
Abstract
Nav1.7 channels are mainly distributed in the peripheral nervous system. Blockade of Nav1.7 channels with small-molecule inhibitors in humans might provide pain relief without affecting the central nervous system. Based on the facts that many reported Nav1.7-selective inhibitors contain aryl sulfonamide fragments, as well as a tricyclic antidepressant, maprotiline, has been found to inhibit Nav1.7 channels, we designed and synthesized a series of compounds with ethanoanthracene and aryl sulfonamide moieties. Their inhibitory activity on sodium channels were detected with electrophysiological techniques. We found that compound 10o potently inhibited Nav1.7 channels stably expressed in HEK293 cells (IC50 = 0.64 ± 0.30 nmol/L) and displayed a high Nav1.7/Nav1.5 selectivity. In mouse small-sized dorsal root ganglion neurons, compound 10o (10, 100 nmol/L) dose-dependently decreased the sodium currents and dramatically suppressed depolarizing current-elicited neuronal discharge. Preliminary in vivo experiments showed that compound 10o possessed good analgesic activity: in a mouse visceral pain model, administration of compound 10o (30−100 mg/kg, i.p.) effectively and dose-dependently suppressed acetic acid-induced writhing.
Similar content being viewed by others


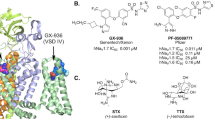
Introduction
Pain severely afflicts almost half a billion people worldwide but has no ideal treatment. There appears to be a correlation between voltage-gated sodium channels (VGSC, Nav) and human pain syndromes, as identified through genetic studies [1, 2]. The human-validated analgesic target sodium channel Nav1.7 is of great interest for the development of new pain medications [3, 4]. It has been reported that gain-of-function mutations in Nav1.7 (SCN9A) lead to several painful conditions, such as inherited erythromelalgia (IEM), paroxysmal extreme pain disorder (PEPD), and small fiber neuropathies, whereas loss-of-function mutations in Nav1.7 lead to congenital insensitivity to pain (CIP) [5,6,7,8]. It has also been reported that people with CIP are characterized by a complete inability to sense pain without disruption in touch or temperature sensing, except for anosmia [9, 10]. Nav1.7 global knockout mice did not respond to pain stimuli [11, 12]. These results have led scientists to believe that Nav1.7 blockade by small-molecule inhibitors in humans might provide pain relief without side effects [3, 4]. One of the challenges in Nav1.7 inhibitor development is subtype selectivity because of the high sequence homology among Nav isoforms. Nonselective sodium channel inhibitors may lead to some adverse side effects [13]. In particular, acceptable selectivity against the cardiac sodium channel Nav1.5 is important for preventing arrhythmic side effects such as long QT syndrome, Brugada syndrome, conduction dysfunction, and atrial fibrillation [14]. Nav1.7 is mainly distributed in the peripheral nervous system (PNS), which provides a possible therapeutic strategy for targeting the PNS without affecting the central nervous system [4, 8, 15]. The development of selective Nav1.7 inhibitors is imperative. To date, a number of structurally diverse and selective Nav1.7 inhibitors have been discovered (Fig. 1) [16,17,18,19,20,21,22].

Known selective Nav1.7 inhibitors and maprotiline
Among those compounds, GNE-131 (1c), with an innovative acyl group replacing a triazole group and a large hydrophobic adamantyl group as its side chain, showed good in vivo efficacy [19]. Based on that design, compound 1d has a simplified adamantane moiety instead of an adamantyl group and maintains good activity while displaying increased liver microsomal stability and improved efficacy (Fig. 1) [20]. Looking at the structure of known Nav1.7 inhibitors, it is not difficult to find that aryl sulfonamide or acyl sulfonamide group is important for activity and selectivity. In particular, compounds containing aryl sulfonamide functionalities have attracted considerable attention. According to the cocrystal structure of GX-936 and Nav1.7, the binding sites of these compounds in the fourth voltage-sensor domain (VSD4) of Nav1.7 can be divided into three parts: anion-binding pocket, selectivity pocket, and lipid-exposed pocket [23]. All the aryl sulfonamide inhibitors have similar warheads, which anchor them in specific regions of the proteins to ensure certain selectivity and activity. The difference between most of these inhibitors is mainly on the other side of the molecule, that is, the part that interacts with the lipid-exposed pocket, which makes it the key moiety for our modification.
Recently, studies showed that several tricyclic antidepressants (TCA), such as amitriptyline, imipramine, and maprotiline (MPT) (Fig. 1), have steady-state affinities for the Nav1.7 inactivated state (Ki) that are much lower than those for the resting state (Kr), demonstrating that they inhibited Nav1.7 channels in a state-dependent manner [24,25,26]. Notably, the Ki values fell in the range of therapeutic plasma concentrations for neuropathic pain relief, suggesting that Nav1.7 channel blocking directly contributed to these TCA-induced analgesic activities [25, 26]. In addition, enhancing channel inactivation and preferentially binding to the channel inactivated state underlie the actions of many identified Nav1.7 channel inhibitors, such as CNV1014802 and PF-05089771 [3, 27, 28]. Therefore, we speculated that these core structures of tricyclic antidepressants might be a good starting point for developing novel Nav1.7 inhibitors. Our group and others have found that MPT is an inhibitor of Nav1.7 with an IC50 value of 1.51 μmol/L for the inactivated state (Fig. 1) [25]. By comparing MPT with other reported Nav1.7 inhibitors, we found that this compound has a unique structure and does not share pharmacophore features with those inhibitors. At the same time, the ethanoanthracene moiety in MPT is a large hydrophobic group that is similar to the adamantyl group in GNE-131, which may provide a key hydrophobic effect and contribute to the potency and selectivity. Considering the good inhibitory activity of MPT against Nav1.7, we speculated that the ethanoanthracene core structure of MPT may facilitate the interaction of the compound with the lipid-exposed pocket of the protein. To improve the inhibition potency, we introduced a warhead, for example, an aryl sulfonamide group, and then we designed and synthesized a series of compounds with ethanoanthracene and aryl sulfonamide moieties.
Materials and methods
Chemistry
The experimental procedures and characterization data of all compounds are provided in the Supplementary Information.
Materials
All compounds were dissolved in dimethyl sulfoxide (DMSO) to produce 20 mmol/L stock solutions and stored at − 20 °C. Before the electrophysiology study, the stock solutions were diluted in the bath solution to obtain the final concentrations. DMSO at the final concentrations (≤ 0.5%) was well-tolerated with no observable toxic effects on cells stably expressing Nav1.7 or Nav1.5 channels. For animal testing, compounds were dissolved in 5% DMSO/95% (0.5% carboxymethyl cellulose sodium).
Cell culture
Human embryonic kidney 293 (HEK293) cells stably expressing hNav1.7 were cultured in high-glucose Dulbecco's Modified Eagle’s Medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and were selected with a 300 μg/mL solution of the antibiotic hygromycin B (Invitrogen, Carlsbad, CA, USA) under standard tissue culture conditions (5% CO2, 37 °C). Human Nav1.5 channels were stably expressed in Chinese hamster lung (CHL) cells that were cultured in high-glucose DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and were selected with a 500 µg/mL solution of the antibiotic G418 (Invitrogen, Carlsbad, CA, USA). Dorsal root ganglion (DRG) neurons were cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. All cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. Before electrophysiological recording, all cells were plated onto poly-L-lysine-coated glass coverslips (Sigma-Aldrich, St Louis, MO, USA) and used within 24 h.
Preparation of dorsal root ganglion neurons
DRG from the thoracic and lumbar sections of C57BL/6 mice (4–5 weeks) were rapidly prepared as described previously [29]. In brief, the isolated ganglia were collected in cold calcium and magnesium-free PBS and cut into pieces with scissors. The fragments were digested with 0.1% collagenase and 0.025% trypsin, which were diluted in DMEM/F12 medium at 37 °C. Then, the digested ganglia were mechanically triturated into single cells with a fire-polished glass Pasteur pipette. The DRG neurons were plated onto poly-D-lysine precoated glass coverslips for the following electrophysiological study.
Electrophysiological study
Whole-cell voltage-clamp and current-clamp recordings were performed at room temperature using an Axopatch 700B patch clamp amplifier (Molecular Devices, Sunnyvale, CA, USA). Pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Sarasota, FL, USA) with an electrode resistance typically ranging from 1.5 to 4 MΩ. To test the inhibitory activities of the compounds on hNav1.7 and hNav1.5 channels, the recording pipette intracellular solution contained the following (in mmol/L): 140 CsF, 10 NaCl, 10 HEPES, 1.1 EGTA and 20 glucose (pH 7.3 adjusted by CsOH); the bath or extracellular solution contained the following (in mmol/L): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and 20 glucose (pH 7.3 adjusted by NaOH). To record the sodium currents in isolated DRG neurons, the pipette solution contained the following (in mmol/L): 120 CsCl, 10 NaCl, 10 TEA-Cl, 10 HEPES, 10 EGTA, 1 CaCl2, and 1 MgCl2 (pH 7.2 adjusted by CsOH); the bath solution contained the following (in mmol/L): 120 NaCl, 1 CaCl2, 20 TEA-Cl, 5 KCl, 1.25 MgCl2, 10 HEPES, and 10 glucose (pH 7.4 adjusted by NaOH). The intracellular and extracellular solutions for the whole-cell current-clamp recording in DRG neurons are as follows: the intracellular solutions contained the following (in mmol/L): 140 KCl, 1 CaCl2, 10 EGTA, 1 MgCl2, and 10 HEPES (pH 7.2 adjusted by KOH); the extracellular solutions contained the following (in mmol/L): 140 NaCl, 5 KCl, 1 CaCl2, 1.25 MgCl2, 10 glucose, 10 HEPES (pH 7.4 adjusted by NaOH). During the recording, the bath solution was continuously perfused using a BPS perfusion system (ALA Scientific Instruments, Westburg, NY, USA). Recording was conducted after a 5 min equilibration period at −80 mV after breaking into the whole-cell configuration. To record sodium currents, the cells were clamped at a holding potential of −40 mV, at which >90% of the sodium channels are inactivated, and then a 20 ms prepulse to −150 mV was applied to partially recover channels from inactivation, followed by a 10 ms pulse to 0 mV to activate the channel at a rate of 1 Hz. Currents were acquired at a 50 kHz sampling frequency and filtered at 2 kHz. Series resistance compensation was used and set to 80%. P/N subtraction was never applied throughout the experiment.
Acetic acid-induced writhing test
All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals under protocols approved by and strictly following guidelines of the Institutional Animal Care and Use Committees (IACUC). The IACUC checked all protocols and approved this study. Male ICR mice, weighing 22–28 g, were obtained from Shanghai SLAC Laboratory Animal Co, Ltd. (Shanghai, China). Mice were housed and assayed under controlled temperature conditions (22 ± 2 °C) and a 12 h light/dark cycle with free access to food and water. Mice were randomly grouped and separately administered the testing compounds or vehicle through the intraperitoneal route 30 min prior to the test. The contralateral abdominal sides of the mice were then intraperitoneally injected with 10 mL/kg of 1.5% acetic acid solution according to the protocol described previously. The number of abdominal writhing movements was counted in the following 30 min. A writhe was defined as a contraction of the abdominal muscles followed by body elongation and hind limb extension. The animal experiment was performed in a blinded manner, i.e., administration of compounds and behavioral assessments were performed by different investigators.
Ligand docking analyses
Nav1.7-VSD4-NavAb in complex with GX-936 (PDB code: 5EK0) and compound 10o was selected for docking studies. The receptor and ligand were prepared using the Protein Preparation Wizard and LigPrep module of the Schrodinger Suite, respectively. The docking analysis was carried out using the Ligand Docking module of Schrodinger’s Glide.
Data and statistical analysis
Patch clamp data were processed using Clampfit 10.3 (Molecular Device, Sunnyvale, CA, USA) and analyzed with GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). To quantify the values of IDrug/IControl, the peak currents obtained from the activation pulse (0 mV) were measured, where IControl is the current magnitude of Nav1.7 channels before the application of the compound and IDrug is the current magnitude of Nav1.7 channels after the perfusion of the compound at the indicated concentration. Dose–response curves were fitted with a 3-parameter Hill equation: Y = Bottom + (Top−Bottom)/(1 + 10^(X−LogIC50)), where Bottom and Top are the minimum and maximum inhibition, respectively, X is the log of concentration, Y is the value of IDrug/IControl, and IC50 is the drug concentration producing a half-maximum response. The results are presented as the mean ± SEM. The significance was determined using the paired two-tailed t test and one-way analysis of variance. A P value ≤ 0.05 was considered statistically significant. Detailed information is provided in the figure legends.
Results
Establishment of an electrophysiological assay for Nav1.7 channel inhibitor
To identify selective channel inhibitors against Nav1.7 through rational design and synthesis, we first established an electrophysiological assay according to a previous study [23]. The inhibitory effects of the derivatives on hNav1.7 channels stably expressed in HEK293 cells were evaluated using the whole-cell voltage-clamp technique. Considering that MPT interacts preferentially with the inactivated state of Nav1.7, the Nav1.7 currents were activated from a holding potential of −40 mV, at which >90% of channels are inactivated; then, a 20 ms prepulse to −150 mV was applied to partially recover channels from inactivation, followed by a 10 ms pulse to 0 mV to activate the channel. The peak currents in the absence and presence of the derivatives were termed IContorl and IDrug, respectively, and the ratios were used to calculate the inhibitory efficacy and IC50 values of the compounds. The measured IC50 value of PF-05089771 is 9.49 nmol/L (Fig. 2b), which is consistent with a previous study (11 nmol/L for the inactivated Nav1.7 channel), suggesting that the recording protocol in the current study is appropriate [30]. Thus, we evaluated the inhibitory activities of the derivatives that were rationally designed and synthesized in the present study under this recording protocol.

The inhibitory activities of 10o and PF-05089771 on hNav1.7 channels. a Representative Nav1.7 currents before and after bath perfusion of 10 nmol/L 10o and PF-05089771. (Inset) The experimental protocol. b Dose–response curves of 10o and PF-05089771 for hNav1.7 channels. The currents were recorded from the HEK293 cell lines stably expressing hNav1.7 channels
Identification of 10o as a potent inhibitor of Nav1.7 channel
Compounds 5a–5c, in which the ethanoanthracene unit and aryl sulfonamide warhead were directly connected with a simple carbon chain, showed moderate inhibitory activity against Nav1.7, which was better than the activity of MPT. This result suggests that the combination of the warhead and ethanoanthracene is feasible, which lays the foundation for the following transformation.
By comparing the structure of compound 5b with that of GX-936, we found that the latter had one ethylazetidine side chain. Therefore, we hoped to further improve the activity by introducing nitrogen-substituted side chains. We obtained compounds 9a–9d by adding these side chains to the linker of 5b (Table 1). The N atom substituents of compounds 9a, 9b, and 9c were methyl, ethyl and isopropyl, respectively. Although their inhibitory activity toward Nav1.7 was not as good as that of MPT, these compounds showed trends in selectivity for Nav1.7/Nav1.5.
The increased activity of compound 9d suggested that the linker between the warhead and ethanoanthracene core had considerable scope for modification. Accordingly, we extended the carbon chain from the O atom to ethanoanthracene and obtained 10a (Table 2). This compound showed increased inhibitory activity against Nav1.7 and improved selectivity between Nav1.7 and Nav1.5. We kept one methyl group on the N atom constant and replaced the other methyl group with ethyl, isopropyl, allyl, propargyl, phenethyl, and p-trifluoromethyl phenyl to obtain compounds 10b–10g, respectively (Table 2). Among those compounds, the inhibitory activities of 10b, 10d, and 10f on Nav1.7 were weak, whereas 10c, 10e, and 10g maintained the activity of 10a. The selectivities of these compounds between Nav1.7 and Nav1.5 were similar to that of 10a but not as good as those of 10c and 10e. Compared with compounds 10a–10g, compound 10h, with an N,N-dimethylethyl side chain, showed better inhibitory activity toward Nav1.7, while its inhibitory activity against Nav1.5 was greatly reduced. Next, we replaced the N,N-dimethyl group in 10a with an ethoxyl or benzoxyl group to obtain compounds 10i and 10j, respectively (Table 2). To our surprise, compared with those of the other compounds studied, the inhibitory activities of these compounds toward Nav1.7 were significantly increased. The activities were 10 times higher than that of 10h, but the selectivities between Nav1.7 and Nav1.5 were decreased to some extent. Based on the activity results of these compounds, we summarized the preliminary structure–activity relationship: the ethanoanthracene and warhead were very important, the O atom on the linker was necessary, and a nitrogen-substituted side chain was indispensable for selectivity.
According to the above structure–activity relationship, we obtained compounds 10k, 10l, and 10m (Table 3) on the basis of 10i by substituting the ethyl group attached to the O atom with N,N-dimethylethyl, N-methylethyl, and N-methylazetidinyl groups, respectively. Among those compounds, the IC50 of 10k and 10l against Nav1.7 reached ~ 10 nmol/L, and the selectivity was well maintained. The Nav1.7 IC50 of 10 m was 2.98 nmol/L. We then obtained compounds 10n–10q by changing the size of the azetidine group or introducing a morpholine moiety (Table 3). Compounds 10n, 10p, and 10q had IC50 values of 5.38 nmol/L, 12.47 nmol/L, and 33.52 nmol/L, respectively, and good selectivity for Nav1.7 over Nav1.5. Compound 10o, substituted by a tetrahydropyrrole group, had an IC50 of 0.64 nmol/L toward Nav1.7 and 33.61 μmol/L toward Nav1.5 (Fig. 2b, Table 3). This compound displayed a selectivity for Nav1.7 over Nav1.5 of >50 000 times. Compound 10o was better than the positive control PF-05089771 in terms of both activity and selectivity and was the most active compound synthesized by us thus far. Since 10o was a racemic compound, the two enantiomers were separated by an HPLC chiral column into (+)−10o and (−)−10o. After testing, the Nav1.7 IC50 of compound (+)−10o was determined to be 0.78 nmol/L, and the IC50 of compound (−)−10o was 106.10 nmol/L (Table 3). This result indicated that compound (+)−10o was the key configuration to maintain inhibitory activity. Overall, our study showed that 10o is a potent Nav1.7 channel inhibitor and that compound (+)−10o is the key configuration for inhibitory activity.
10o inhibits the native sodium currents and dampens neuronal excitability
Nav1.7 is heavily expressed in small-diameter mouse DRG neurons [11]. The inhibitory effects of 10o on native sodium currents were then evaluated in mouse DRG neurons. We found that the peak currents decreased as the concentration of 10o in the perfusion solution increased (Fig. 3a). The current amplitudes were reduced by 24% ± 6% and 40% ± 6% in the presence of 10 nmol/L and 100 nmol/L 10o, respectively (Fig. 3b). To identify the TTX-sensitive (TTX-S) currents in each neuron, 500 nmol/L TTX was applied after 10o. Nav1.7 is one of the TTX-S isoforms. Notably, the reduction in peak currents caused by 500 nmol/L TTX was 44% ± 4%, suggesting that the Nav1.7 channels expressed in the DRG neurons could be completely suppressed by 100 nmol/L 10o. Encouragingly, a similar level of inhibition on native sodium currents was also observed after treatment with 100 nmol/L PF-05089771, a previously reported selective Nav1.7 inhibitor (Fig. 3c, d). As Nav1.7 is considered a threshold channel and has a critical role in the generation and conduction of action potentials, we further examined the inhibitory activities of 10o and PF-05089771 on neuronal excitability. The firing frequency of DRG neurons was induced by a step of a 500 ms current injection. As expected, both 10o and PF-05089771 dose-dependently suppressed action potentials in the DRG neurons (Fig. 3e). Interestingly, the inhibitory effect of 10o was stronger than that of PF-05089771. The reduction in the firing frequency caused by 10 nmol/L 10o was almost equal to that induced by 100 nmol/L PF-05089771 (Fig. 3f, g, h). Overall, 10o inhibits the native sodium currents and suppresses the neuronal excitability of small-diameter DRG neurons.

Characterization of the inhibitory activities of 10o and PF-05089771 in DRG neurons. Representative sodium current traces recorded from DRG neurons after sequential application of 10 nmol/L and 100 nmol/L 10o a and PF-05089771 c. 500 nmol/L TTX was finally perfused to indicate the total TTX-S sodium currents. Bar graph showing the inhibitory values of 10o b and PF-05089771 d summarized from panel a and c, respectively. Representative voltage traces following current injection step of 100 pA before and after perfusion of 10o e and PF-05089771 g at the indicated concentrations. The DRG neurons were clamped at −40 mV (depolarized state). Bar graph showing changes in firing frequency in the absence and presence of 10o f and PF-05089771 h at the indicated concentrations summarized from panel e and g, respectively. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, paired two-tailed t test
Analgesic activities of 10o in the acetic acid-induced writhing test
The discovery of 10o as a potent inhibitor in both Nav1.7-expressing cells and native DRG neurons prompted us to explore its analgesic activity in vivo. Acetic acid-induced writhing in mice is a typical model for inflammatory pain and is widely used for the assessment of the peripheral analgesic activity of compounds [31, 32]. Intraperitoneal injection of acetic acid in mice produces a localized inflammatory response, sensitizes the peripheral nociceptive neurons and ultimately causes writhing behaviors along the abdominal wall [33]. It has been reported that in addition to distribution in peripheral somatic neurons, Nav1.7 is also distributed in visceral sensory neurons and sympathetic ganglion neurons [8, 15]. Some Nav1.7 channel inhibitors, such as μ-SLPTX-Ssm6a, μ-TRX-Hhn1b and HWTX-IV, potently attenuate acetic acid-induced pain behaviors in mice [34,35,36]. Thus, the acetic acid-induced writhing mouse model was adopted for evaluating the analgesic activity of 10o. PF-05089771, a potent and selective aryl sulfonamide inhibitor of Nav1.7 in phase II clinical trials, was selected as a positive control and directly compared with 10o [27, 30]. Male ICR mice weighing 22–28 g were injected intraperitoneally with vehicle, PF-05089771 or 10o at the indicated doses 30 min before intraperitoneal injection of 1.5% acetic acid solution (Fig. 4a). The abdominal writhing times were counted cumulatively over the following period of 30 min. As shown in Fig. 4b, the pain behaviors dose-dependently decreased in the groups that were administered 10o and PF-05089771. Moreover, the reduction in the pain behavior produced by 60 mg/kg 10o is comparable to that produced by 100 mg/kg PF-05089771, suggesting that 10o has an excellent analgesic activity in vivo (Fig. 4b). The attenuation of acetic acid-induced writhing by 10o suggested that it might be beneficial for visceral pain relief.

Effects of indomethacin, PF-05089771 and 10o on acetic acid-induced visceral pain. a The writhing times caused by intraperitoneal injection of acetic acid solution at the indicated doses (n = 3–4). b The analgesic activities of PF-05089771 and 10o on acetic acid-induced pain (n = 6–12). All Nav1.7 inhibitors (PF-05089771 and 10o) and vehicle were administered at the indicated doses through i.p. route (0.2 mL/10 g) 30 min prior to the test. Indomethacin (30 mg/kg) was applied through p.o. route 60 min before the test. Diluted acetic acid solution (1.5%) was intraperitoneally injected to induce writhing behavior in ICR mice weighing 22–28 g. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, one-way ANOVA
Molecular docking study of 10o
Compound 10o has a highly hydrophobic ethanoanthracene core, which is different from known Nav1.7 inhibitors. To understand its mode of action against Nav1.7, we performed a docking study using known structural information. Figure 5 shows the docking analysis of compound 10o in the aryl sulfonamide-binding site in the VSD4 of chimeric human Nav1.7 (PDB ID: 5EK0) [23, 37, 38]. Compound 10o has two enantiomers, among which (S)−10o has a higher binding score than (R)−10o, as calculated by Schrodinger. By analyzing the docking result for compound (S)−10o, we find that compound (S)−10o forms van der Waals interactions with residues Tyr1537, Trp1538, Val1541, Met1582, Phe1583, and Arg1605. As illustrated in Fig. 5d, e, the sulfonamide thiadiazole headgroup of compound (S)−10o sits in the anion-binding pocket deep in the binding site. This moiety interacts with the guanidinium group of Arg1608 via a salt bridge and forms a bidentate hydrogen bond with the guanidinium group of Arg1602. The benzonitrile ring occupies the selectivity pocket comprising Tyr1537 and Trp1538, which are the key drivers of Nav isoform selectivity. Tyr1537 forms a π-stacking interaction with the benzonitrile ring, and Trp1538 participates in the π-stacking interaction with one phenyl ring of the ethanoanthracene moiety, which, we infer, accounts for the excellent isoform selectivity of compound (S)−10o. The pyrrolidine nitrogen of compound (S)−10o forms a salt bridge with the side chain of Glu1534. The ethanoanthracene moiety occupies the lipid-exposed pocket quite well (Fig. 5d) and forms π-stacking interactions with both Trp1538 and Phe1583, which contribute to the significant improvement in the activity of compound (S)−10o compared with GX-936.

Molecular docking analysis of 10o. a Top-view model showing the binding site of compound 10o on VSD4 of Nav1.7. Compound 10o is shown in magenta globe model. b GX-936 and compound 10o. c Compound 10o (magenta) superimposed with GX-936 (green) in the binding pocket of Nav1.7 VSD4. d Compound 10o docked into Nav1.7 VSD4 (PDB code 5EK0). Compound 10o is shown in magenta sticks. Dashed lines indicate H-bonding interactions or electrostatic interactions (red) and π-stacking interactions (green) between compound 10o and VSD4. e Compound 10o–VSD4 interaction diagram
Discussion
The important roles of the Nav1.7 channel in pain sensing have been widely studied, especially the finding that dysfunctional mutations of its coding gene, SCN9A, lead to CIP [7, 39]. Patients with Nav1.7-related CIP do not generally display motor, cognitive, or automatic deficits except for olfactory function damage [9, 10]. Moreover, Nav1.7 is preferentially expressed in the peripheral system. These findings suggested that selective blocking of Nav1.7 could effectively provide pain relief without other potential side effects [3, 4]. As a promising analgesic target, many Nav1.7 channel-selective inhibitors have been identified, such as GX-936, PF-05089771, AM-0466, and GNE-131 (Fig. 1) [17, 19, 23, 37]. Among them, PF-05089771, a highly selective and potent Nav1.7 inhibitor, showed good effects on IEM patients and was used in clinical studies to treat painful diabetic peripheral neuropathy; however, the study was terminated in clinical phase II because PF-05089771 failed to achieve the therapeutic effect [37]. Thus, the rational design and synthesis of sulfonamide-based derivatives through combination with other functional moieties would be necessary and worthwhile to identify novel inhibitors against the Nav1.7 channel.
In the present study, we designed and synthetized a novel selective Nav1.7 inhibitor 10o by combining the chemical structures of MPT and GX-936 [23]. Because (+)−10o exhibited a higher activity than (−)−10o (Table 3), (+)−10o was the main configuration binding to Nav1.7. Consistent with its intense inhibition of Nav1.7 in the heterologous system, we found that this compound decreases the native sodium currents in isolated small-diameter DRG neurons in a dose-dependent manner (Fig. 3a, b). Furthermore, 10o intensively suppressed the firing frequency of the DRG neurons (Fig. 3e). In addition to the reduction in the number of action potentials, the depolarized amplitude also decreased after application of 10o compared with the control. This phenomenon was also observed in the presence of PF-05089771 (Fig. 3g). The mechanism underlying these results could be ascribed to the fact that Nav1.7 also contributes to the rising phase of an action potential [40]. Thus, these results obtained in heterologous cell lines and native neurons supported that 10o is a potent Nav1.7 channel inhibitor.
Previous studies have shown that Nav1.7 is widely expressed in the visceral neuron system, including sympathetic, vagal, and pelvic nerves [41, 42]. Patients with PEPD, which is caused by gain-of-function mutations of Nav1.7, display severe episodic perirectal pain syndrome and tend to be triggered by bowel movements or stimulation of the rectal or perineal areas. Recently, abdominal pain syndrome caused by Nav1.7 mutations was also reported in IEM patients. In contrast to the visceral pain syndrome observed in patients with IEM and PEPD, no visceral pain behavior was detected in CIP patients [3]. These studies suggested that inhibition of Nav1.7 might be useful for the treatment of visceral pain. In the present study, we evaluated the analgesic activities of 10o in the acetic acid-induced writhing test, a classic model of visceral pain that is widely used in the evaluation of analgesics. We found that 10o and PF-05089771 could relieve acetic acid-induced visceral pain in a dose-dependent manner. Notably, the analgesic effects of compound 10o were stronger than those of PF-05089771. The reduction in writhing produced by 60 mg/kg 10o was even larger than that caused by 100 mg/kg PF-05089771. These results demonstrated to us that compound 10o might have some therapeutic action on visceral pain, providing a direction for further exploration.
Compound 10o contained an anionic warhead of the VSD inhibitor aryl sulfonamide, so we speculated that 10o was a VSD inhibitor. Another specific fragment of 10o is the highly hydrophobic ethanoanthracene core. We speculated that this moiety acted on the lipid-exposed pocket of VSD4 through its strong hydrophobic property, further increasing the binding force between the inhibitor and the pocket. Similar hydrophobic fragments also exist in the inhibitor GNE-131 and compound 1d (Fig. 1) [19, 20]. In addition, compound 10o also has an alkaline tetrahydropyrrole side chain, which is similar to the ethylazetidine group in GX-936. The interaction with three binding pockets enables 10o to bind to VSD4 stably, thus inhibiting the conformational change in the Nav1.7 channel and blocking the channel current.
In conclusion, taking MPT as a starting point, we designed and synthesized a novel series of bridged-ring sulfonamide Nav1.7 inhibitors with high Nav1.7/Nav1.5 selectivity. Through varied modification and exploration, we discovered compound 10o, which has good activity and selectivity. The binding mode of 10o with Nav1.7 was illustrated by computer simulation. The hydrophobic effect and an extra π-stacking interaction between the ethanoanthracene core and Phe1583 may provide new evidence for developing novel Nav1.7 inhibitors with high potency and selectivity. Further research focusing on improving the physical and chemical properties of these compounds is ongoing.
References
Bennett DL, Woods CG. Painful and painless channelopathies. Lancet Neurol. 2014;13:587–99.
Waxman SG, Merkies ISJ, Gerrits MM, Dib-Hajj SD, Lauria G, Cox JJ, et al. Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol. 2014;13:1152–60.
Vetter I, Deuis JR, Mueller A, Israel MR, Starobova H, Zhang A, et al. Nav1.7 as a pain target - from gene to pharmacology. Pharmacol Ther. 2017;172:73–100.
Yang Y, Mis MA, Estacion M, Dib-Hajj SD, Waxman SG. Nav1.7 as a pharmacogenomic target for pain: moving toward precision medicine. Trends Pharmacol Sci. 2018;39:258–75.
Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–8.
de Lera Ruiz M, Kraus RL. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem. 2015;58:7093–118.
Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71:311–9.
Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Nav1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62.
Weiss J, Pyrski M, Jacobi E, Bufe B, Willnecker V, Schick B, et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature. 2011;472:186–90.
Zufall F, Pyrski M, Weiss J, Leinders-Zufall T. Link between pain and olfaction in an inherited sodium channelopathy. Arch Neurol. 2012;69:1119–23.
Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. 2012;3:791–99.
Shields SD, Deng L, Reese RM, Dourado M, Tao J, Foreman O, et al. Insensitivity to pain upon adult-onset deletion of Nav1.7 or its blockade with selective inhibitors. J Neurosci. 2018;38:10180–201.
Emery EC, Luiz AP, Wood JN. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets. 2016;20:975–83.
Erdemli G, Kim AM, Ju H, Springer C, Penland RC, Hoffmann PK. Cardiac safety implications of hNav1.5 blockade and a framework for pre-clinical evaluation. Front Pharmacol. 2012;3:1–9.
Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA. 1997;94:1527–32.
DiMauro EF, Altmann S, Berry LM, Bregman H, Chakka N, Chu-Moyer M, et al. Application of a parallel synthetic strategy in the discovery of biaryl acyl sulfonamides as efficient and selective Nav1.7 inhibitors. J Med Chem. 2016;59:7818–39.
Graceffa RF, Boezio AA, Able J, Altmann S, Berry LM, Boezio C, et al. Sulfonamides as selective Nav1.7 inhibitors: optimizing potency, pharmacokinetics, and metabolic properties to obtain atropisomeric quinolinone (AM-0466) that affords robust in vivo activity. J Med Chem. 2017;60:5990–6017.
Focken T, Liu S, Chahal N, Dauphinais M, Grimwood ME, Chowdhury S, et al. Discovery of aryl sulfonamides as isoform-selective inhibitors of Nav1.7 with efficacy in rodent pain models. ACS Med Chem Lett. 2016;7:277–82.
Focken T, Chowdhury S, Zenova A, Grimwood ME, Chabot C, Sheng T, et al. Design of conformationally constrained acyl sulfonamide isosteres: identification of N-([1,2,4]Triazolo[4,3-a]pyridin-3-yl)methane-sulfonamides as potent and selective hNav1.7 inhibitors for the treatment of pain. J Med Chem. 2018;61:4810–31.
Sun S, Jia Q, Zenova AY, Wilson MS, Chowdhury S, Focken T, et al. Identification of selective acyl sulfonamide-cycloalkylether inhibitors of the voltage-gated sodium channel Nav1.7 with potent analgesic activity. J Med Chem. 2019;62:908–27.
Wu YJ, Guernon J, Shi J, Ditta J, Robbins KJ, Rajamani R, et al. Development of new benzenesulfonamides as potent and selective Nav1.7 inhibitors for the treatment of pain. J Med Chem. 2017;60:2513–25.
Luo G, Chen L, Easton A, Newton A, Bourin C, Shields E, et al. Discovery of indole- and indazole-acylsulfonamides as potent and selective Nav1.7 inhibitors for the treatment of pain. J Med Chem. 2019;62:831–56.
Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science. 2015;350:aac5464.
Mico JA, Ardid D, Berrocoso E, Eschalier A. Antidepressants and pain. Trends Pharmacol Sci. 2006;27:348–54.
Dick IE, Brochu RM, Purohit Y, Kaczorowski GJ, Martin WJ, Priest BT. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J Pain. 2007;8:315–24.
Wang SY, Calderon J, Kuo Wang G. Block of neuronal Na+ channels by antidepressant duloxetine in a state-dependent manner. Anesthesiology. 2010;113:655–65.
Theile JW, Fuller MD, Chapman ML. The selective Nav1.7 inhibitor, PF-05089771, interacts equivalently with fast and slow inactivated Nav1.7 channels. Mol Pharmacol. 2016;90:540–8.
Zheng YM, Wang WF, Li YF, Yu Y, Gao ZB. Enhancing inactivation rather than reducing activation of Nav1.7 channels by a clinically effective analgesic CNV1014802. Acta Pharmacol Sin. 2018;39:587–96.
Zheng Y, Xu H, Zhan L, Zhou X, Chen X, Gao Z. Activation of peripheral KCNQ channels relieves gout pain. Pain. 2015;156:1025–35.
Alexandrou AJ, Brown AR, Chapman ML, Estacion M, Turner J, Mis MA, et al. Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS One. 2016;11:e0152405.
Gawade SP. Acetic acid induced painful endogenous infliction in writhing test on mice. J Pharm Pharm. 2012;3:348.
Gregory NS, Harris AL, Robinson CR, Dougherty PM, Fuchs PN, Sluka KA. An overview of animal models of pain: disease models and outcome measures. J Pain. 2013;14:1255–69.
Ribeiro RA, Vale ML, Thomazzi SM, Paschoalato AB, Poole S, Ferreira SH, et al. Involvement of resident macrophages and mast cells in the writhing nociceptive response induced by zymosan and acetic acid in mice. Eur J Pharmacol. 2000;387:111–8.
Yang S, Xiao Y, Kang D, Liu J, Li Y, Undheim EA, et al. Discovery of a selective Nav1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc Natl Acad Sci USA. 2013;110:17534–9.
Liu Y, Tang J, Zhang Y, Xun X, Tang D, Peng D, et al. Synthesis and analgesic effects of mu-TRTX-Hhn1b on models of inflammatory and neuropathic pain. Toxins (Basel). 2014;6:2363–78.
Liu Y, Wu Z, Tang D, Xun X, Liu L, Li X, et al. Analgesic effects of Huwentoxin-IV on animal models of inflammatory and neuropathic pain. Protein Pept Lett. 2014;21:153–8.
Swain NA, Batchelor D, Beaudoin S, Bechle BM, Bradley PA, Brown AD, et al. Discovery of clinical candidate 4-[2-(5-Amino-1H-pyrazol-4-yl)-4- chlorophenoxy]-5-chloro-2-fluoro-N-1,3-thiazol-4-ylbenzenesulfonamide (PF-05089771): design and optimization of diaryl ether aryl sulfonamides as selective inhibitors of Nav1.7. J Med Chem. 2017;60:7029–42.
McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci USA. 2013;110:E2724–32.
McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, et al. Defining the functional role of Nav1.7 in human nociception. Neuron. 2019;101:905–19.
Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev. 2019;99:1079–151.
Muroi Y, Ru F, Kollarik M, Canning BJ, Hughes SA, Walsh S, et al. Selective silencing of Nav1.7 decreases excitability and conduction in vagal sensory neurons. J Physiol. 2011;589:5663–76.
Evans MS, Maglinger GB, Fletcher AM, Johnson SR. Benzonatate inhibition of voltage-gated sodium currents. Neuropharmacology. 2016;101:179–87.
Acknowledgements
This work was supported by the National Science Fund for Distinguished Young Scholars (81825021) and the National Natural Science Foundation of China (81603096, 81773707).
Author information
Authors and Affiliations
Contributions
FJN and ZBG designed and conceived the experiments; JTW and YMZ conducted the experiments and contributed equally to the project; YTC conducted the docking analysis; MG performed the HPLC analysis; all authors analyzed the data; JTW, YMZ, ZBG, and FJN wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, Jt., Zheng, Ym., Chen, Yt. et al. Discovery of aryl sulfonamide-selective Nav1.7 inhibitors with a highly hydrophobic ethanoanthracene core. Acta Pharmacol Sin 41, 293–302 (2020). https://doi.org/10.1038/s41401-019-0267-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0267-z
