrogate the immunosuppression mediated by granulocytic MDSCs (gMDSCs) and facilitate the infiltration and activation of CD8+ T cells [99]. Similarly, the PI3Kγ inhibitor IPI-549 sensitized tumors rich in myeloid cells to anti-PD-1 or anti-CTLA-4 antibodies [87]. A phase I/Ib study (NCT02637531) is in the process of evaluating the activity of IPI-549 as monotherapy or in combination with nivolumab in patients with advanced solid tumors. Notably, analysis of blood samples from subjects treated with IPI-549 and nivolumab showed immune activation and reduced immune suppression, including the upregulation of IFN-γ-responsive factors and restoration of exhausted CD8+ T cells [100]. Recruitment of MDSCs rendered head and neck cancers more resistant to checkpoint inhibitors, while IPI-145 facilitated CD8+ T cell-dependent immune responses to anti-PD-L1 and improved outcome, which may be attributed to IPI-145-mediated inhibition of MDSCs and upregulation of PD-1 and PD-L1 [99].
Chemotherapy and molecularly targeted therapy have been used as first-line treatments for cancer for many years. In addition to their activity against tumor cells, their effect on immune modulation has been increasingly appreciated. Conventional cytotoxic drugs increase tumor immunogenicity by elevating MHC or antigen expression, making the recognition and elimination of tumor cells by immune cells easier [101]. Oncogenic pathways such as RAS/RAF/MAPK signaling were shown to impair antitumor immune responses [102]. Accordingly, inhibitors targeting these pathways could improve the immune landscape in tumor tissue. Studies have suggested that simultaneous treatment with chemotherapy or oncogenic inhibitors and PI3K inhibitors could achieve a stronger immune response in several preclinical studies (Table 1). For example, the PI3Kγ/δ inhibitor TG100-115 altered the suppressive transcriptional program in macrophages and enhanced the response of pancreatic ductal adenocarcinoma to gemcitabine [88]. The pan-PI3K inhibitor LY294002 and RAF inhibitor sorafenib triggered a stronger immune response against PTEN-loss and BRAF-mutated melanoma [67]. The anti-HER2/neu antibody trastuzumab and PI3Kα inhibitor A66 displayed potent activity against HER2+ breast cancer by recruiting and activating CD8+ T cells, which might be a solution to overcome resistance to trastuzumab [103].
Summary and perspective
Class I PI3Ks have pleiotropic roles in immune cells and stromal components of the peritumoral environment, providing a rationale for the utilization of PI3K inhibitors to boost antitumor host immunity. The mechanisms that contribute to the proinflammatory effect of PI3K inhibitors include the following: the enhanced infiltration of immune cells into tumor tissue, particularly T cells; abrogated tumor progression mediated by regulatory immune cells, such as Tregs, MDSCs and TAMs, and the augmented antitumor responses of T cells; modification of the transcriptional program of CD8+ T cells, converting them from a shorter-lived effector phenotype to a memory phenotype with more persistence; interference with tumor angiogenesis; and promotion of the normalization of blood vessels to improve the delivery of chemotherapy and immunotherapy. Hence, PI3K inhibitors not only target the proliferation and metastasis of tumor cells but also enhance immune surveillance. In light of these new findings, several preclinical studies have revealed that PI3K inhibitors synergize with other therapies, including immune checkpoint inhibitors, to restore antitumor immunity. A few combinatorial regimens have been extended to clinical trials, and preliminary synergetic effects have been observed.
Despite these promising results, a number of challenges need to be addressed. First, the functions of different class I PI3K isoforms in immune cells remain elusive. Further clarification of the roles of these four PI3K isoforms in the development, differentiation, expansion, and function of immune cells in the tumor microenvironment will lay the foundation to illustrate the exact roles of PI3Ks in regulating the tumor-immune interaction. Second, the mechanisms of action of various types of PI3K inhibitors in tumor cells, as well as adjacent cells need to be fully dissected. Given the complexity and heterogeneity of the tumor microenvironment, newly developed technologies such as single-cell RNA-seq (scRNA-seq) will help to provide a more comprehensive understanding of the context in which PI3K inhibitors affect tumor cells and the microenvironment. The immunomodulatory effects of PI3K inhibitors may be further developed as biomarkers to indicate the efficacy of PI3K inhibitors. Third, a multitude of issues, such as the optimal dose and schedule to achieve the maximal effect in combination therapy based on PI3K inhibitors, must be cogitated. Although efforts have been dedicated to mostly PI3Kγ and PI3Kδ inhibitors, it should be noted that other isoform-selective inhibitors display the potential to be used as an immune modulator. Moving forward, the efficacy of these PI3K inhibitors in monotherapy and combination therapy needs to be further studied in clinical trials.
References
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501.
Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507.
Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24.
Robbins HL, Hague A. The PI3K/Akt pathway in tumors of endocrine tissues. Front Endocrinol. 2015;6:188.
Aziz SA, Davies M, Pick E, Zito C, Jilaveanu L, Camp RL, et al. Phosphatidylinositol-3-kinase as a therapeutic target in melanoma. Clin Cancer Res. 2009;15:3029.
Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019;59:125–32.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9.
Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170:605–35.
Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol. 2013;31:675–704.
Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3:317–30.
So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442:465–81.
Suzuki H, Terauchi Y, Fujiwara M, Aizawa S, Yazaki Y, Kadowaki T, et al. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science. 1999;283:390–2.
Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 1999;283:393–7.
Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI3-kinase mutant mice. Science. 2002;297:1031–4.
Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Carpino N, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22:8580–91.
Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Scholvinck L, Kalina T, et al. Genetic defects in PI3Kdelta affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol. 2017;176:77–86.
Lougaris V, Faletra F, Lanzi G, Vozzi D, Marcuzzi A, Valencic E, et al. Altered germinal center reaction and abnormal B cell peripheral maturation in PI3KR1-mutated patients presenting with HIGM-like phenotype. Clin Immunol. 2015;159:33–6.
Shiroki F, Matsuda S, Doi T, Fujiwara M, Mochizuki Y, Kadowaki T, et al. The p85alpha regulatory subunit of class IA phosphoinositide 3-kinase regulates beta-selection in thymocyte development. J Immunol. 2007;178:1349–56.
Janas ML, Varano G, Gudmundsson K, Noda M, Nagasawa T, Turner M. Thymic development beyond beta-selection requires phosphatidylinositol 3-kinase activation by CXCR4. J Exp Med. 2010;207:247–61.
Webb L., Vigorito E., Wymann M., Hirsch E., Turner M. Cutting edge: T cell development requires the combined activities of the p110γ and p110δ catalytic isoforms of phosphatidylinositol 3-kinase1. J Immunol. 2005;175:2783–7.
Swat W, Montgrain V, Doggett T, Douangpanya J, Puri K, Vermi W, et al. Essential role of PI3Kδ and PI3Kγ in thymocyte survival. Blood. 2006;107:2415–22.
Ward SG, Cantrell DA. Phosphoinositide 3-kinases in T lymphocyte activation. Curr Opin Immunol. 2001;13:332–8.
Okkenhaug K, Patton D, Bilancio A, Garcon F, Rowan W, Vanhaesebroeck B. The p110 isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol. 2006;177:5122–8.
Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity. 2011;34:224–36.
Martin A, Schwartz M, Jameson S, Shimizu Y. Selective regulation of CD8 effector T cell migration by the p110γ isoform of phosphatidylinositol 3-kinase. J Immunol. 2008;180:2081–8.
Thomas MS, Mitchell JS, DeNucci CC, Martin AL, Shimizu Y. The p110gamma isoform of phosphatidylinositol 3-kinase regulates migration of effector CD4 T lymphocytes into peripheral inflammatory sites. J Leukoc Biol. 2008;84:814–23.
McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23.
Lim EL, Okkenhaug K. PI3Kδ is a Treg target in cancer immunotherapy. Immunology. 2019;157:210–8.
Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI3K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510:407–11.
Dong S, Harrington BK, Hu EY, Greene JT, Lehman AM, Tran M, et al. PI3K p110δ inactivation antagonizes chronic lymphocytic leukemia and reverses T cell immune suppression. J Clin Investig. 2018;129:122–36.
Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta. 2015;1851:882–97.
Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82.
Hirsch E, Katanaev V, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–53.
Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–49.
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040.
Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immunol Methods. 1999;232:3–14.
Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, et al. PI3Kbeta plays a critical role in neutrophil activation by immune complexes. Sci Signal. 2011;4:ra23.
Mace E. Phosphoinositide-3-kinase signaling in human natural killer cells: new insights from primary immunodeficiency. Front Immunol. 2018;9:445.
Kerr WG, Colucci F. Inositol phospholipid signaling and the biology of natural killer cells. J Innate Immun. 2011;3:249–57.
Zebedin E, Simma O, Schuster C, Putz EM, Fajmann S, Warsch W, et al. Leukemic challenge unmasks a requirement for PI3Kdelta in NK cell-mediated tumor surveillance. Blood. 2008;112:4655–64.
Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, et al. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol. 2000;1:419–25.
al-Aoukaty A, Rolstad B, Maghazachi A. Recruitment of pleckstrin and phosphoinositide 3-kinase gamma into the cell membranes, and their association with G beta gamma after activation of NK cells with chemokines. J Immunol. 1999;162:3249–55.
Saudemont A, Garçon F, Yadi H, Roche-Molina M, Kim N, Segonds-Pichon A, et al. p110gamma and p110delta isoforms of phosphoinositide 3-kinase differentially regulate natural killer cell migration in health and disease. Proc Natl Acad Sci USA. 2009;106:5795–800.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. ELife. 2016;5:e11612.
Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, et al. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–74.
Sahin-Heco E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal A, et al. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol. 2014;193:1717–27.
Yue S, Rao J, Zhu J, Busuttil R, Kupiec-Weglinski J, Lu L, et al. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J Immunol. 2014;192:5343–53.
Linton MF, Moslehi JJ, Babaev VR. Akt signaling in macrophage polarization, survival, and atherosclerosis. Int J Mol Sci. 2019;20:2703.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016;539:437–42.
Aksoy E, Vanden Berghe W, Detienne S, Amraoui Z, Fitzgerald KA, Haegeman G, et al. Inhibition of phosphoinositide 3-kinase enhances TRIF-dependent NF-κB activation and IFN-β synthesis downstream of Toll-like receptor 3 and 4. Eur J Immunol. 2005;35:2200–9.
Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–81.
Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32.
Wynn TA. Myeloid-cell differentiation redefined in cancer. Nat Immunol. 2013;14:197–9.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–20.
Gyori D, Chessa T, Hawkins PT, Stephens LR. Class (I) phosphoinositide 3-kinases in the tumor microenvironment. Cancers. 2017;9:24.
Soler A, Angulo-Urarte A, Graupera M. PI3K at the crossroads of tumor angiogenesis signaling pathways. Mol Cell Oncol. 2015;2:e975624.
Castel P, Carmona FJ, Grego-Bessa J, Berger MF, Viale A, Anderson KV, et al. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci Transl Med. 2016;8:332ra42.
Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12:86.
Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–91.
Jablonska-Trypuc A, Matejczyk M, Rosochacki S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J Enzym Inhib Med Chem. 2016;31:177–83.
Awad AE, Kandalam V, Chakrabarti S, Wang X, Penninger JM, Davidge ST, et al. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma-dependent manner. Am J Physiol Cell Physiol. 2010;298:C679–92.
Li Z, Zhou J, Zhang J, Li S, Wang H, Du J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int J Cancer. 2019;145:1946–57.
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–16.
Dong Y, Richards JA, Gupta R, Aung P, Emley A, Kluger Y, et al. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene. 2013;33:4632–42.
Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009;29:881–8.
Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res. 2017;23:587.
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–56.
Stamatkin C, Ratermann KL, Overley CW, Black EP. Inhibition of class IA PI3K enzymes in non-small cell lung cancer cells uncovers functional compensation among isoforms. Cancer Biol Ther. 2015;16:1341–52.
Borcoman E, De La Rochère P, Richer W, Vacher S, Chemlali W, Krucker C, et al. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. Oncoimmunology. 2019;8:1–17.
Sai J, Owens P, Novitskiy S, Hawkins O, Vilgelm A, Yang J, et al. PI3K inhibition reduces mammary tumor growth and facilitates antitumor immunity and anti-PD1 responses. Clin Cancer Res. 2017;23:3371–84.
Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1α and HIF2α stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res. 2014;12:1520.
Abu Eid R, Samara RN, Ozbun L, Abdalla MY, Berzofsky JA, Friedman KM, et al. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res. 2014;2:1080–9.
Zheng W, O’Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia. 2018;32:1157–67.
Marshall N, Galvin K, Corcoran AM, Boon L, Higgs R, Mills K. Immunotherapy with PI3K inhibitor and toll-like receptor agonist induces IFN-gamma+IL-17+ polyfunctional T cells that mediate rejection of murine tumors. Cancer Res. 2011;72:581–91.
Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36:1170–6.
Soler A, Figueiredo AM, Castel P, Martin L, Monelli E, Angulo-Urarte A, et al. Therapeutic benefit of selective inhibition of p110α PI3-kinase in pancreatic neuroendocrine tumors. Clin Cancer Res. 2016;22:5805.
Shi JJ, Xing H, Wang YX, Zhang X, Zhan QM, Geng MY, et al. PI3Kalpha inhibitors sensitize esophageal squamous cell carcinoma to radiation by abrogating survival signals in tumor cells and tumor microenvironment. Cancer Lett. 2019;459:145–55.
Sivaram N, McLaughlin PA, Han HV, Petrenko O, Jiang YP, Ballou LM, et al. Tumor-intrinsic PIK3CA represses tumor immunogenecity in a model of pancreatic cancer. J Clin Invest. 2019;129:3264–76.
Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16:601–20.
Pan Y, Jia LP, Liu Y, Han Y, Deng Q. Alteration of tumor associated neutrophils by PIK3CA expression in endometrial carcinoma from TCGA data. J Ovarian Res. 2019;12:81.
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI3K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–9.
Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA. 2008;105:7797–802.
Rausch M, Tchaicha J, Tibbitts T, Henau OD, Sharma S, Pink M, et al. Abstract B032: The PI3K-γ inhibitor, IPI-549, increases antitumor immunity by targeting tumor-associated myeloid cells and remodeling the immune-suppressive tumor microenvironment. Cancer Immunol Res. 2016;4:B032.
De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443–7.
Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6:870–85.
Peluso M, Faia K, Winkler D, Patel N, Brophy E, White K, et al. Duvelisib (IPI-145) inhibits malignant b-cell proliferation and disrupts signaling from the tumor microenvironment through mechanisms that are dependent on PI3K-δ and PI3K-γ. Blood. 2014;124:328.
Abu-Eid R, Samara RN, Ozbun L, Abdalla MY, Berzofsky JA, Friedman KM, et al. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res. 2014;2:1080–9.
Chellappa S, Kushekhar K, Munthe LA, Tjønnfjord GE, Aandahl EM, Okkenhaug K, et al. The PI3K p110δ isoform inhibitor idelalisib preferentially inhibits human regulatory T cell function. J Immunol. 2019;202:1397–405.
Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced therapeutic efficacy and memory of tumor-specific CD8 T cells by ex vivo PI3K-delta inhibition. Cancer Res. 2017;77:4135–45.
Bowers J, Majchrzak K, Nelson M, Aksoy B, Wyatt M, Smith A, et al. PI3Kδ inhibition enhances the antitumor fitness of adoptively transferred CD8+ T cells. Front Immunol. 2017;8:1221.
O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91–103.
Zhang H, Chen J. Current status and future directions of cancer immunotherapy. J Cancer. 2018;9:1773–81.
Sambi M, Bagheri L, Szewczuk M. Current challenges in cancer immunotherapy: multimodal approaches to improve efficacy and patient response rates. J Oncol. 2019;2019:1–12.
Alatrash G, Jakher H, Stafford PD, Mittendorf EA. Cancer immunotherapies, their safety and toxicity. Expert Opin Drug Saf. 2013;12:631–45.
Tawbi HAH, Peng W, Milton D, Amaria RN, Glitza IC, Hwu WJ, et al. Phase I/II study of the PI3Kβ inhibitor GSK2636771 in combination with pembrolizumab (P) in patients (pts) with PD-1 refractory metastatic melanoma (MM) and PTEN loss. J Clin Oncol. 2018;36:TPS9596.
Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kdelta/gamma. Cancer Res. 2017;77:2607–19.
Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol. 2018;36:3013.
Weir GM, Liwski RS, Mansour M. Immune modulation by chemotherapy or immunotherapy to enhance cancer vaccines. Cancers. 2011;3:3114–42.
Kobayashi Y, Lim SO, Yamaguchi H. Oncogenic signaling pathways associated with immune evasion and resistance to immune checkpoint inhibitors in cancer. In: Seminars in cancer biology. Academic Press; 2019.
Choi JH, Kim KH, Roh KH, Jung H, Lee A, Lee JY, et al. A PI3K p110α-selective inhibitor enhances the efficacy of anti-HER2/neu antibody therapy against breast cancer in mice. Oncoimmunology. 2018;7:e1421890.
Acknowledgements
This work was supported by “Personalized Medicines-Molecular Signature-based Drug Discovery and Development”, Strategic Priority Research Program of the Chinese Academy of Sciences [XDA12020111, XDA12020235, and XDA12050407]; National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program” [2018ZX09711002-011-014 and 2018ZX09711002-004-004] and National Natural Science Foundation of China [81773760 and 81973345]. It was also partially supported by the Fudan-SIMM Joint Research Fund [FU-SIMM20172005].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Sun, P., Meng, Lh. Emerging roles of class I PI3K inhibitors in modulating tumor microenvironment and immunity. Acta Pharmacol Sin 41, 1395–1402 (2020). https://doi.org/10.1038/s41401-020-00500-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00500-8
Keywords
This article is cited by
-
Small-Molecule Compounds Boost CAR-T Cell Therapy in Hematological Malignancies
Current Treatment Options in Oncology (2023)
-
Immune Checkpoint Inhibitors and Novel Immunotherapy Approaches for Breast Cancer
Current Oncology Reports (2022)
