
Abstract
Renal fibrosis is an inevitable outcome of all kinds of progressive chronic kidney disease (CKD). Recently, asiatic acid (AA), a triterpenoid compound from Chinese medicine Centella asiatica, has been found to attenuate renal fibrosis. In the current study, we explored the mechanisms underlying antifibrotic effect of AA on UUO model. SD rats and ICR mice were subjected to unilateral ureteral occlusion (UUO) surgery. Prior the surgery, rats were administered AA (10 mg·kg−1 per day, ig) for 7 days, whereas the mice received AA (15 mg·kg−1 per day, ig) for 3 days. UUO group displayed significant degree of renal dysfunction, interstitial fibrosis, oxidative stress, and activation of the TGF-β/Smad and Wnt/β-catenin signaling pathway in the kidney, these pathological changes were greatly ameliorated by pretreatment with AA. In addition, we found that co-treatment with GW9662, a selective PPAR-γ antagonist (1 mg·kg−1 per day, ip) for 7 days, abolished the protective effects of AA. We further revealed that AA pretreatment did not significantly change the expression levels of PPAR-γ in the kidney, but markedly increase the plasma levels of 15d-PGJ2, an endogenous ligand of PPAR-γ. In UUO mice, pretreatment with 15d-PGJ2 (24 μg·kg−1 per day, ip, for 7 days) produced similar protective effect as AA. Moreover, AA pretreatment upregulated the expression levels of active, nuclear-localized SREBP-1 (nSREBP-1), whereas fatostatin, a specific inhibitor of SREBP-1, decreased the expression of nSREBP-1, as well as the level of 15d-PGJ2. These results provide new insight into the antifibrotic mechanism of AA and endogenous metabolites might become a new clue for investigation of drug mechanism.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD) affects nearly 10% of adults in the United States, and the incidence and prevalence of this disease are increasing worldwide [1]. CKD progresses to end-stage renal disease as renal function declines gradually when a critical number of functional nephrons are lost. Treatment options for CKD are limited and only offer partial protection against CKD progression [2, 3]. The development of more effective drugs to halt CKD progression is therefore a critical challenge for public health.
Natural small molecules remain promising drug sources. Asiatic acid (AA), a triterpenoid compound, is isolated from Centella asiatica, which is a traditional Chinese medicine. Previous studies have shown that AA has a variety of pharmacological effects, including anti-inflammation [4], antioxidation [5], and antitumor effects [6, 7]. Few studies have evaluated the mechanism of AA for the treatment of renal fibrosis. Although previous studies have demonstrated that AA produces a significant effect on the inhibition of renal fibrosis by regulating TGF-β/Smad signaling [8, 9], the details remain elusive. It is unclear whether AA regulates key proteins in the TGF-β/Smad signaling pathway by direct binding or through other mediators. It has been reported that thiazolidinediones, peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists, attenuate renal interstitial fibrosis, and inflammation through a reduction in TGF-β [10]. We hypothesize that AA attenuates renal injury and fibrosis by activating PPAR-γ. To test this hypothesis, we investigated whether the selective and irreversible PPAR-γ antagonist GW9662 counteracts the protective effects against renal injury and fibrosis afforded by AA treatment in animals. Our results showed that AA upregulated the expression of nuclear-localized sterol regulatory element-binding proteins-1 (nSREBP-1), enhanced 15d-PGJ2, activated PPAR-γ, and consequentially attenuated renal damage in unilateral ureteral occlusion (UUO) models.
Materials and methods
Chemicals and reagents
LC-grade methanol and chloroform were purchased from Merck (Darmstadt, Germany). Internal standards (myristic acid), N, O-bis (trimethylsilyl) trifluoroacetamide (BSTFA), methoxyamine hydrochloride, and pyridine were supplied by Sigma-Aldrich (St. Louis, MO, USA). 15-Deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2) was supplied by Cayman Chemical (Ann Arbor, MI, USA). AA was obtained from FeiYu Biotech Ltd Co (Nantong, China), and GW9662 was purchased from AbMol BioScience (Houston, TX, USA). Fatostatin was supplied by TargetMol (Shanghai, China). The chemicals used in this study were of analytical grade, and their purity was above 99.5%.
Animal models
All animal experiments were approved by the Ethics Committee for Animal Experiments of China Pharmaceutical University. After acclimatization for 1 week, rats and mice were used in the experiments. Male Sprague-Dawley rats (weight 180–200 g) and male ICR mice (weight 18–20 g) were given free access to water and regular chow. An animal model of UUO was established as described previously [11]. In brief, under chloral hydrate anesthesia, the left ureter was double-ligated with a 4-0 silk suture following a midline abdominal incision. Sham-operated animals underwent the same surgical intervention except for ureteral ligation.
To investigate the effect of AA, the rats were randomly separated into the following four groups: the UUO group (n = 6), the sham-operated group (control group, n = 6), the UUO + AA group (n = 6) (rats that were orally administered AA (10 mg·kg−1 per day, once a day continuously for 7 days) 12 h prior to UUO surgery), and the UUO + AA + GW9662 group (n = 6) (rats that were administered GW9662 (1 mg·kg−1 per day, ip, once a day continuously for 7 days) a half hour prior to the oral administration of AA). All rats were operated in the morning and sacrificed at 9:00 A.M. 7 days later. For the verification test, male ICR mice were used and randomly separated into the following four groups: the UUO group (n = 3), the control group (n = 3), the UUO + AA group (n = 3) (UUO mice that were given AA (15 mg·kg−1 per day, once a day continuously for 3 days)), and the control + AA group (n = 3) (sham-operated mice that were given AA (15 mg·kg−1 per day, once a day continuously for 3 days)). All mice were operated on in the morning and sacrificed at 9:00 A.M. 3 days later. To verify the role of the alterative metabolite 15d-PGJ2 in renal injury, we also used a mouse model, and mice were randomly separated into the UUO group (n = 3), the control group (n = 3), the UUO + 15d-PGJ2 group (n = 3) (mice that were administered 15d-PGJ2 (24 μg·kg−1 per day, ip, once a day continuously for 7 days)), and the UUO + 15d-PGJ2 + GW9662 group (n = 3) (mice were administered GW9662 (2 mg·kg−1 per day, ip, once a day continuously for 7 days) a half hour prior to the administration of 15d-PGJ2). All mice were operated on in the morning and sacrificed at 9:00 A.M. 7 days later. To investigate the relationship between AA and the generation of 15d-PGJ2, a mouse model was used, and male ICR mice were randomly separated into the UUO group (n = 3), the control group (n = 3), the UUO + AA group (n = 3) (mice that were given AA (15 mg·kg−1 per day, once a day continuously for 3 days)), and the UUO + AA + fatostatin group (n = 3) (mice that were given fatostatin (30 mg·kg−1 per day, ip, once a day continuously for 3 days) a half hour prior to the administration of AA). All mice were operated on in the morning and sacrificed at 9:00 A.M. 3 days later. Under chloral hydrate anesthesia, plasma was collected for biochemical detection or GC/MS analysis, and the left kidney was harvested and stored at −80 °C until analysis.
Blood parameters, histology, and Western blotting
Blood analyses were performed on an Olympus AU640 automatic analyzer. The histological protocol was described in detail previously [12]. The Western blot protocol was described previously [13]. The following primary antibodies were used: anti-collagen I (ab34710, Abcam, Cambridge, UK), anti-fibronectin (ab45688, Abcam), anti-α-SMA (ab124964, Abcam), anti-GAPDH (HRP-60004, Proteintech, Rosemont, IL, USA), anti-PPAR-γ (ab209350, Abcam), anti-SREBP-1 (ab28481, Abcam), anti-TGF-β1 (21898-1-AP, Proteintech), anti-Smad2 (#5339, CST, Danvers, MA, USA), anti-Smad3 (#9523, CST), anti-Smad7 (MAB2029, R&D, Minneapolis, MN, USA), anti-β-catenin (#8480, CST), anti-12-LO (C-5) (sc-365194, Santa Cruz, Santa Cruz, CA, USA), anti-Rac1 (66122-1-Ig, Proteintech), and anti-NOX4 (14347-1-AP, Proteintech) antibodies, and the Wnt Signaling Antibody Sampler Kit (#2915, CST). Semiquantitative analysis of each protein was performed by using ImageJ software (version 1.48), and the band densities were normalized to those of GAPDH.
mRNA isolation and qRT-PCR
Total mRNA was extracted using a High Pure RNA Isolation Kit (RNAiso Plus, Takara Bio, Japan) according to the manufacturer’s instructions. Total RNA was reverse transcribed by a HiScript II Q RT SuperMix for qPCR according to the manufacturer’s instructions (Vazyme, Nanjing, China). Quantitative real-time PCR (qRT-PCR) was carried out by the Step One System (A&B, Waltham, MA, USA) using AceQ qPCR SYBR Green Master Mix (Vazyme). The mRNA levels of the genes were calculated by normalization to the levels of GAPDH. The following primers were used: SREBP-1: (forward) 5′-CAGCAGCAGTGGTGGCAGTG-3′, (reverse) 5′-GGTTGCAGGTCAGACACAGGAAG-3′; GAPDH: (forward) 5′-TCCTGCACCACCAACTGCTTAG-3′, (reverse) 5′-AGTGGCAGTGATGGCATGGACT-3′.
GC-MS sample preparation
Internal standard solutions (10 µL of myristic acid in methanol, 1 mg/mL) were added to 100 μL of plasma. The mixed solution was extracted with 300 µL of methanol and chloroform (3:1, v/v) and vortexed for 30 s. The mixture was stored at room temperature for 10 min and centrifuged at 15 000 × g for 10 min at 4 °C. The resulting 300 μL of supernatant was transferred to a sample vial for vacuum drying at room temperature. The residue was redissolved in 80 μL of a methoxyamine solution (15 mg/mL in pyridine) and vortexed for 1 min. An oximation reaction was performed at 37 °C for 1.5 h. Then, 80 μL of BSTFA (containing 1% TMCS) was added to the solution, and the solution was vortexed for 30 s. The sample was kept at 70 °C for 1 h and vortexed for 10 s. The supernatant was transferred to a sample vial for GC-MS analysis.
The samples were analyzed using an Agilent 7890 chromatograph coupled with a 5977B MS system (Agilent Technologies, Santa Clara, CA, USA). Separation was achieved on a DB-5 ms capillary column coated with 95% dimethyl 5% diphenyl polysiloxane (30 m × 0.25 mm i.d., 0.25-µm film). The initial GC oven temperature was set at 60 °C for 1 min, followed by a 10 °C/min oven temperature ramp to 325 °C, which was maintained for 10 min. The temperature of the inlet, transfer line, and ion source was set to 250, 290, and 250 °C, respectively. The injection volume was 1 μL with a split ratio of 1:10. Helium was used as the carrier gas with a constant flow rate of 0.87 mL/min. Measurements were made with electron impact ionization (70 eV) in full scan mode (m/z 50−650).
Data analysis
The raw GC-MS data were converted and prepared for multivariate analysis according to the previously published methods [14]. The peak area was normalized to the internal standard. PCA analysis was used to visualize general clustering, trends and outliers among the observations. OPLS-DA was used to validate the PCA model and identify the differential metabolites. Fold changes of the arithmetic mean values (each group/the control group) were calculated. Student’s t test was used to analyze the statistical significance of the results. The P values obtained from Student’s t test were further adjusted by a false discovery rate method based on the Hochberg–Benjamini method. Differential metabolites were identified by a library search (NIST and Fiehn) and confirmed by available references.
Results
GW9662 abolished the protective effects of AA in UUO rats
Representative photographs of the left kidneys of the rats are shown in Fig. 1a. As shown in Fig. 1b, the UUO rats exhibited significant increases in serum creatinine (Scr), blood urea nitrogen (BUN), and triglyceride (TG) levels, suggesting a significant degree of renal dysfunction. Treatment with AA group significantly attenuated the increase in BUN and Scr levels. However, the attenuation of BUN and Scr levels observed after AA treatment was reversed by the administration of GW9662, an antagonist of PPAR-γ (Fig. 1b). Histological analysis suggested that kidney tissues from the UUO rats exhibited severe tubular dilatation, tubular atrophy, and widened interstitial space with severe inflammatory cell infiltration (Fig. 1c). The oral administration of AA attenuated the interstitial injury, whereas the attenuation of interstitial damage was reversed by GW9662 (Fig. 1d, e).

a Representative photographs of left kidneys. b Biochemical parameters including serum creatinine (Scr), blood urea nitrogen (BUN), total cholesterol (TC), triglyceride (TG), and total protein (TP) in each group. c Representative photomicrographs of the H&E staining and Masson’s trichrome staining from left kidneys of control, UUO, UUO + AA, UUO + AA + GW9662 rats. d Bar graph depicts renal injury scores based on H&E staining. e Bar graph depicts renal interstitial fibrosis scores based on Masson’s trichrome staining. (H&E staining; scale bar, 100 µm; Masson staining; scale bar, 50 µm; magnification, ×200). *P < 0.05, **P < 0.01, ***P < 0.001 (compared with control group); #P < 0.05, ##P < 0.01 (compared with UUO group)
UUO resulted in a significant increase in Collagen I (Col I), fibronectin (FN), and alpha-smooth muscle actin (α-SMA) levels, as well as Collagen III levels, suggesting interstitial fibrosis (Fig. 2a, b). The treatment of rats with AA produced a significant attenuation of the UUO-mediated increase in Col I, FN, and α-SMA levels, indicating improvement in interstitial fibrosis (Fig. 2b). The administration of GW9662 in AA-treated rats produced an increase in the expression of the abovementioned proteins similar to that in UUO rats and thus abolished the protective effect mediated by AA (Fig. 2a, b).

a Immunohistochemical staining of α-SMA and collagen Ш. scale bar, 50 µm. b Western blots showing fibrotic protein expression in kidneys from all groups. c Western blots showing protein expression in the Wnt/β-catenin signaling pathway in kidneys from all groups. d Western blots showing protein expression in the TGF-β/Smad signaling pathway in kidneys from all groups. e Western blots showing PPAR-γ expression in kidneys from control, UUO, UUO + AA and UUO + AA + GW9662 rats. f Western blots showing protein expression of Nox4, Rac1, and 12-LOX in kidneys from all groups. Quantitative measurements of protein expression in kidneys from each group as indicated. *P < 0.05, **P < 0.01, or ***P < 0.001
Wnt/β-catenin signaling is closely associated with renal fibrosis [15] as well as the classic TGF-β/Smad signaling axis. We next examined the effects of GW9662 on the Wnt/β-catenin and TGF-β/Smad signaling pathways in UUO rats treated with AA. UUO caused a significant increase in Dvl2, Dvl3, and β-catenin expression, which indicated the activation of Wnt/β-catenin signaling. Treatment with AA mediated a significant attenuation of the UUO-induced increase in Dvl2, Dvl3, and β-catenin expression, while the administration of GW9662 abolished the protective effect of AA (Fig. 2c). Meanwhile, we found that UUO caused a significant increase in TGF-β1, Smad2, and Smad3 expression and a decrease in Smad7 expression, which indicated the activation of TGF-β/Smad signaling (Fig. 2d). After treatment with AA, the upregulation of TGF-β1, Smad2, and Smad3 expression was significantly reduced; the downregulation of Smad7 expression was significantly increased (Fig. 2d). The administration of GW9662 in AA-treated rats produced a marked increase in TGF-β1, Smad2, and Smad3 expression as well as a marked decrease in Smad7 expression, suggesting that it abolished the improvement in the expression of these proteins mediated by AA (Fig. 2d).
In addition to fibrosis-related proteins and pathways, AA also affected oxidative stress in UUO rats. As shown in Fig. 2f, the expression levels of reactive oxygen species (ROS)-generating molecules including NOX4, Rac1, and 12-LOX were significantly increased in UUO rats. Treatment with AA produced a significant attenuation of the UUO-mediated increase in NOX4, Rac1, and 12-LOX expression, and the administration of GW9662 abolished the antioxidant effect mediated by AA (Fig. 2f).
Expression of PPAR-γ in rat kidneys
Since an antagonist of PPAR-γ, GW9662, can reverse the renal protective effects of AA, we hypothesized that AA may ameliorate renal fibrosis via activating PPAR-γ. Therefore, we investigated the expression of PPAR-γ in kidneys from different group of rats. UUO rats induced a significant increase in the expression of PPAR-γ compared with that in sham-operated rats (Fig. 2e). When compared with that in UUO rats, the expression of PPAR-γ did not significantly change in rat kidneys after treatment with AA or GW9662 (Fig. 2e), which indicates that AA activates PPAR-γ through other processes instead of directly increasing its protein expression.
Untargeted GC-MS-based metabolomics identified important differential plasma metabolites
Typical total ion chromatograms of the representative samples obtained are presented in Supplementary Fig. S1. A PCA score plot of the plasma metabolites from the control and UUO groups is shown in Fig. 3a. There was a clear separation observed in the plot of the plasma samples from the control and UUO groups, and a supervised method, OPLS-DA, was also used to show the metabolic distinction between the two groups (Fig. 3b). The OPLS-DA model parameters R2Y and Q2 (cum) were 0.885 and 0.701, suggesting good fitness and predictive ability of the OPLS-DA model. PCA and OPLS-DA score plots of plasma metabolites in the control, UUO and UUO + AA groups are shown in Fig. 3c, d. The UUO + AA group was more similar to the control groups than the UUO group was.

a PCA scores plot from control and UUO groups. b OPLS-DA scores plot from control and UUO groups. c PCA scores plot from control, UUO, and UUO + AA groups. d OPLS-DA scores plot from control, UUO, and UUO + AA groups. e Alteration of the level of 15d-PGJ2 and linoleic acid in plasma samples of rats using GC-MS. Asterisk denotes UUO + AA vs UUO, **P < 0.01. f The plasma level of 15d-PGJ2 in mice treated with AA. *P < 0.05, **P < 0.01
Twenty-two plasma metabolites were identified (Table 1). Out of the 22 metabolites, 15 were differentially expressed in UUO rats. Then, a heatmap was created to visualize the relative levels of the 22 differential metabolites in each group (Supplementary Fig. S2). Notably, of these 22 identified metabolites, there were two PPAR-γ ligands, namely, 15d-PGJ2 and linoleic acid. Both of them were significantly increased in AA-treated rats compared with UUO rats (Fig. 3e).
Targeted GC-MS-based metabolomics validated the production of 15d-PGJ2
To validate whether AA can enhance the production of 15d-PGJ2, the plasma level of 15d-PGJ2 in mice was measured by GC-MS. Mice treated with AA showed a higher level of 15d-PGJ2 than that exhibited by controls. In addition, UUO mice treated with AA showed an enhanced level of 15d-PGJ2 compared with that shown by UUO mice (Fig. 3f). These results demonstrate that AA indeed increases the plasma level of 15d-PGJ2.
15d-PGJ2 mediated similar therapeutic effects as those of AA in UUO mice
To prove our hypothesis that AA ameliorates renal fibrosis via enhanced formation of 15d-PGJ2, we administered 15d-PGJ2 to UUO mice to test whether 15d-PGJ2 shows similar therapeutic effect as those of AA. UUO mice exhibited significant increases in BUN and Scr levels, suggesting a significant reduction in renal function. Treatment with 15d-PGJ2 significantly attenuated the increase in BUN and Scr levels. However, the attenuation of BUN and Scr levels observed after 15d-PGJ2 treatment was reversed by the administration of GW9662 (Fig. 4a, b). Kidney tissues from UUO mice showed severe interstitial fibrosis and inflammatory cell infiltration. The administration of 15d-PGJ2 significantly attenuated the interstitial damage, whereas the attenuation was reversed by GW9662 (Figs. 4c and 5a). In addition, UUO mediated a significant increase in the expression of type I collagen (Col I), FN, and α-SMA, which are marker proteins in fibrosis (Fig. 5b). The treatment of mice with 15d-PGJ2 produced a significant attenuation of the UUO-induced increase in Col I, FN, and α-SMA expression, indicating an improvement in interstitial fibrosis, while the administration of GW9662 abolished the protective effect mediated by 15d-PGJ2 (Fig. 5b). Similar to AA, 15d-PGJ2 inhibited the activation of Wnt/β-catenin and TGF-β/Smad signaling pathways, and the effect was blocked by GW9662 (Fig. 5c, d). Taken together, these data suggest that 15d-PGJ2 shows similar therapeutic effects as those of AA in the UUO animal model and that the renoprotective effects of 15d-PGJ2 can be abolished by GW9662.

Biochemical parameters including serum creatinine (Scr) (a) and blood urea nitrogen (BUN) (b) in each group of mice. c Representative photomicrographs of the H&E staining and Masson’s trichrome staining from left kidneys of control, UUO, UUO + 15d-PGJ2, UUO + 15d-PGJ2 + GW9662 mice (H&E and Masson staining; scale bar, 100 µm; magnification, ×200). *P < 0.05, **P < 0.01, ***P < 0.001 (compared with control group); #P < 0.05, ##P < 0.01 (compared with UUO group)

a Immunohistochemical staining of α-SMA and Collagen Ш. scale bar, 50 µm. b Western blots showing fibrotic protein expression in kidneys from all groups. c Western blots showing protein expression in the Wnt/β-catenin signaling pathway in kidneys from all groups. d Western blots showing protein expression in the TGF-β/Smad signaling pathway in kidneys from all groups. Quantitative measurements of protein expression in kidneys from each group as indicated. *P < 0.05, **P < 0.01, or ***P < 0.001
AA regulated the formation of endogenous 15d-PGJ2 through SREBP-1
It was demonstrated that AA activates PPAR-γ by promoting the production of endogenous 15d-PGJ2, the natural ligand of PPAR-γ. To investigate how AA regulates 15d-PGJ2, we measured the expression of SREBP-1, which is of importance in the synthesis of fatty acids since 15d-PGJ2 contains a long-chain fatty acid structure. We found that the expression of active nSREBP-1 was significantly increased in UUO rats treated with AA compared with model rats, while the expression of full-length SREBP-1 was slightly decreased (Fig. 6a), and total SREBP-1 mRNA levels remained unchanged between the two groups (Fig. 6b), suggesting that AA mediated SREBP-1 activity at the posttranscriptional level. To validate whether SREBP-1 regulates the formation of 15d-PGJ2, we gave UUO + AA mice fatostatin to test whether the level of 15d-PGJ2 was decreased. The treatment of mice with AA significantly increased the expression of nSREBP-1 compared with that in UUO mice, while the administration of fatostatin, which is an inhibitor of SREBP-1, in AA-treated mice produced a decrease in the expression of nSREBP-1 (Figs. 6c and S3). The plasma level of 15d-PGJ2 in the mice was measured by GC-MS. UUO mice treated with AA showed a higher level of 15d-PGJ2 compared with that in the UUO group, while fatostatin significantly decreased the level of 15d-PGJ2 compared with that in UUO mice treated with AA (Fig. 6d), which was consistent with the expression of nSREBP-1. Moreover, the expression of fibrotic proteins was decreased in AA-treated mice and upregulated after the administration of fatostatin (Supplementary Fig. S4). These results demonstrated that AA might regulate the level of endogenous 15d-PGJ2 in mice through SREBP-1 and play an antifibrotic role.

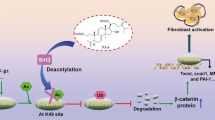
The protein (a) and mRNA expression (b) of SREBP-1 in kidneys from control, UUO, and UUO + AA rats. c Western blots showing nSREBP-1 expressions in kidneys from Control, UUO, UUO + AA, and UUO + AA + fatostatin mice. Quantitative measurements of protein expression in kidneys from each group as indicated. *P < 0.05, **P < 0.01. d Alteration of the plasma level of 15d-PGJ2 in mice using GC-MS. *P < 0.05, **P < 0.01. e Suggested antifibrotic mechanism of asiatic acid
Discussion
In this study, we demonstrated that the upregulation of nSREBP-1 mediated by AA enhances the level of endogenous 15d-PGJ2, which activates PPAR-γ and protects against renal damage and renal fibrosis (Fig. 6e). Our major novel findings include the following: (1) AA attenuates renal injury, oxidative stress, and fibrosis induced by the activation of PPAR-γ through increasing its natural ligand 15d-PGJ2 in UUO rats; (2) 15d-PGJ2 attenuates interstitial fibrosis in UUO mice; and (3) AA regulates the production of endogenous 15d-PGJ2, which may be through SREBP-1. This study demonstrates for the first time that the administration of the selective PPAR-γ antagonist GW9662 abolishes the renoprotective effects observed upon treatment with AA in rats. This result indicates that the renoprotective effects of AA are closely associated with PPAR-γ.
PPAR-γ, which is expressed in many organs, including the kidney [16], is a member of the ligand-activated transcription factor superfamily. PPAR-γ is involved in the expression of many genes and regulates glucose metabolism, insulin sensitivity, lipid metabolism, fibrosis, inflammation, and oxidative stress [17, 18]. It has been reported that the PPAR-γ agonist pioglitazone reduces proteinuria and preserves renal structure and function in passive Heymann nephritis rats [19]. In addition, a PPAR-γ agonist can inhibit the generation of ROS and the oxidative stress response [20, 21], and oxidative stress plays an important role in the pathogenesis of CKD and its complications [22, 23]. In our study, AA attenuated oxidative stress and fibrosis via the activation of PPAR-γ in UUO rats. PPAR-γ expression was upregulated in UUO rats compared to control rats, which is consistent with previous studies of a renal ischemia reperfusion injury model [24, 25]. Compared with that in UUO rats, the expression of PPAR-γ did not change upon treatment with AA, implying that the renoprotective effects afforded by AA may be due to the overproduction of endogenous PPAR-γ agonists instead of the upregulation of the protein expression of PPAR-γ.
To verify whether endogenous PPAR-γ ligands are overproduced, we applied untargeted GC-MS-based metabolomics to investigate plasma samples from the control, UUO, and UUO + AA groups. Twenty-two plasma metabolites were identified and are shown in Table 1; we found two ligands of PPAR-γ, namely, 15d-PGJ2, and linoleic acid. They were significantly increased in AA-treated rats compared with UUO rats (Fig. 3e). Endogenous 15d-PGJ2 produced by the nonenzymatic dehydration of PGD2 has an affinity for PPAR-γ of approximately 2 μM [26]. This metabolite is the most potent natural ligand of PPAR-γ. PPAR-γ is also activated by a variety of long-chain polyunsaturated fatty acids. Linoleic acid has been reported to bind to and activate PPAR-γ with relatively low affinity (Kd = 10–100 μM) [27]. Then, we further validated that AA indeed enhances the plasma level of 15d-PGJ2 in mice using targeted GC-MS-based metabolomics (Fig. 3f). Moreover, the administration of 15d-PGJ2 attenuated (1) renal dysfunction (reduced the increase in Scr and BUN levels, Fig. 4a, b); (2) interstitial damage (H&E staining, Fig. 4c); (3) the expression of fibrotic proteins (Fig. 5a, b); and (4) the disorder of TGF-β/Smad and Wnt/β-catenin signaling pathways (Fig. 5c, d). Overall, 15d-PGJ2 showed similar therapeutic effects as those of AA in UUO animal model, and their renoprotective effects were abolished by GW9662. These data indicate that AA ameliorates renal fibrosis, at least in part via enhanced formation of endogenous 15d-PGJ2 in a UUO animal model.
SREBPs play a central role in cell metabolism by controlling the synthesis of fatty acids, TGs, and cholesterol [28]. SREBP-1 is one of the most important SREBPs, which are synthesized as inactive precursors bound to the ER [28, 29]. When sterols are depleted in the ER membrane, SREBP-1 is transported to the Golgi, proteolytically released by two proteases and then shuttled to the nucleus to induce the expression of genes involved in fatty acid synthesis. A previous study reported that the expression of an activated form of SREBP-1 causes cells to produce ligand(s) of PPAR-γ [30]. However, the ligand that is produced is still unknown. Our results show that UUO animals treated with AA exhibit significant increases in the levels of activated SREBP-1 and 15d-PGJ2 compared to those in the UUO group (Figs. 3e and 6a), while the administration of fatostatin in AA-treated mice produces a decrease in nSREBP-1 and 15d-PGJ2 levels (Fig. 6c, d). The data presented here indicate clearly that nSREBP-1 triggers the generation of endogenous 15d-PGJ2.
In summary, we demonstrated that AA increases the expression of activated SREBP-1, upregulates nSREBP-1, enhances the level of endogenous 15d-PGJ2, which activates PPAR-γ, attenuates oxidative stress, rebalances the disorder of the TGF-β/Smad and Wnt/β-catenin signaling pathways, and ameliorates renal fibrosis (Fig. 6e). Our study, for the first time, reveals, in part, the mechanism by which AA can treat renal fibrosis through regulating endogenous metabolites. This study also provides a paradigm for mechanistic studies of molecules with similar characteristics as those of AA.
References
Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. Prevalence of chronic kidney disease in the United States. J Am Med Assoc. 2007;298:2038–47.
Brenner BM, Cooper ME, De Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–9.
De Zeeuw D. Unmet need in renal protection–do we need a more comprehensive approach? Contrib Nephrol. 2011;171:157–60.
Yun KJ, Kim JY, Kim JB, Lee KW, Jeong SY, Park HJ, et al. Inhibition of LPS-induced NO and PGE2 production by asiatic acid via NF-kappa B inactivation in RAW 264.7 macrophages: possible involvement of the IKK and MAPK pathways. Int Immunopharmacol. 2008;8:431–41.
Pittella F, Dutra RC, Junior DD, Lopes MT, Barbosa NR. Antioxidant and cytotoxic activities of Centella asiatica (L) Urb. Int J Mol Sci. 2009;10:3713–21.
Lee YS, Jin DQ, Kwon EJ, Park SH, Lee ES, Jeong TC, et al. Asiatic acid, a triterpene, induces apoptosis through intracellular Ca2+ release and enhanced expression of p53 in HepG2 human hepatoma cells. Cancer Lett. 2002;186:83–91.
Park BC, Bosire KO, Lee ES, Lee YS, Kim JA. Asiatic acid induces apoptosis in SK-MEL-2 human melanoma cells. Cancer Lett. 2005;218:81–90.
Xu C, Wang W, Xu M, Zhang J. Asiatic acid ameliorates tubulointerstitial fibrosis in mice with ureteral obstruction. Exp Ther Med. 2013;6:731–6.
Meng XM, Zhang Y, Huang XR, Ren GL, Li J, Lan HY. Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget. 2015;6:36984–97.
Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, et al. PPAR-gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-beta. Lab Investig. 2009;89:47–58.
Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, Sautter J, et al. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc Natl Acad Sci U S A. 2009;106:14518–23.
Zhang ZH, Vaziri ND, Wei F, Cheng XL, Bai X, Zhao YY. An integrated lipidomics and metabolomics reveal nephroprotective effect and biochemical mechanism of Rheum officinale in chronic renal failure. Sci Rep. 2016;6:22151.
Zhao YY, Wang HL, Cheng XL, Wei F, Bai X, Lin RC, et al. Metabolomics analysis reveals the association between lipid abnormalities and oxidative stress. inflammation, fibrosis, and Nrf2 dysfunction in aristolochic acid-induced nephropathy. Sci Rep. 2015;5:12936.
Zhang ZH, He JQ, Qin WW, Zhao YY, Tan NH. Biomarkers of obstructive nephropathy using a metabolomics approach in rat. Chem Biol Interact. 2018;296:229–39.
He W, Tan RJ, Li Y, Wang D, Nie J, Hou FF, et al. Matrix metalloproteinase-7 as a surrogate marker predicts renal Wnt/β-catenin activity in CKD. J Am Soc Nephrol. 2012;23:294–304.
Broeders N, Abramowicz D. Peroxisome proliferator-activated receptors (PPARs): novel therapeutic targets in renal disease. Kidney Int. 2002;61:354–5.
Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Thermodynamics in gliomas: interactions between the canonical Wnt/beta-catenin pathway and PPAR gamma. Front Physiol. 2017;8:352.
Lakatos HF, Thatcher TH, Kottmann RM, Garcia TM, Phipps RP, Sime PJ. The role of PPARs in lung fibrosis. PPAR Res. 2007;7:13–23.
Benigni A, Zoja C, Tomasoni S, Campana M, Corna D, Zanchi C, et al. Transcriptional regulation of nephrin gene by peroxisome proliferator-activated receptor-gamma agonist: molecular mechanism of the antiproteinuric effect of pioglitazone. J Am Soc Nephrol. 2006;17:1624–32.
Sener G, Sehirli AO, Gedik N, Dülger GA. Rosiglitazone, a PPAR-gamma ligand, protects against burn-induced oxidative injury of remote organs. Burns. 2007;33:587–93.
Mohideen P, Bornemann M, Sugihara J, Genadio V, Sugihara V, Arakaki R. The metabolic effects of troglitazone in patients with diabetes and end-stage renal disease. Endocrine. 2005;28:181–6.
Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–36.
Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013;83:1029–41.
Yoshimura R, Matsuyama M, Segawa Y, Tsuchida K, Takemoto Y, Kuratsukuri K, et al. Study of peroxisome proliferator-activated receptor (PPAR)-gamma in renal ischemia-reperfusion injury. Transpl Proc. 2004;36:1946–8.
Collino M, Patel NS, Lawrence KM, Collin M, Latchman DS, Yaqoob MM, et al. The selective PPAR-gamma antagonist GW9662 reverses the protection of LPS in a model of renal ischemia-reperfusion. Kidney Int. 2005;68:529–36.
Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–9.
Krey G, Braissant O, L’Horset F, Kalkhoven E, Perroud M, Parker MG, et al. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11:779–91.
Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46.
Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Investig. 2002;109:1125–31.
Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci USA. 1998;95:4333–7.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant 81703691) and the Program for Jiangsu Province Innovative Research Team. The authors would like to thank Zhe Wang for the discussion of the manuscript.
Author information
Authors and Affiliations
Contributions
ZHZ, HCC and NHT designed the experiments. ZHZ and JQH collected and analyzed the data. YYZ and NHT contributed analytical tools. ZHZ wrote the manuscript. NHT revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhang, Zh., He, Jq., Zhao, Yy. et al. Asiatic acid prevents renal fibrosis in UUO rats via promoting the production of 15d-PGJ2, an endogenous ligand of PPAR-γ. Acta Pharmacol Sin 41, 373–382 (2020). https://doi.org/10.1038/s41401-019-0319-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0319-4
Keywords
This article is cited by
-
PDGFβ receptor-targeted delivery of truncated transforming growth factor β receptor type II for improving the in vitro and in vivo anti-renal fibrosis activity via strong inactivation of TGF-β1/Smad signaling pathway
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
The gut microbe Bacteroides fragilis ameliorates renal fibrosis in mice
Nature Communications (2022)
-
Dihydroartemisinin suppresses renal fibrosis in mice by inhibiting DNA-methyltransferase 1 and increasing Klotho
Acta Pharmacologica Sinica (2022)
-
Selenium Deficiency Leads to Changes in Renal Fibrosis Marker Proteins and Wnt/β-Catenin Signaling Pathway Components
Biological Trace Element Research (2022)
-
Asiatic acid alleviates Ang-II induced cardiac hypertrophy and fibrosis via miR-126/PIK3R2 signaling
Nutrition & Metabolism (2021)
