
Abstract
23,24-Dihydrocucurbitacin B (designated as C95 in this article) is a cucurbitane triterpenoid that has been shown to possess a variety of pharmacological activities, such as anti-inflammatory and anti-HIV-1 activities etc. In this study, we investigated the effects of 23,24-dihydrocucurbitacin B on lipid regulation. We showed that 23,24-dihydrocucurbitacin B (1–5 μM) dose-dependently promoted DiI-LDL uptake in HepG2 cells by upregulating low-density lipoprotein receptor (LDLR) protein. In HepG2 cells, 23,24-dihydrocucurbitacin B (1–10 μM) dose-dependently enhanced LDLR promoter activity by elevating the mature form of SREBP2 (sterol regulatory element binding protein 2) protein levels on one hand, and inhibited PCSK9 (proprotein convertase subtilisin/kexin type 9) promoter activity by attenuating HNF1α (hepatocyte nuclear factor-1α) protein levels in nuclei on the other hand. Consequently, the expression of LDLR protein markedly increased, whereas the PCSK9-mediated LDLR protein degradation decreased. In a high-cholesterol LVG golden Syrian Hamster model, administration of 23,24-dihydrocucurbitacin B (30 mg · kg−1⋅ d−1, intragastric, for 3 weeks) significantly decreased the serum LDL-cholesterol (LDL-C) levels. PCSK9 protein levels in the serum and liver tissues were significantly decreased, whereas LDLR protein levels in liver tissues were significantly increased in the treated animals as compared with the control animals. In conclusion, our study demonstrates for the first time that 23,24-dihydrocucurbitacin B exhibits dual transcriptional regulation of LDLR and PCSK9 in HepG2 cells by increasing SREBP2 protein levels and decreasing HNF1α protein levels in the nuclei. These results propose a new strategy to simultaneously manage LDLR and PCSK9 protein expression and provide a promising lead compound for drug development.
Similar content being viewed by others


Introduction
LDL-cholesterol (LDL-C) has been considered a key predictor for cardiovascular disease, which is presently the leading cause of mortality and morbidity worldwide [1,2,3]. At present, statins, which are HMG-CoA reductase (HMGCR) inhibitors, are the mainstays for lipid-lowering treatment. HMGCR is the rate-limiting enzyme in the cholesterol synthesis pathway. Depletion of intracellular cholesterol activates the proteolytic cleavage of sterol regulatory element binding protein 2 (SREBP2) and releases the mature form of SREBP2 into the cell nucleus. By binding with sterol regulatory element 1 (SRE1) in the LDLR promoter region, SREBP2 upregulates LDLR transcription, resulting in LDL-C reductions in the blood [4].
In addition to low-density lipoprotein receptor (LDLR), statins also promote the expression of the proprotein convertase subtilisin/kexin type 9 (PCSK9) protein [5]. PCSK9 acts as a natural eliminator of LDLR by binding to the EGF-A extracellular domain of the LDLR protein, triggering its degradation in lysosomes and thus increasing the levels of circulating LDL-C [6]. Both the LDLR and PCSK9 genes harbor the positive regulatory motif SRE1 in their promoter regions [5, 7, 8]. Statins upregulate the expression of LDLR and PCSK9 through the SREBP2-SRE1 pathway in HepG2 cells [5]. Upregulation of PCSK9 is believed to counteract the effects of statins to further reduce LDL-C levels in patients [5]. Another positive regulator of PCSK9 transcription is the hepatocyte nuclear factor-1α (HNF1α) protein. HNF1α promotes PCSK9 transcription by binding with the HNF1 motif, which is located upstream of SRE1 in the PCSK9 promoter [9]. The plant-derived compound berberine (BBR) has been reported to transcriptionally suppress PCSK9 expression by attenuating nuclear SREBP2 and HNF1α protein abundance [9]. Given the posttranslational regulation of LDLR by PCSK9, PCSK9 elimination and interruption of LDLR-PCSK9 interactions have been demonstrated to be effective strategies to combat dyslipidemia [10]. The first humanized PCSK9 antibodies, alirocumab, and evolocumab, significantly reduce cardiovascular mortality [11, 12]. However, their daunting costs and troublesome subcutaneous administration are evident drawbacks. Therefore, small orally available compounds acting as PCSK9 regulators with excellent efficacy and low cost are desired.
23,24-Dihydrocucurbitacin B, designated as C95 in this article, is a cucurbitane triterpenoid extracted from the roots of Trichosanthes cucumeroides. C95 has been reported to possess multiple pharmacological activities, such as delayed type hypersensitivity-inhibiting [13], antitumor [14,15,16], anti-inflammatory [17], and anti-HIV-1 activities [18]. However, few reports are available about the lipid-regulating effects of C95.
Herein, we evaluated the effects of C95 on LDL uptake in HepG2 cells in vitro, explored the underlying mechanisms, and assessed the lipid-lowering efficacy of C95 in high-fat diet (HFD)-fed hamsters in vivo. We found that C95 dose-dependently promoted DiI-LDL uptake through an LDLR-dependent pathway in HepG2 cells. Further investigations revealed that C95 dose-dependently enhanced LDLR protein levels by promoting LDLR mRNA expression and attenuating PCSK9-mediated LDLR protein degradation. Furthermore, C95 potently improved the lipid profile in a high-cholesterol golden Syrian hamster model. Our findings demonstrate, for the first time, that 23,24-dihydrocucurbitacin B exerts dual regulation of LDLR and PCSK9 through a unique mechanism.
Materials and methods
Materials and chemicals
Dulbecco’s modified Eagle’s medium (DMEM, catalog number: SH30243.01) was purchased from HyClone Laboratories (Logan, UT, USA). Fetal bovine serum (FBS, catalog number: 10091–148) and Lipofectamine 3000 (catalog number: L3000–015) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Atorvastatin (atorvastatin calcium salt trihydrate, catalog number: PZ0001, ≥98%-HPLC) and the β-actin antibody (catalog number: A5441, 1:3000) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies for human LDLR (catalog number: ab52818, 1:1000) and PCSK9 (catalog number: ab181142, 1:2000) were purchased from Abcam (Cambridge, UK), and the Lamin B1 antibody (catalog number: 66095–1, 1:2000) was purchased from Proteintech (Chicago, IL, USA).
Antibodies for hamster proteins: the LDLR antibody was purchased from BioVision (Mountain View, CA, USA, catalog number: 3839, 1:2000). The PCSK9 antibody was a kindly gift from the laboratory of Prof. Jing-wen Liu (Department of Veterans Affairs, Palo Alto Health Care System, Palo Alto, CA, USA). The HNF1α antibody was obtained from Santa Cruz (catalog number: sc-6547, 1:2000). The SREBP2 antibody was obtained from Abcam (catalog number: ab30682, 1:2000).
Hamster serum triglyceride (TG), total cholesterol (TC), high density lipoprotein cholesterol (HDL-C), LDL-C, and the activities of aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were measured with commercial kits (Shino-Test Co., Ltd., Shanghai, China) and the Hitachi 7020 autoanalyzer (Japan).
Extraction and isolation
C95 was extracted and isolated from the roots of T. cucumeroides, which were collected from Yunnan (China) and identified by professor He-ming Yang (Shanghai Institute of Materia Medica). The air-dried, powdered roots (10.0 kg) of T. cucumeroides were extracted three times with 95% aqueous ethanol at room temperature. The combined extracts were concentrated to yield a crude extract, which was partitioned between EtOAc and H2O. The EtOAc layer was added to a silica gel column and eluted with petroleum ether-acetone (1:0 to 0:1). Nine major fractions (A-I) were obtained, and fraction D was chromatographed on a C18 column. C95 (2320 mg) was eluted with 80% MeOH-H2O, further purified with the Sephadex LH-20 column, and eluted with MeOH. C95 was identified as the known compound 23,24-dihydrocucurbitacin B by comparison of the 13C NMR spectra with data reported previously [19].
23,24-Dihydrocucurbitacin B (C95)
13C NMR (100 MHz, pyridine-d5) δ: 19.5, 20.6, 21.0, 22.4, 22.8, 24.7, 26.1, 26.6, 26.7, 30.0, 32.8, 34.8, 35.9, 37.5, 43.4, 46.9, 49.3, 49.4, 49.9, 51.5, 51.6, 59.7, 70.9, 73.0, 80.7, 82.2, 120.7, 141.9, 170.7, 213.4, 213.8, 215.7. HR-ESI-MS: m/z 583.3243 [M + Na]+ (calculated for C32H48O8Na+, 583.3241).
High-performance liquid chromatography (HPLC) analysis
MeOH-H2O (0.1% formic acid) (65:35), eluted at 5.45 min, 95.6% purity. C95 was dissolved in dimethyl sulfoxide and serially diluted to predetermined concentrations.
Cell culture
HepG2 cells (catalog number: HB-8065, ATCC, Manassas, VA, USA) were maintained in DMEM with 10% FBS (v/v) and incubated under a humidified atmosphere of 95% O2 and 5% CO2 at 37 °C. The cells were subcultured once every 2 days. Cells within 4–11 passages were used for experiments.
Cell viability analysis
HepG2 cells were seeded in 96-well plates (1.0 × 104 cells/well) for 24 h before treatment with the indicated concentrations of C95. After another 24 h, cell viability was detected with the Enhanced Cell Counting Kit-8 (Enhanced CCK-8, catalog number: C0043) according to the manufacturer’s instructions (Beyotime Biotechnology, Shanghai, China). The viability of cells treated with vehicle control dimethyl sulfoxide (DMSO) was expressed as 1.0.
Lipoprotein isolation and DiI-LDL preparation
Human plasma was obtained from Shanghai Xuhui Central Hospital, China, after informed consent was obtained and approval was granted by the Ethics Committee. The procedures conformed to the principles outlined in the Declaration of Helsinki [20]. LDL and lipoprotein-deficient serum (LPDS) were separated from the pooled plasma of healthy volunteers by ultracentrifugation and were then dialyzed in dialysis buffer and phosphate-buffered saline (PBS). LDL was then labeled with the fluorescent probe DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate, catalog number: 60010, Biotium, CA, USA) as previously described [21], with some modifications. In brief, DiI was dissolved in DMSO (15 mg/mL) and added into the LDL/LPDS mixture (v/v, 1:2) to a final concentration of 300 mg DiI/mg LDL protein, and the mixture was incubated for 18 h at 37 °C. The mixture was subjected to ultracentrifugation to obtain DiI-labeled LDL (DiI-LDL) and then dialyzed in dialysis buffer and PBS. The obtained DiI-LDL was sterilized using 0.45 μm filters (Millipore, MA, USA).
DiI-LDL uptake assay
DiI-LDL uptake assays were conducted as described previously [21], with minor modifications. In brief, HepG2 cells were seeded in 24-well plates. After specific treatments, the culture medium was changed to DiI-LDL DMEM (20 μg/mL), and the cells were incubated for 3 h at 37 °C in the dark. Then, the cells were rinsed twice with ice-cold PBS buffer containing 0.4% albumin (Sigma-Aldrich) and washed twice with ice-cold PBS buffer. Then, 500 μL of isopropanol was added into each well, and the cells were incubated for 20 min in the dark at room temperature with constant shaking. Aliquots of 200 μL were used for fluorescence detection with the SpectraMax M2e Microplate Reader (520–578 nm, Molecular Devices, San Jose, CA, USA).
Western blot analysis of proteins in hamster liver tissues and HepG2 cells
HepG2 cells were cultured in six-well plates for 12 h. Then, the medium was changed to 2% LPDS for 24 h, after which the cells were subjected to different treatments for the indicated time points. Then, the cells were washed three times with ice-cold PBS buffer, and total cellular protein was extracted using 100 μL/well lysis buffer containing a protease and phosphatase inhibitor cocktail (catalog number: 539134, Calbiochem, Frankfurt, HE, Germany) and centrifuged at 12000 × g at 4 °C for 10 min. Cell nuclear and cytoplasmic proteins were extracted using the Nuclear and Cytoplasmic Protein Extraction Kit according to the manufacturer’s instructions (catalog number: P0013B, Beyotime Biotechnology).
Proteins in liver tissues were extracted using 200 μL of lysis buffer containing the protease and phosphatase inhibitor cocktail from 20–40 mg of frozen liver tissues.
The protein concentrations were determined with the BCA Protein Assay kit (catalog number: P0010, Beyotime Biotechnology). In total, 30 μg/lane of proteins were subjected to 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto PVDF membranes (catalog number: 162–0177, Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% skim milk for 2 h at room temperature and then incubated with primary antibodies overnight at 4 °C. The membranes were then washed three times in TBST solution, incubated with secondary antibodies (catalog number: 1706515 or 1706516, Bio-Rad) for 2 h and visualized using Clarity Western ECL Blotting Substrates (catalog number: 170–5061, Bio-Rad). The values were normalized to those for the housekeeping protein Lamin B1 or β-actin.
RNA interference
Small interfering RNA (siRNA) was transfected into HepG2 cells at 50%–60% confluence using Lipofectamine 3000 reagent according to the manufacturer’s instructions (Invitrogen, Thermo Fisher Scientific). SiRNAs against human LDLR (forward: 5′-CGGCUUAAGAACAUCAACAdTdT-3′, reverse: 5′-UGUUGAUGUUCUUAAGCCGdTdT-3′), PCSK9 (forward: 5′-GGUCUGGAAUGCAAAGUCAdTdT-3′, reverse: 5′-UGACUUUGCAUUCCAGACCdTdT-3′) and a scrambled siRNA were synthesized by GenePharma Inc. (Shanghai, China). After transfection, the cells were cultured in fresh medium for another 24 h before treatment.
RNA isolation and real-time QPCR
Total RNA from HepG2 cells or hamster liver tissues was extracted using TRIzol Reagent (catalog number: 15596–018, Thermo Fisher Scientific) and reverse transcribed into cDNA (Takara, Dalian, China). Real-time QPCR was performed using SYBR Green PCR Supermix (catalog number: 172–5125, Bio-Rad) with the specific primers listed in Table 1. The relative signal intensities were measured with the CFX Real-Time PCR Detection System (Bio-Rad). The values were normalized to those for the housekeeping gene GAPDH and then analyzed in Bio-Rad CFX Manager software using the mode for normalized expression (2−ΔΔCt). The results are presented as fold changes in expression relative to the expression in the control group.
Transient transfections of luciferase reporter constructs
Wild-type or mutant LDLR and PCSK9 promoter reporter constructs were co-transfected with the Renilla luciferase vector, which was constitutively expressed as a transfection control, into HepG2 cells using Lipofectamine 3000 reagent. Luciferase activity was evaluated according to the manufacturer’s instructions (Promega Inc., Madison, WI, USA). The data were normalized to the Renilla luciferase activity.
Animals and drug treatment
Male LVG golden Syrian hamsters (strain code: 501), with body weights of 80–90 g, were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd., China. All hamsters were housed in ventilated cages under a 12 h light–dark cycle with free access to water. The animals were fed a normal diet for the first week to allow them to become acclimated to the environment, and the normal diet was then switched to a HFD containing 0.5% cholesterol and 10% lard oil until the end of the experiment. After feeding of the HFD for another week, blood was collected via the retro-orbital sinus from hamsters fasted for 16 h and subjected to centrifugation at 4000 × g for 10 min at room temperature to obtain serum. Then, lipid profile analyses, including analyses of TC, HDL-C, LDL-C, and TG, were performed. The hamsters were then grouped into two groups with similar lipid profiles: a HF group (n = 6) and a C95 group (n = 6), which were treated with vehicle and C95 (30 mg· kg−1⋅ d−1), respectively, in an intragastric (i.g.) manner for 21 days (D1–21). The serum lipid profiles and body weights were measured at four time points: the day before the switch to the HFD, one week after the switch to the HFD, on D10 after treatment and on D21 after treatment. Food intake was monitored every day during treatment (D1–21). At the end of the administration period (D21), the hamsters were anaesthetized with inhaled isoflurane (2%) and sacrificed to obtain liver samples and serum. The liver samples were weighed and stored at −80 °C in parts. The serum PCSK9 protein levels and serum ALT and AST activities were assessed.
During the experiments, all procedures were conducted under the guidelines of Directive 2010/63/EU of the European Parliament for the protection of animals used for scientific purposes [22] and were approved by the Institutional Ethics Committee of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Enzyme-linked immunosorbent assay (ELISA)
At the end of the administration period (D21), the hamsters were anaesthetized with inhaled isoflurane (2%). Blood was collected via the retro-orbital sinus from hamsters and subjected to centrifugation at 4000 × g for 10 min at room temperature to obtain serum. The PCSK9 protein levels in serum were measured using the Quantikine ELISA Kit for Mouse Proprotein Convertase 9/PCSK9 according to the manufacturer’s instructions (catalog number: MPC900, R&D Systems Inc., Minneapolis, MN, USA).
Statistical analysis
All statistical analyses were performed following the recommendations for experimental design and analyses in pharmacology [23]. The values are presented as the mean ± S.D. All data were obtained from at least three independent experiments.
Student’s t test was used to determine two sets of data which were significantly different from each other. One-way ANOVA was used to analyze differences among at least three groups of data, and Dunnett’s multiple comparisons test was used to compare all groups with the control group. Statistical analysis was performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). Linear regression analysis was used to evaluate the dose-dependent relationship. Differences were considered significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
C95 elevates DiI-LDL uptake by increasing LDLR protein levels in HepG2 cells
C95 exhibited little effect on the viability of HepG2 cells within the experimental concentrations (1–20 μM), and the viability of cells in all groups was above 95% (Fig. 1b). DiI-LDL uptake assays were then conducted to evaluate the effects of C95 on LDL uptake in vitro. C95 dose-dependently promoted DiI-LDL uptake in HepG2 cells (Fig. 1c). Accordingly, LDLR protein levels increased dose-dependently (Fig. 1d). As LDL uptake can be mediated in both LDLR-dependent and LDLR-independent ways [24], the involvement of LDLR in C95-induced LDL uptake was evaluated by siRNA-mediated knockdown. Knockdown of LDLR with the specific siRNA abolished the C95-induced elevation of DiI-LDL uptake (Fig. 1e, f).

C95 elevates LDL uptake by increasing LDLR protein levels in HepG2 cells. a Chemical structure of C95. b The effect of C95 on the viability of HepG2 cells. c C95 dose-dependently promoted LDL uptake in HepG2 cells. Atorvastatin (ATOR, 5 μM) was used as a positive control. d C95 increased LDLR protein levels in HepG2 cells. Atorvastatin (ATOR, 5 μM) was used as a positive control. e Efficacy of si-LDLR. f Knockdown of LDLR protein by siRNA abolished the effect of C95 on LDL uptake activity in HepG2 cells. Results are presented as means ± S.D., n ≥ 5. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle control by one-way ANOVA (Dunnett’s multiple comparisons test)
Therefore, we concluded that C95 regulated DiI-LDL uptake by increasing LDLR protein levels in HepG2 cells.
C95 upregulates LDLR mRNA levels and attenuates PCSK9 mRNA levels in HepG2 cells
As mentioned above, PCSK9 has an important role in the posttranslational regulation of the LDLR protein. We further assessed whether PCSK9 was involved in the C95-induced LDLR elevation in HepG2 cells. To this end, after treatment with C95, whole-cell lysates and cell culture medium were subjected to Western blot assays. As expected, LDLR protein levels increased dose-dependently in response to C95 treatment. In addition, PCSK9 protein levels were reduced in both HepG2 cells and cell culture medium in a dose-dependent manner (Fig. 2a). We then sought to determine whether the increases in LDLR protein levels were wholly induced by C95-mediated reductions in PCSK9 protein levels. To remove the influence of PCSK9 on LDLR protein abundance, we knocked out PCSK9 protein with a specific siRNA. To our surprise, C95-induced additional increases in LDLR protein levels and LDL uptake activity in PCSK9-siRNA transfected HepG2 cells (Fig. 2b). In addition, C95 dose-dependently increased LDLR mRNA levels (Fig. 2c, left). To the best of our knowledge, PCSK9 promotes the degradation of LDLR protein but has little effect on LDLR mRNA. Knockdown of PCSK9 in human primary hepatocytes [25], mouse livers [26], hamster livers [27], and nonhuman primates [28] has little effect on LDLR mRNA expression. The results implied that C95 upregulated LDLR protein levels not only by decreasing PCSK9 protein levels, but also by increasing LDLR mRNA levels in HepG2 cells. Moreover, C95 dose-dependently decreased PCSK9 mRNA levels in HepG2 cells (Fig. 2c, right).

C95 upregulates LDLR mRNA levels and attenuates PCSK9 mRNA levels in HepG2 cells. a C95 downregulated PCSK9 protein levels in both HepG2 cell lysates (C-PCSK9) and culture medium (M-PCSK9). b The effects of C95 on LDLR protein abundance and LDL uptake activity in HepG2 cells transfected with PCSK9-siRNA. HepG2 whole-cell lysates were subjected to Western blot assays to detect the LDLR and PCSK9 protein levels. *P < 0.05, **P < 0.01, ***P < 0.001 vs. untreated and un-transfected cells group, #P < 0.05, ###P < 0.001 vs. untreated cells transfected with PCSK9-siRNA by one-way ANOVA (Dunnett’s multiple comparisons test). c The effects of C95 on LDLR mRNA (left column) and PCSK9 mRNA (right column) levels in HepG2 cells. Results are presented as means ± S.D., n ≥ 5. ***P < 0.001 vs. vehicle control by one-way ANOVA (Dunnett’s multiple comparisons test)
These results showed that consistent with its effects on protein levels, C95 upregulated LDLR mRNA levels and attenuated PCSK9 mRNA levels in HepG2 cells.
C95 inhibits PCSK9 promoter activity through the HNF1 motif and upregulates LDLR promoter activity through the SRE1 motif
To determine the mechanism by which C95 downregulated PCSK9 mRNA levels and simultaneously upregulated LDLR mRNA levels in HepG2 cells, luciferase reporter assays were carried out to analyze the effects of C95 on PCSK9 and LDLR promoter activities. In line with its effects on protein and mRNA levels, C95 dose-dependently inhibited PCSK9 promoter activity and enhanced LDLR promoter activity (Fig. 3a, b).

C95 inhibits PCSK9 promotor activity through HNF1 motif and upregulates LDLR promotor activity through SRE1 motif. a, b C95 inhibited PCSK9 and enhanced LDLR promoter activities. c, d Schematic presentation of the mutation constructs of human PCSK9 and LDLR promoter luciferase reporters. e, f Relative luciferase activities of transfected cells treated or untreated with C95. Atorvastatin (ATOR) served as a positive control. Results are presented as means ± S.D., n = 5. *P < 0.05, #P < 0.05, ***P < 0.001 vs. vehicle control by one-way ANOVA (Dunnett’s multiple comparisons test)
To define the specific sites in the PCSK9 and LDLR promoters that were responsible for C95-induced regulation, we transfected HepG2 cells with PCSK9 and LDLR promoter luciferase reporter constructs (Fig. 3c, d) to evaluate promoter activity changes. Consistent with previous research [9], mutations in the HNF1 or SRE1 motif in the PCSK9 promoter induced dramatic reductions in promoter activity. Moreover, C95 induced additional decreases in cells transfected with the SRE-mut construct (P < 0.05 vs. vehicle control in the same group) but not in cells transfected with the HNF-mut construct (P > 0.05, Fig. 3e), which indicated that the HNF1 motif was the major functional site through which C95 decreased PCSK9 protein expression in HepG2 cells. In cells transfected with LDLR promoter luciferase reporter constructs, increases in promoter activity induced by atorvastatin or C95 were abolished by mutation of the SRE1 motif in the LDLR promoter (Fig. 3f), which proved that C95 upregulated LDLR promoter activity through the SRE1 motif.
These results implied that C95 inhibits PCSK9 promoter activity mainly through the HNF1 motif and upregulates LDLR promoter activity mainly through the SRE1 motif.
C95 influences both HNF1α and SREBP2 protein abundance in HepG2 cell nuclei
Considering that the SRE1 and HNF1 motifs are the functional sites for SREBPs and HNF1α, which are transcription factors, to regulate their target genes, the protein levels of HNF1α, SREBP2, LDLR, and PCSK9 in HepG2 cell lysates were monitored after exposure to C95 at 5 μM for various durations (0–24 h). Intriguingly, we found that the levels of the mature form of the SREBP2 protein were elevated within the first 3 h of treatment and then returned to normal afterwards. However, the HNF1α protein levels declined until the end of treatment (24 h), and at 24 h, the protein level was approximately 0.51 times that at 0 h (P < 0.05). Accordingly, the PCSK9 protein levels decreased until the end of treatment (24 h), at which time the protein abundance was ~ 0.44 times that at 0 h (P < 0.05). In contrast, the LDLR protein levels continuously increased within this period (Fig. 4a). In addition, LDLR and PCSK9 mRNA levels were quantified in HepG2 cells with the same treatments during the same time periods (1–24 h). Similarly, C95 induced a continuous increase in LDLR mRNA in the first 12 h of treatment, with a slight decrease at 24 h. Interestingly, the PCSK9 mRNA levels slightly but nonsignificantly increased within the first 3 h of treatment and then tended to decrease until the end of treatment (24 h), at which time it was only 0.59 times that at 0 h (P < 0.05) (Fig. 4b). Based on the time-related changes in the HNF1α and SREBP2 proteins during the treatment process, we next detected these two proteins in HepG2 cells after exposure to different concentrations of C95 for 24 h and 3 h, respectively, at which time the two protein levels were changed significantly (Fig. 4a). The results showed that C95 (1–10 μM) dose-dependently decreased HNF1α protein levels in both the cytoplasm and nuclei after treatment for 24 h (Fig. 4c) and dose-dependently (1–10 μM) elevated the levels of the mature form of the SREBP2 protein after treatment for 3 h (Fig. 4d). BBR, which has been demonstrated to decrease HNF1α protein levels after 24 h of treatment in HepG2 cells [9], was used as a positive control (Fig. 4c).

C95 influences both HNF1α and SREBP2 protein abundance in HepG2 cell nuclei. a LDLR, PCSK9, SREBP2 (mature form), HNF1α and β-actin protein levels in HepG2 cell lysates were quantified by Western blot assays after exposed to 5 μM C95 for various time points (0–24 h). b LDLR and PCSK9 mRNA levels in HepG2 cells were quantified by real-time QPCR after exposed to 5 μM C95 for various time points (0–24 h). c HNF1α protein levels were quantified in HepG2 cytoplasmic (C-HNF1α) and nuclear lysates (N-HNF1α) after treatment with different concentrations of C95 for 24 h. BBR (10 μM) served as a positive control. d SREBP2 (mature form) protein levels were quantified in HepG2 cell lysates after treatment with different concentrations of C95 for 3 h. Results are presented as means ± S.D., n ≥ 5. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle control by one-way ANOVA (Dunnett’s multiple comparisons test)
C95 improves lipid profiles in HFD-fed hamster serum
To evaluate the lipid-lowering effects of C95 in vivo, HFD-fed golden Syrian hamsters were treated with C95 (i.g., 30 mg · kg−1⋅ d−1, n = 6) or vehicle (n = 6) for 21 days (D1–21).
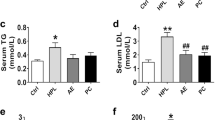
After 10 days of treatment, TC (Fig. 5a), TG (Fig. 5b), and LDL-C (Fig. 5c) levels in the treatment group were significantly decreased compared with those in the HF group (P < 0.01 or P < 0.001). These effects were even more significant at the end of the treatment period (D21, P < 0.001 vs. HF group). No significant differences were induced in serum HDL-C (Fig. 5d). The activities of serum ALT and AST were assessed to evaluate the possible damage to hamster liver tissues. We found that the activities of the two enzymes were similar between the two groups (P > 0.05, Fig. 5e, f), as were the body weights and food intake (P > 0.05, Supplementary Figure S1).

C95 improves lipid profiles in HFD-hamsters serum. a–d Serum total cholesterol (TC), triglyceride (TG), LDL-C, and HDL-C of HFD-hamsters were measured at indicated time points. Serum TC, TG, and LDL-C were significantly attenuated at D10 in the treatment group compared with the HF group, and these effects were even more significant at the end of the treatment period (D21). No significant differences were induced in serum HDL-C levels between the two groups. e, f The activities of ALT and AST had no significant difference between the two groups. g, h, l LDLR, PCSK9, HMGCR, and HMGCS1 mRNA levels in hamsters liver tissues. i PCSK9 protein levels in serum were quantified by ELISA at the end of the experiment. j, k LDLR, PCSK9, HNF1α, and SREBP2 proteins levels in hamster liver tissue lysates were detected with Western blot assays. Results are presented as means ± S.D., n = 6. *P < 0.05, **P < 0.01, ***P < 0.001 vs. HF group by one-way ANOVA (Dunnett’s multiple comparisons test, Fig. 5a–d), or by Student’s t test (Fig. 5e–l)
In addition, consistent with the results obtained from HepG2 cells, LDLR mRNA (Fig. 5g), LDLR protein (Fig. 5j), and SREBP2 protein (mature form, Fig. 5k) levels in hamster liver tissues were significantly elevated after treatment. PCSK9 mRNA (Fig. 5h), PCSK9 protein (Fig. 5j), and HNF1α protein (Fig. 5k) levels in hamster liver tissues significantly declined after administration of C95 at 30 mg · kg−1⋅ d−1 for 3 weeks, as were PCSK9 protein levels in serum at D21 (Fig. 5i).
Furthermore, in addition to those of LDLR, the mRNA levels of HMGCR and HMGCS1, two other target genes of SREBP2, were detected by real-time QPCR to assess whether the SREBP2 pathway was activated by C95 in hamster liver tissues. Compared with those in the HF group, the mRNA levels of both HMGCR and HMGCS1 in hamster liver tissues were significantly elevated in the C95 treatment group (Fig. 5l), which confirmed that the SREBP2 pathway was indeed activated by C95 in hamster liver tissues. These results proved that C95 improves lipid profiles in HFD-fed hamster serum and validated that C95 is a promising lipid-lowering compound in vivo.
Discussion
In this study, for the first time, we evaluated the effects of 23,24-dihydrocucurbitacin B on LDL uptake in vitro and the lipid-lowering effects in vivo and explored the mechanism of action of this compound in lipid regulation. We demonstrated that 23,24-dihydrocucurbitacin B promotes LDLR expression and diminishes PCSK9 expression at the transcriptional level by increasing SREBP2 protein while decreasing HNF1α protein in HepG2 cell nuclei.
BBR has been reported to dually regulate LDLR and PCSK9 [9]; BBR upregulates LDLR expression by stabilizing LDLR mRNA and reduces the protein levels of both HNF1α and SREBP2 in the nucleus to suppress transcription of PCSK9. In contrast, 23,24-dihydrocucurbitacin B upregulates LDLR expression by increasing LDLR mRNA levels through promotion of SREBP2 protein levels and downregulates PCSK9 expression by decreasing HNF1α protein levels in the cell nucleus. We found that the levels of the mature form of the SREBP2 protein were elevated within the first 3 h of treatment, whereas HNF1α protein levels decreased throughout the 24 h of treatment (Fig. 4a) in HepG2 cells. The elevations in the levels of the SREBP2 protein (mature form) induced by C95 at 3 h were dose-dependent (Fig. 4d), as was the inhibition of HNF1α protein at 24 h (Fig. 4c). Moreover, the C95-mediated upregulation of SREBP2 protein and downregulation of HNF1α protein were also confirmed in hamster liver tissues (Fig. 5k). However, in the current study, we did not focus on why and how these time-related changes occurred in HepG2 cells or whether the changes in SREBP2 and HNF1α proteins in hamster liver tissues were time-related. We will try to answer these questions in our future studies.
However, we noticed that the 1.5- to 2.0-fold increases in SREBP2 protein and LDLR promoter activity (Figs. 4a, 3b) were inconsistent with the increase in LDLR mRNA after exposure to 5 μM C95, which reached a level approximately six-fold greater than that in the control group (Fig. 4b). Moreover, the change trends between LDLR mRNA and SREBP2 protein were inconsistent as well. The decline in LDLR mRNA (Fig. 4b) occurred several hours later than the decline in SREBP2 protein (Fig. 4a). Considering that LDLR mRNA is unstable because of the three AU-rich elements (AREs) in its 3′-UTR and that the half-life of LDLR mRNA is approximately 30 min [29], we suspected that C95 might also induce posttranscriptional regulation of LDLR mRNA, such as regulation of the stability of LDLR mRNA, so that the half-life of LDLR mRNA was lengthened. To comprehensively evaluate the effects of C95 on LDLR expression, more experiments are warranted and should be conducted in future studies. Consistent with the decline in HNF1α protein, PCSK9 protein levels decreased during the treatment (Figs. 4a, c, 5j, k). This decline in PCSK9 protein may have contributed to an elevation in LDLR protein such that LDLR protein levels increased continuously during the 24 h of treatment (Fig. 4a), even though the LDLR mRNA levels were decreased at 24 h (Fig. 4b).
The mRNA levels of HMGCR and HMGCS1, the target genes of the SREBP2 protein, were also promoted by C95 in hamster liver tissues, which indicated that the SREBP2 pathway was activated by C95. Given that LDLR and PCSK9 share the common SRE1 motif in their promoter regions, the activation of the SREBP2 pathway might also have promoted the transcription of PCSK9 mRNA. However, the slight increase in PCSK9 mRNA within the first 3 h of treatment did not produce a statistically significant difference in the treatment group compared with the control group (Fig. 4b). We speculated that the possible increase in PCSK9 mRNA caused by SREBP2 activation may have been counteracted by the attenuation of HNF1α protein levels. It has been reported that the upregulation of PCSK9 promoter activity mediated by the SREBP2 protein relies at least partially on the HNF1α protein, since SREBP2 induces no significant increases in PCSK9 promoter activity if the HNF1 motif is mutated [9].
In conclusion, our study evaluated the lipid regulation efficacy of 23,24-dihydrocucurbitacin B both in vitro and in vivo and demonstrated that 23,24-dihydrocucurbitacin B transcriptionally regulates both LDLR and PCSK9 expression. Our study also revealed that 23,24-dihydrocucurbitacin B exhibits a remarkable lipid-lowering effect in vivo. This study expands understanding of the bioactivities of 23,24-dihydrocucurbitacin B, proposes another strategy to regulate LDLR and PCSK9 protein expression and provides a promising lead compound for drug development.
References
Wadhera RK, Steen DL, Khan I, Giugliano RP, Foody JM. A review of low-density lipoprotein cholesterol, treatment strategies, and its impact on cardiovascular disease morbidity and mortality. J Clin Lipidol. 2016;10:472–89.
Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;2014:S1–45.
Grundy SM, Arai H, Barter P, Bersot TP, Betteridge DJ, Carmena R, et al. An International Atherosclerosis Society Position Paper: global recommendations for the management of dyslipidemia-full report. J Clin Lipidol. 2014;8:29–60.
Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161:161–72.
Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–9.
McNutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. 2009;284:10561–70.
Dawson PA, Hofmann SL, van der Westhuyzen DR, Sudhof TC, Brown MS, Goldstein JL. Sterol-dependent repression of low density lipoprotein receptor promoter mediated by 16-base pair sequence adjacent to binding site for transcription factor Sp1. J Biol Chem. 1988;263:3372–9.
Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. 2008;49:399–409.
Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem. 2009;284:28885–95.
Fitzgerald G, Kiernan T. PCSK9 inhibitors and LDL reduction: pharmacology, clinical implications, and future perspectives. Expert Rev Cardiovasc Ther. 2018;16:567–78.
Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:331–40.
Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99.
Escandell JM, Recio MC, Manez S, Giner RM, Cerda-Nicolas M, Gil-Benso R, et al. Dihydrocucurbitacin B inhibits delayed type hypersensitivity reactions by suppressing lymphocyte proliferation. J Pharmacol Exp Ther. 2007;322:1261–8.
Siqueira JM, Gazola AC, Farias MR, Volkov L, Rivard N, de Brum-Fernandes AJ, et al. Evaluation of the antitumoral effect of dihydrocucurbitacin-B in both in vitro and in vivo models. Cancer Chemother Pharmacol. 2009;64:529–38.
Yang L, Wu S, Zhang Q, Liu F, Wu P. 23,24-Dihydrocucurbitacin B induces G2/M cell-cycle arrest and mitochondria-dependent apoptosis in human breast cancer cells (Bcap37). Cancer Lett. 2007;256:267–78.
Lang KL, Silva IT, Zimmermann LA, Machado VR, Teixeira MR, Lapuh MI, et al. Synthesis and cytotoxic activity evaluation of dihydrocucurbitacin B and cucurbitacin B derivatives. Bioorg Med Chem. 2012;20:3016–30.
Recio MC, Prieto M, Bonucelli M, Orsi C, Manez S, Giner RM, et al. Anti-inflammatory activity of two cucurbitacins isolated from Cayaponia tayuya roots. Planta Med. 2004;70:414–20.
Chen JC, Zhang GH, Zhang ZQ, Qiu MH, Zheng YT, Yang LM, et al. Octanorcucurbitane and cucurbitane triterpenoids from the tubers of Hemsleya endecaphylla with HIV-1 inhibitory activity. J Nat Prod. 2008;71:153–5.
Wu PL, Lin FW, Wu TS, Kuoh CS, Lee KH, Lee SJ. Cytotoxic and anti-HIV principles from the rhizomes of Begonia nantoensis. Chem Pharm Bull (Tokyo). 2004;52:345–9.
World Med A. World medical association declaration of Helsinki ethical principles for medical research involving human subjects. J Am Med Assoc. 2013;310:2191–4.
Stephan ZF, Yurachek EC. Rapid fluorometric assay of LDL receptor activity by DiI-labeled LDL. J Lipid Res. 1993;34:325–30.
Wells DJ. Animal welfare and the 3Rs in European biomedical research. In: Annals of the New York academy of sciences; v1245. Animal models: their value in predicting drug efficacy and toxicity. Oxford: Blackwell Science Publishing; 2011. p 14–26.
Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al. Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol. 2015;172:3461–71.
Ye ZJ, Go GW, Singh R, Liu W, Keramati AR, Mani A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J Biol Chem. 2012;287:1335–44.
Zhang L, McCabe T, Condra JH, Ni YG, Peterson LB, Wang W, et al. An anti-PCSK9 antibody reduces LDL-cholesterol on top of a statin and suppresses hepatocyte SREBP-regulated genes. Int J Biol Sci. 2012;8:310–27.
Luo Y, Warren L, Xia D, Jensen H, Sand T, Petras S, et al. Function and distribution of circulating human PCSK9 expressed extrahepatically in transgenic mice. J Lipid Res. 2009;50:1581–8.
Dong B, Singh AB, Azhar S, Seidah NG, Liu J. High-fructose feeding promotes accelerated degradation of hepatic LDL receptor and hypercholesterolemia in hamsters via elevated circulating PCSK9 levels. Atherosclerosis. 2015;239:364–74.
Lindholm MW, Elmen J, Fisker N, Hansen HF, Persson R, Moller MR, et al. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol Ther. 2012;20:376–81.
Wilson GM, Vasa MZ, Deeley RG. Stabilization and cytoskeletal-association of LDL receptor mRNA are mediated by distinct domains in its 3’ untranslated region. J Lipid Res. 1998;39:1025–32.
Acknowledgements
We gratefully acknowledge grants from the Drug Innovation Major Project of China (No. 2018ZX09735001-002-005), the Personalized Medicines—Molecular Signature-based Drug Discovery and Development Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA12040335), the National Natural Science Foundation of China (No. 81773863), and the Youth Innovation Promotion Association. We thank Professor Jing-wen Liu for kindly providing the anti-hamster PCSK9 antibody as a gift. We also express our sincere gratitude to our mentor Professor Yi-ping Wang for his enlightening mentoring and strong support throughout this study, and our deep sorrow for the loss of him, who passed away on 11 April 2018.
Author information
Authors and Affiliations
Contributions
LJX designed this research and revised the manuscript; HHL performed the research and drafted the article; JL and XJZ extracted and isolated the compound used in this article; JML and CX contributed analytical tools and data analysis; WQW analyzed the structure and purity of the compound used in this article; and YLL contributed critical reagents.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, Hh., Li, J., Zhang, Xj. et al. 23,24-Dihydrocucurbitacin B promotes lipid clearance by dual transcriptional regulation of LDLR and PCSK9. Acta Pharmacol Sin 41, 327–335 (2020). https://doi.org/10.1038/s41401-019-0274-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0274-0
Keywords
This article is cited by
-
Targeting proprotein convertase subtilisin/kexin type 9 (PCSK9): from bench to bedside
Signal Transduction and Targeted Therapy (2024)
-
Research on Hepatocyte Regulation of PCSK9-LDLR and Its Related Drug Targets
Chinese Journal of Integrative Medicine (2023)
-
Association of genetic polymorphisms of PCSK9 with type 2 diabetes in Uygur Chinese population
BMC Cardiovascular Disorders (2022)
