
Abstract
Acute sympathetic stress causes excessive secretion of catecholamines and induces cardiac injuries, which are mainly mediated by β-adrenergic receptors (β-ARs). However, α1-adrenergic receptors (α1-ARs) are also expressed in the heart and are activated upon acute sympathetic stress. In the present study, we investigated whether α1-AR activation induced cardiac inflammation and the underlying mechanisms. Male C57BL/6 mice were injected with a single dose of α1-AR agonist phenylephrine (PE, 5 or 10 mg/kg, s.c.) with or without pretreatment with α-AR antagonist prazosin (5 mg/kg, s.c.). PE injection caused cardiac dysfunction and cardiac inflammation, evidenced by the increased expression of inflammatory cytokine IL-6 and chemokines MCP-1 and MCP-5, as well as macrophage infiltration in myocardium. These effects were blocked by prazosin pretreatment. Furthermore, PE injection significantly increased the expression of NOD-like receptor protein 3 (NLRP3) and the cleavage of caspase-1 (p20) and interleukin-18 in the heart; similar results were observed in both Langendorff-perfused hearts and cultured cardiomyocytes following the treatment with PE (10 μM). Moreover, PE-induced NLRP3 inflammasome activation and cardiac inflammation was blocked in Nlrp3-/- mice compared with wild-type mice. In conclusion, α1-AR overactivation induces cardiac inflammation by activating NLRP3 inflammasomes.
Similar content being viewed by others

Introduction
Sympathetic overactivation causes excessive release of catecholamines, including norepinephrine and epinephrine, which can induce or promote cardiac remodeling and dysfunction in many cardiovascular diseases [1,2,3]. The overactivation of adrenergic receptors (ARs), especially β-ARs, mediates sympathetic stress-induced cardiac injury [4]. Cardiac inflammation is an important pathological process involved in sympathetic stress-induced cardiac injury [5]. Our previous study showed that activation of the inflammasome in the myocardium induces the rapid induction of inflammatory cytokines, macrophage infiltration, and subsequent pathological cardiac remodeling elicited by acute β-AR stimulation [6]. However, the α1-AR subtype is also expressed and activated in cardiomyocytes during sympathetic stress in addition to β-AR. It remains unclear whether and how acute α1-AR activation induces cardiac inflammation following sympathetic overactivation.
Nod-like receptor protein 3 (NLRP3) inflammasomes consist of three main components, namely, NLRP3, ASC, and precursor caspase-1, and drive sterile inflammation in different pathologies [7, 8]. Upon activation of the inflammasome, the precursor caspase-1 is cleaved into caspase-1, which further cleaves and activates the downstream proinflammatory cytokines, including interleukin-1β (IL-1β) and IL-18 [7, 8]. The NLRP3 inflammasome plays important roles in the development and progression of cardiovascular diseases and is the main inflammatory sensor activated by a variety of stimuli [9, 10]. Our previous study found that activated β-ARs induced the activation of the NLRP3 inflammasome and the cleavage of downstream IL-18 in cardiomyocytes and triggered heart inflammation and cardiac injury [6]. Nonetheless, it is unknown whether α1-AR overactivation also causes NLRP3 inflammasome activation and whether the NLRP3 inflammasome mediates α1-AR overactivation-induced cardiac inflammation.
In the present study, we treated mice with the selective α1-AR agonist phenylephrine (PE) and determined cardiac inflammation and dysfunction to test the role of α1-ARs in cardiac inflammation induced by acute sympathetic stress. The activation of the NLRP3 inflammasome was detected in Langendorff-perfused mouse hearts and cultured neonatal mouse cardiomyocytes, and the role of NLRP3 in PE-induced heart inflammation was further explored in NLRP3-deficient mice to clarify the underlying mechanism of α1-AR-induced cardiac inflammation.
Materials and methods
Antibodies and reagents
The Mac-3 antibody was purchased from BD Biosciences (San Jose, CA, USA). NLRP3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Caspase-1 (P20) was purchased from Sigma (St. Louis, MO, USA). The IL-18 antibody and enzyme-linked immunosorbent assay (ELISA) kits were purchased from MBL (Nagoya, Japan). Interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and CC chemokine (MCP-1, MCP-5) ELISA kits were purchased from R&D Systems Incorporated (Minneapolis, MN, USA). Inositol triphosphate (IP3) ELISA kits were purchased from Cloud-Clone (Katy, TX, USA). The PE and prazosin reagents were purchased from Sigma (St. Louis, MO, USA).
Animals
All animal experimental protocols were approved by the Committee of Peking University on the Ethics of Animal Experiments (LA2016-018). The study was conducted in accordance with the Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 2011) and the Guidelines from Peking University Health Science Center. Wild-type (WT) male C57BL/6J mice were purchased from the Department of Laboratory Animal Science, Peking University. Male NLRP3-deficient (Nlrp3-/-) mice (C57BL/6N background) and their WT littermates were kindly provided by Dr. Ai-hua Zhang (Institute of Pediatrics, Nanjing Medical University, Nanjing, China). All mice were used for experiments for 12 weeks and were housed in a specific pathogen-free environment (temperature 20–24 °C under relative humidity of 30%–70%) under a 12 h/12 h light–dark cycle and fed a rodent diet ad libitum. PE at different doses was subcutaneously injected into mice to determine its dose–response relationship with the infiltration of macrophages in hearts, and a single dose of 5 or 10 mg/kg was chosen for further investigation to activate α1-ARs. The α-AR antagonist prazosin (5 mg/kg) was given 30 min prior to PE injection. The effects of PE and prazosin were confirmed by detecting the level of IP3. The cardiac samples were collected on day 1 or day 3 following PE treatment using lysis buffer (10 mM Tris-HCl, pH 7.4; 100 mM NaCl; 1 mM EDTA; 1 mM EGTA; 1 mM NaF; 20 mM Na4P2O7; 2 mM Na3VO4; 1% Triton X-100; 10% glycerol; 0.1% sodium dodecyl sulfate (SDS); 1% deoxycholic acid; 1 mM PMSF; and 1 g/mL aprotinin).
Echocardiographic measurements
Mice were placed in the supine position on a movable, heated platform maintained at 37 °C and anesthetized with 1.0%–1.5% isoflurane (Baxter Healthcare Corp, New Providence, RI, USA) to stabilize the heart rate at 400–500 beats per minute. Using the Vevo 2100 system (VISUALSONICS Inc., Toronto, Canada), the thickness and chamber dimensions were determined from M-mode images acquired at the mid-papillary level in the parasternal short-axis view and B-mode images acquired in the parasternal long- and short-axis views. The ejection fraction (EF%) was calculated based on the parameters from M-mode images to reflect the systolic cardiac function. An apical four-chamber view was acquired, and the peak flow velocities during early diastole (E wave) were measured across the mitral valve. The peak early diastolic velocity (E’ wave) of the mitral valve ring was also measured in this view. Then, E/E’ was calculated, reflecting the left ventricular diastolic function.
Blood pressure (BP) and heart rate (HR) monitoring using the radio telemetry method
A radio telemetry system was used to detect the BP and HR of mice. Transmitter implantation surgery, software operation, device operation, and BP measurements were conducted according to the experimental protocol [11]. After the surgery, the mice typically need ~1 week to resume normal circadian blood pressure rhythm and return to their pre-implantation body weight. PE was then administered to mice to examine its effect on mouse BP and HR.
Langendorff perfusion of isolated hearts
Hearts were isolated from anesthetized mice and perfused with a Langendorff apparatus (Harvard Apparatus, Germany). The operation was conducted following the instruction of the device and related literature [12]. The hearts were retrograde perfused with Krebs–Henseleit (K–H) buffer under constant perfusion pressure (120 mmHg). After equilibration for 15 min, the hearts were perfused with K–H buffer containing PE for 15 min and then perfused with K–H buffer without PE for 3 h. The dose–response relationship between PE and the NLRP3 expression level was analyzed, and a dose of 10 μM was chosen for further investigation. Cardiac samples were collected with lysis buffer at 15 min, 1 h or 3 h following PE treatment for Western blotting experiments and IP3 determination.
Isolation and culture of primary neonatal mouse cardiomyocytes (NMCMs)
Cardiomyocytes were isolated and cultured from 1-day-old neonatal C57BL/6 mice as described previously [13]. In brief, cardiomyocytes were obtained using trypsinization and collagenase type II and kept at 37 °C in humidified air containing 5% CO2 for 2 h. The nonattached cardiomyocyte-rich fraction was plated on plastic dishes (5 × 105/dish), and 100 μM bromodeoxyuridine was added to prevent fibroblast proliferation. The cells were incubated in serum-free medium for 4 h prior to drug or mock treatments. The dose–response relationship between PE and the NLRP3 expression level was analyzed, and a dose of 10 μM was chosen for further investigation.
Immunohistochemistry
Mouse hearts were collected on day 1 or day 3 after PE treatment, harvested, washed with cold phosphate buffered saline, fixed with 4% paraformaldehyde for 6–8 h, and embedded in paraffin. Serial sections (5 μm thick) were stained with antibodies against the macrophage marker Mac-3 (1:200 dilution, BD Biosciences, #550292, San Jose, CA, USA). Tissue sections were imaged by the NanoZoomer-SQ (Hamamatsu, Japan). Ten fields were randomly selected from the image of each section, and the size of the Mac-3-positive area was quantified with Image-Pro Plus 6.0. The ratio of positive-stained area to total myocardial area was calculated to determine the infiltration of macrophages.
Enzyme-linked immunosorbent assay (ELISA)
The protein levels of IL-6, TNF-α, MCP-1, and MCP-5 in heart lysate were measured by ELISA kits (R&D Systems, Minneapolis, MN, USA). The IL-18 cytokine protein in serum was measured by the MBL ELISA kit (MBL, Nagoya, Japan). The levels of IP3 were detected by an ELISA kit (Cloud-Clone, Katy, TX, USA). Procedures were conducted according to the manufacturer’s instructions, and absorbances were read at 450 nm (Microplate Reader Model 550, Bio-Rad, Hercules, CA, USA). All concentration values were in the linear range of the standard curve, and the final results are shown as the ratio of total protein concentrations, or pg/mL.
Western blotting analysis
The levels of NLRP3, caspase-1 (p20), and cleaved IL-18 were examined by Western blotting. Protein samples (60 μg) were separated by electrophoresis on 10% or 15% SDS polyacrylamide gels and transferred to polyvinylidene fluoride membranes. The membranes were incubated with primary antibodies for at least 8 h at 4 °C. After incubating the membranes with corresponding HRP-conjugated secondary antibodies, protein bands were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore Corporation, Burlington, MA, USA). Blotting bands were obtained using the GeneSys software system and quantified by calculating the gray value of each band using ImageJ software.
Cell counting kit-8 assay
The cellular viability following PE treatment was determined with a Cell Counting Kit-8 (CCK-8) (Solarbio, Beijing, China). The operation was conducted according to the instructions of the kit. Cardiomyocytes were cultured in 96-well plates and treated with PE. CCK-8 solutions were added and incubated for 2 h. The OD450 nm was detected in a Microplate Reader Model 550 (Bio-Rad, Hercules, CA, USA), and the viability of cardiomyocytes was calculated.
Statistical analysis
Data are expressed as the mean ± SEM from 5–6 independent experiments. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA) and SPSS 22 (IBM Co., Armonk, NY, USA). For parametric data with equal variances, analysis of variance (ANOVA) combined with the Least Significant Difference (LSD) post hoc test was used to analyze the differences among groups, and Student’s unpaired two-tailed t-test was used to analyze differences between two groups. For data with unequal variances, Welch's ANOVA with the Games-Howell post hoc test was used. P < 0.05 was considered statistically significant.
Results
PE-induced cardiac inflammation and dysfunction
Mice were treated with PE at different doses to selectively activate α1-ARs. On day 3 following the treatment, PE at 5 mg/kg began to increase the infiltration of macrophages in hearts, and the infiltration reached approximately the highest level at 10 mg/kg (Supplementary Fig. S1). The doses of 5 mg/kg and 10 mg/kg were chosen for further investigation. Following PE treatment, macrophage infiltration occurred on day 1 after PE injection and was further aggravated on day 3 (Fig. 1a). PE induced the increased expression of the inflammatory cytokine IL-6 and the chemokines MCP-1 and MCP-5 in a dose-dependent manner on both day 1 and day 3 (Fig. 1b). However, the expression of the proinflammatory cytokine TNF-α was not affected by PE. Echocardiography found that PE treatment through day 3 induced structural and functional cardiac changes (Supplementary Table S1). In particular, EF% was decreased and E/E’ was increased, suggesting that PE induced cardiac systolic and diastolic dysfunction (Fig. 1c).

Phenylephrine (PE) induced cardiac inflammation and dysfunction. a Immunostaining of Mac-3 (macrophage marker) in the heart on day 1 or day 3 after single-dose PE treatment. Positive stains are indicated by arrows (n = 6). b Concentrations of proinflammatory cytokines (IL-6, TNF-α) and chemokines (MCP-1, MCP-5) at different time points (open bars represent day 1, filled bars represent day 3) after PE treatment, as measured by ELISA (n = 6–8). c Representative echocardiographic images (M-mode, pulsed-wave Doppler, and tissue Doppler), EF%, and E/E’ values on day 3 after PE treatment (n = 6). The results are presented as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, as determined by one-way ANOVA with the LSD post hoc test or Kruskal–Wallis ANOVA with post hoc Dunn’s multiple comparison tests
To verify the role of α1-ARs in PE-induced cardiac inflammation and dysfunction, mice were pretreated with prazosin (5 mg/kg), an α-AR antagonist, prior to PE treatment. Macrophage infiltration, proinflammatory cytokine upregulation, and cardiac dysfunction were prevented by prazosin (Fig. 2 and Supplementary Table S2). PE is well known to increase BP by activating α1-ARs in vascular smooth muscle. Prazosin prevented the PE-induced increases in systolic and diastolic BP but not heart rate (Supplementary Fig. S2). Prazosin also inhibited the PE-induced increase in IP3 (Supplementary Fig. S3), indicating that α1-ARs were effectively blocked by prazosin at 5 mg/kg in this study.

α1-AR-mediated cardiac inflammation and dysfunction induced by PE. a Immunostaining of Mac-3 (macrophage marker) in the heart on day 1 and day 3 after PE treatment with or without prazosin (Praz) pretreatment. Positive stains are indicated by arrows (n = 6). b Concentrations of proinflammatory cytokines (IL-6) and indicative chemokines (MCP-1, MCP-5) at different time points (open bars represent day 1, filled bars represent day 3) (n = 6–8). c Representative echocardiographic images, EF%, and E/E’ values of the mice treated with prazosin and PE (n = 6). *P < 0.05; **P < 0.01; ***P < 0.001, as determined by one-way ANOVA with the LSD post hoc test or Kruskal–Wallis ANOVA with post hoc Dunn’s multiple comparison tests
PE activated NLRP3 inflammasomes in hearts and cardiomyocytes
NLRP3 inflammasomes are crucial to innate immunity and inflammation. The NLRP3 expression level, cleaved active form of caspase-1 (p20), and downstream IL-18 were increased by PE treatment in mice on day 1 (Fig. 3a). Consistently, IL-18 levels in serum were increased (Fig. 3b). On day 3, although the NLRP3 protein level remained upregulated, the cleavages of caspase-1 and IL-18 were not increased compared with the control group (Supplementary Fig. S4), suggesting that the cardiac NLRP3 inflammasome was activated in the early phase following PE treatment.

PE induced cardiac NLRP3/caspase-1/IL-18 pathway activation in mice. a Protein levels of NOD-like receptor protein 3 (NLRP3), caspase-1 (p20), and cleaved interleukin-18 (IL-18) in the heart of WT mice on day 1 after PE treatment with or without prazosin (Praz) pretreatment (n = 6). b IL-18 levels in the mouse serum after PE treatment through day 1 or day 3 (n = 6). *P < 0.05; **P < 0.01; ***P < 0.001, as determined by one-way ANOVA with the LSD post hoc test or Kruskal–Wallis ANOVA with post hoc Dunn’s multiple comparison tests
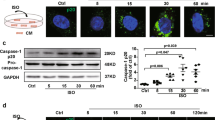
To observe the direct effects of PE on NLRP3 inflammasomes, Langendorff-perfused hearts were treated with PE. The concentration-response experiments revealed that PE at 10 μM induced the highest protein expression level of NLRP3 (Supplementary Fig. S5). In addition to NLRP3 expression, PE at 10 μM caused the cleavage of caspase-1 and IL-18 in hearts at 3 h following PE perfusion compared to those with vehicle perfusion (Fig. 4a). The PE-induced increase in NLRP3 expression was inhibited by blocking α1-ARs with prazosin (Supplementary Fig. S6). The effect of PE and prazosin on α1-AR signaling was confirmed by detecting IP3 (Supplementary Fig. S7). The activation of NLRP3 upon α1-AR activation was further validated in isolated NMCMs. PE increased the protein level in a concentration-dependent manner, and the highest level was reached at 10 μM (Supplementary Fig. S8). NLRP3, cleaved caspase-1, and IL-18 were rapidly increased at 5, 15, and 30 min following PE exposure (Fig. 4b). The increase in NLRP3 was suppressed by α1-AR blockade (Supplementary Fig. S9). The blockade of α1-AR can be indicated by the inhibition of IP3 (Supplementary Fig. S10). In addition, PE treatment did not cause severe cell death within 2 h, as indicated by the CCK-8 assay (Supplementary Fig. S11).

PE induced NLRP3/caspase-1/IL-18 pathway activation in isolated mouse hearts and cardiomyocytes. a Protein levels of NLRP3, caspase-1 (p20), and cleaved IL-18 in Langendorff-perfused hearts with 10 μM PE at the indicated time points, as revealed by Western blotting analysis (n = 6). b Protein levels of NLRP3, caspase-1 (p20), and cleaved IL-18 in NMCMs after 10 μM PE treatment at the indicated time points (n = 6). The results are presented as the mean ± SEM. *P < 0.05; **P < 0.01, as determined by one-way ANOVA with the LSD post hoc test and Student’s unpaired two-tailed t-test or Kruskal–Wallis ANOVA with post hoc Dunn’s multiple comparison tests. Ctrl, control
NLRP3 deficiency prevented PE-induced cardiac inflammation and dysfunction
The roles of the NLRP3 inflammasome in PE-induced cardiac inflammation and dysfunction were examined using Nlrp3-/- mice. NLRP3 expression was undetected in cardiac samples from Nlrp3-/- mice. Cleaved caspase-1 (p20), IL-18 and IL-18 serum levels were not increased by PE in Nlrp3-/- mice compared with levels in WT mice (Fig. 5a, b), suggesting that NLRP3 deficiency blocked activation of the NLRP3/caspase-1/IL-18 pathway.

PE-induced NLRP3/caspase-1/IL-18 pathway activation was blocked in NLRP3-deficient mice. a Protein levels of NLRP3, caspase-1 (p20), and cleaved IL-18 in the heart of WT and NLRP3-deficient (Nlrp3-/-) mice on day 1 following PE treatment (n = 5). b IL-18 levels in mouse serum on day 1 or day 3 following PE treatment (n = 6). The results are presented as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, as determined by one-way ANOVA with the LSD post hoc test or Kruskal–Wallis ANOVA with post hoc Dunn’s multiple comparison tests
As a result, PE treatment-induced macrophage infiltration was reduced in Nlrp3-/- mouse hearts compared with that in WT mice (Fig. 6a). NLRP3 deficiency also decreased the elevation of proinflammatory IL-6 and the chemokine factors MCP-1 and MCP-5 induced by PE treatment (Fig. 6b). PE-induced cardiac systolic and diastolic dysfunction was also ameliorated in Nlrp3-/- mice, as revealed by the EF% and E/E’ determined by echocardiography (Fig. 6c and Supplementary Table S3).

NLRP3 deficiency prevented cardiac inflammation and dysfunction induced by PE. a Immunostaining of Mac-3 (macrophage marker) in the heart on day 1 or day 3 following PE treatment. Positive stains are indicated by arrows (n = 6). b Concentrations of proinflammatory cytokines (IL-6) and chemokines (MCP-1, MCP-5) at the indicated time points after PE treatment, as measured by ELISA (open bars represent day 1, filled bars represent day 3) (n = 6). c Representative echocardiographic images, EF%, and E/E’ values of the PE-treated Nlrp3-/- mice. (n = 6). The results are presented as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, as determined by one-way ANOVA with the LSD post hoc test or Kruskal–Wallis ANOVA with post hoc Dunn’s multiple comparison tests
Discussion
In addition to β-AR, α1-ARs are also expressed in the heart and are activated during acute sympathetic stress. The present study provides the first evidence that the acute activation of α1-ARs causes cardiac inflammation and dysfunction. Further results showed that the underlying mechanism is via the NLRP3/caspase-1/IL-18 pathway. Knockout of NLRP3 blocked PE-induced cardiac inflammation and dysfunction, indicating that NLRP3 plays an important role in α1-AR-mediated cardiac inflammation. Therefore, the NLRP3 inflammasome can be a therapeutic target for both β-AR- and α1-AR-mediated cardiac inflammation during acute sympathetic stress.
Our previous study found that acute β-AR activation induced macrophage infiltration and cytokine upregulation in hearts, as well as cardiac dysfunction [6]. In this study, the acute activation of α1-ARs, another important AR subtype, can also cause elevated proinflammatory cytokines and macrophage infiltration in hearts. Among these cytokines, MCP-1 and MCP-5 are chemokines that can promote macrophage recruitment to inflammation sites [14, 15], and IL-6 is considered to occur downstream of inflammatory cell infiltration [16]. However, in this study, although cardiac macrophage infiltration induced by acute α1-AR activation was rare on day 1, IL-6 expression was increased. On day 3, the macrophage infiltration was increased, but the IL-6 expression was decreased compared with day 1. This may be because α1-ARs can directly stimulate IL-6 messenger RNA (mRNA) expression and protein secretion from cardiomyocytes by regulating transcription through p38 and nuclear factor κB (NF-κB) signaling as well as mRNA stability [17]. Notably, TNF-α, another important proinflammatory cytokine, was unaffected by acute α1-AR activation. In our previous study, β-AR activation induced the upregulation of TNF-α. This result suggests that there are discrepancies between α1-AR- and β-AR-mediated cardiac inflammation. Nonetheless, the overall effect of acute α1-AR activation is the promotion of cardiac inflammation. Moreover, acute α1-AR activation caused cardiac dysfunction in our study. Therefore, acute sympathetic stress induced cardiac inflammation and dysfunction through both α1-ARs and β-ARs.
Chronic α1-AR activation has been found to induce the upregulation of cytokines and cardiac remodeling by increasing pressure overload [18]. NLRP3 inflammasome activation mediated cardiac dysfunction and myocardial remodeling in chronic pressure-overloaded hearts [19]. This study clearly found that acute α1-AR activation can also cause cardiac inflammation and dysfunction through NLRP3 inflammasome activation in vivo. Furthermore, α1-AR activation can directly activate the NLRP3 inflammasome in Langendorff-perfused hearts and cultured cardiomyocytes independent of the effects caused by the increase in BP. An acute β-AR insult induced rapid NLRP3 inflammasome activation through the reactive oxygen species (ROS) pathway, as shown in our previous study [6]. ROS are crucial for NLRP3 inflammasome activation [20], and NADPH oxidase 4, a source of cellular superoxide anions, promotes NLRP3 inflammasome activation [21]. Interestingly, α1-AR agonists can also promote ROS elevation by NADPH in ventricular cardiac myocytes of rats [22]. Thus, ROS may contribute to NLRP3 inflammasome activation following acute overactivation of both α1-AR and β-AR.
The inhibition of β-AR overactivation by β-blockers has been recommended for various heart diseases, including acute coronary syndromes and stress cardiomyopathy [3, 23]. However, β-blockers have limitations, as they also antagonize physiological effects caused by β-AR, such as positive chronotropic, dromotropic, and inotropic effects. This study provided another limitation of the application of β-blockers in acute sympathetic stress in that the α1-AR insult can also induce cardiac inflammation that cannot be blocked by β-blockers. Furthermore, our studies found that activation of the NLRP3 inflammasome mediated cardiac inflammation induced by both α1-ARs and β-ARs. Thus, the NLRP3 inflammasome can be a potential target for suppressing inflammation induced by overactivation of both α1-AR and β-AR during acute sympathetic stress.
In conclusion, α1-AR overactivation induces cardiac inflammation and dysfunction by activating the NLRP3 inflammasome in cardiomyocytes. Our findings suggest that the NLRP3 inflammasome is a potential target for inhibiting cardiac inflammation caused by an acute α1-AR insult during sympathetic stress.
References
Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352:539–48.
Anand IS, Fisher LD, Chiang YT, Latini R, Masson S, Maggioni AP, et al. Changes in brain natriuretic peptide and norepinephrine over time and mortality and morbidity in the Valsartan Heart Failure Trial (Val-HeFT). Circulation. 2003;107:1278–83.
Brodde OE. Beta 1- and beta 2-adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol Rev. 1991;43:203–42.
Huang CJ, Webb HE, Zourdos MC, Acevedo EO. Cardiovascular reactivity, stress, and physical activity. Front Physiol. 2013;4:314.
Manolis AJ, Poulimenos LE, Kallistratos MS, Gavras I, Gavras H. Sympathetic overactivity in hypertension and cardiovascular disease. Curr Vasc Pharmacol. 2014;12:4–15.
Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon beta-adrenergic insult. Eur Heart J. 2018;39:60–9.
Pasqua T, Pagliaro P, Rocca C, Angelone T, Penna C. Role of NLRP-3 Inflammasome in hypertension: a potential therapeutic target. Curr Pharm Biotechnol. 2018;19:708–14.
Wang Z, Hu W, Lu C, Ma Z, Jiang S, Gu C, et al. Targeting NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome in cardiovascular disorders. Arterioscler Thromb Vasc Biol. 2018;38:2765–79.
Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW, Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22:1146–61.
Westman PC, Lipinski MJ, Luger D, Waksman R, Bonow RO, Wu E, et al. Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. 2016;67:2050–60.
Wang Y, Thatcher SE, Cassis LA. Blood pressure monitoring using radio telemetry method in mice. Methods Mol Biol. 2017;1614:75–85.
Liao R, Podesser BK, Lim CC. The continuing evolution of the Langendorff and ejecting murine heart: new advances in cardiac phenotyping. Am J Physiol Heart Circ Physiol. 2012;303:H156–67.
Iwatsubo K, Minamisawa S, Tsunematsu T, Nakagome M, Toya Y, Tomlinson JE, et al. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem. 2004;279:40938–45.
Chen D, Carpenter A, Abrahams J, Chambers RC, Lechler RI, McVey JH, et al. Protease-activated receptor 1 activation is necessary for monocyte chemoattractant protein 1-dependent leukocyte recruitment in vivo. J Exp Med. 2008;205:1739–46.
Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–26.
Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–31.
Perez DM, Papay RS, Shi T. alpha1-Adrenergic receptor stimulates interleukin-6 expression and secretion through both mRNA stability and transcriptional regulation: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB. Mol Pharmacol. 2009;76:144–52.
Koren L, Barash U, Zohar Y, Karin N, Aronheim A. The cardiac maladaptive ATF3-dependent cross-talk between cardiomyocytes and macrophages is mediated by the IFNgamma-CXCL10-CXCR3 axis. Int J Cardiol. 2017;228:394–400.
Li R, Lu K, Wang Y, Chen M, Zhang F, Shen H, et al. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem Biophys Res Commun. 2017;485:69–75.
Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22:1111–29.
Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabon MA, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. 2016;22:1002–12.
Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol. 2002;282:C926–34.
Roffi M, Patrono C, Collet JP, Mueller C, Valgimigli M, Andreotti F, et al. [2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Task force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). G Ital Cardiol (Rome). 2016;17:831–72.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81260028 to Li Wang, No. 81530009 to You-yi Zhang and No. 81670205 to Han Xiao), the Fund for Fostering Young Scholars of Peking University Health Science Center (No. BMU2017PY016 to Han Xiao) and the Open Foundation from Beijing Key Laboratory of Hypertension Research (No. 2017GXY-KFKT-05 to Han Xiao).
Author information
Authors and Affiliations
Contributions
YYZ, HX, and LW conceived the project and designed the study. JZX, JMW, GMH, HJG, SXW, WWC, MZL, WLX, and YS performed the experiments. JZX, JMW, and YNF analyzed the data. JZX and JMW wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Xin, Jz., Wu, Jm., Hu, Gm. et al. α1-AR overactivation induces cardiac inflammation through NLRP3 inflammasome activation. Acta Pharmacol Sin 41, 311–318 (2020). https://doi.org/10.1038/s41401-019-0305-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0305-x
Keywords
This article is cited by
-
Cardioprotective Potential of Cymbopogon citratus Essential Oil against Isoproterenol-induced Cardiomyocyte Hypertrophy: Possible Involvement of NLRP3 Inflammasome and Oxidative Phosphorylation Complex Subunits
Current Medical Science (2024)
-
Dual-omics reveals temporal differences in acute sympathetic stress-induced cardiac inflammation following α1 and β-adrenergic receptors activation
Acta Pharmacologica Sinica (2023)
-
Gefapixant, a Novel P2X3 Antagonist, Protects against Post Myocardial Infarction Cardiac Dysfunction and Remodeling Via Suppressing NLRP3 Inflammasome
Current Medical Science (2023)
-
Psychological stress: neuroimmune roles in periodontal disease
Odontology (2023)
-
Ainsliadimer C, a disesquiterpenoid isolated from Ainsliaea macrocephala, ameliorates inflammatory responses in adipose tissue via Sirtuin 1-NLRP3 inflammasome axis
Acta Pharmacologica Sinica (2022)
