
Abstract
Type 2 diabetes (T2D) and Alzheimer’s disease (AD) share several common pathophysiological features. Huperzine A (Hup A), a Lycopodium alkaloid extracted from the Chinese herb moss Huperzia serrata, is a specific and reversible inhibitor of acetylcholinesterase, which is clinically used for the treatment of AD. In this study, we investigated whether Hup A improved the metabolic and cognitive functions in the high fat-induced (HFD) obese mice and genetic ob/ob mice. HFD and ob/ob mice were treated with Hup A (0.1, 0.3 mg · kg−1 · d−1, ig) for 3 months. Body weight was monitored and glucose tolerance tests were performed. Novel object recognition test and Morris water maze assay were conducted to evaluate the cognitive functions. We found that the Hup A treatment had no significant effect on peripheral metabolism of obese mice, whereas Hup A (0.1, mg · kg−1 · d−1) improved both the abilities of object recognition and spatial memory in HFD-fed mice, but not in ob/ob mice. Furthermore, Hup A treatment significantly upregulated the insulin and phosphorylated Akt levels in the cortex of HFD-fed mice, but not ob/ob mice. In addition, Hup A (0.3, mg · kg−1 · d−1) significantly decreased cortical β-secretase (BACE1) expression. In conclusion, these results demonstrate that treatment with Hup A (0.1, mg · kg−1 · d−1) can effectively improve the cognitive functions, at least in diet-induced obese mice.
Similar content being viewed by others


Introduction
The incidence of dementia and type 2 diabetes (T2D) in the world is increasing at an alarming rate and has become a major public health concern [1,2,3]. Alzheimer’s disease (AD) is the most common form of dementia, accounting for 50%–60% of all cases [4, 5]. The proteolytic cleavage of amyloid precursor protein (APP) by beta-site APP-cleaving enzyme 1 (β-secretase, BACE1) and γ-secretase complex is one of the key features of AD pathology development, increasing the levels of Aβs and leading to their aggregation and formation of extracellular amyloid plaques [6,7,8]. Moreover, mice deficient in BACE1 exhibit a normal phenotype but abolished amyloid generation and enhanced insulin sensitivity and obesity resistance [9, 10]. Mounting evidence has demonstrated that AD patients are prone to T2D; however, diabetic patients also have a higher risk of developing AD than patients without diabetes [11,12,13]. Being obese in midlife is also a risk factor for AD later in life [14, 15]. Interestingly, T2D and AD share many common age-related pathophysiological features. Patients with T2D exhibit lower cognitive scores [16], and db/db mice, an animal model of T2D, exhibit spatial learning deficiencies [17]. In turn, AD brains show insulin resistance, impaired glucose metabolism and hyperinsulinemia [18, 19]. Furthermore, the insulin concentration of AD patients is decreased in the cerebrospinal fluid but increased in the plasma [20, 21]. However, the mechanism that links T2D and AD is still unclear.
Huperzine A (Hup A), a novel Lycopodium alkaloid originally extracted from the Chinese herb moss Huperzia serrata, is a potent, specific, selective and reversible inhibitor of acetylcholinesterase (AChE) [22, 23]. Previous studies have reported that Hup A improves cognitive dysfunction, attenuates behavioral disturbance, and slows the progression of AD in a broad range of animal models as well as in AD patients [24,25,26,27,28]. Hup A protects neurons from harm in AD by not only reducing the loss of acetylcholine [24] but also preventing Aβ-induced apoptosis [29, 30]. In addition, Hup A mitigates brain injury and cognitive deficits after repetitive traumatic damage and attenuates oxidative injury by various modes of action [28, 31].
Hup A may possibly improve neuronal insulin signaling and cognitive decline related to diabetes. Studies have reported that Hup A increases phospho-Akt levels in murine hippocampal HT22 cells after glutamate toxicity [32]. Moreover, in streptozotocin (STZ)-induced diabetic rats, Hup A not only reduces body weight and oxidative stress but also improves blood glucose levels and cognitive deficits [33]. However, whether Hup A attenuates obesity-related cognitive declines remains unclear. Here, we aimed to explore the role of Hup A in the regulation of peripheral metabolism and cognitive dysfunctions in diet-induced obese mice and genetically obese ob/ob mice.
Materials and methods
Reagents
Huperzine A (white powder, purity >99%, provided by Professor Hai-yan Zhang) was dissolved in distilled water that contained 40% volume of 0.1 M hydrochloric acid at 5 mg/mL as a stock solution and was then diluted to the proper concentration with distilled water for administration [34]. The antibodies applied in this study targeted the following proteins: Akt (CST, 4691), phospho-Akt (Ser473) (CST, 4060), BACE1 (CST, 5606), and β-actin (CST, 4970).
Animals and diets
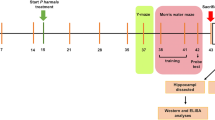
Male C57 BL/6 mice aged 8 weeks were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd., housed in wood chip-bedded plastic cages at constant room temperature (23 ± 2 ℃) under a 12–h light/dark cycle (lights on 07:00–19:00), and fed with water and food available ad libitum. After 2 weeks of acclimatization to living conditions, mice were divided into two groups, fed a high-fat diet (HFD, 60% energy from fat, 20% from protein and 20% from carbohydrates, D12492, Research Diets, Inc.) or low-fat diet (LFD, 10% energy from fat, 20% from protein and 70% carbohydrates, D12450B, Research Diets, Inc.). Heterozygotic male and female ob+/− mice were obtained by crossing homozygotic leptin gene knockout (ob/ob) mice and WT mice. HFD-induced obese mice and ob/ob mice were randomly divided into three groups and intragastrically administered 0.1 mg or 0.3 mg Hup A per kilogram of body weight per day or vehicle for three mouths. Simultaneously, LFD mice were treated with the vehicle of Hup A. This study was approved by the Animal Care and Research Committee of Chongqing Medical University, China.
Glucose tolerance test (GTT)
Mice were fasted for 16 h, and fasted blood glucose was measured by a glucometer (ACCU-CHEK Performa, Roche) through tail bleeding. Subsequently, mice were intraperitoneally injected with glucose (2 g/kg body weight), and blood glucose was measured at 15, 30, 60, 120 min after glucose injection.
Novel object recognition test
The recognition ability of mice was assessed using the new object recognition test (Fig. 2a) as described [35, 36]. Mice were trained in the task for three days. On the first day, mice were submitted to a 5-min acclimatization session in the apparatus (an open field: long × width × height: 40 cm × 40 cm × 50 cm, made up of transparent plastic sheets) without objects. Then, 24 h later, mice were allowed to explore two identical objects (A1 and A2) for 5 min. The objects were fixed in two adjacent corners (~9 cm from the wall). Two hours later, mice underwent a short-term memory test, during which they were allowed to explore the apparatus for 5 min in the presence of two objects (the familiar object A1 and the new object B). On the third day, to examine long-term memory, the same groups of mice were allowed to explore two objects for 5 min (the familiar object A1 and the new object B). Object exploration was tracked by the Any-maze tracking system (Stoelting, USA), which can record the time spent on the objects during the experimental sessions. Exploration was defined as sniffing or touching the object with the nose. A “recognition index” was calculated for each animal as expressed by the ratio: TB/(TA + TB) (TA = time spent exploring the familiar object; TB = time spent exploring the novel object).
Morris water maze
Spatial learning and memory were assessed using the Morris water maze task as previously described [37, 38]. The water maze consisted of a circular stainless-steel pool 150 cm in diameter filled with water (25 ± 1 °C) made opaque with nontoxic white dye. The pool was surrounded by light blue curtains with three distal visual cues fixed to the curtains and was artificially divided into four quadrants: NE, NW, SW, and SE. A CCD camera was suspended above the center of pool to record the swimming paths of the mice, and the video output was digitized with the Any-maze tracking system (Stoelting, USA). This test included spatial training and probe testing. Twenty-four hours before spatial training, mice were allowed to adapt to the pool for 120 s with free swimming. Then, mice were trained in the spatial learning session for six trials per day for 5 consecutive days. In each trial, mice were placed in the water at one of four positions (NE, NW, SW, and SE) facing the pool wall. Then, the mice were required to swim to find a hidden platform (13 cm in diameter, located in the SW quadrant) submerged 1 cm below the water. During each trial, mice were allowed to swim until they found the hidden platform and stayed on the platform for 20 s before being returned to a holding cage. Mice that failed to find the hidden platform in the 120 s were guided to the platform and allowed to remain there for 20 s. Twenty-four hours after the last trial, the probe test was performed. Mice were returned to the pool without the platform at a new point for 120 s, and their swimming paths were recorded.
Enzyme-linked immunosorbent assay (ELISA) analysis
Insulin levels in the cortex were measured with the ELISA kit for insulin (CEA448Mu, Cloud- Clone Corp.). Aβ42 levels in the cortex were measured with the ELISA kit (Invitrogen Corporation, KMB3441).
Western blotting
Frozen mouse tissue was lysed in lysis buffer (10 mL lysis buffer; cOmplete Lysis-M, REF 04719956001, Roche) containing one tablet of protein inhibitor (cOmplete™ ULTRA Tablets, Mini, EDTA-free, EASYpack Protease Inhibitor Cocktail, Roche) and one tablet of phosphatase inhibitor (PhosSTOP™, PHOSS-RO, Roche) to obtain total protein supernatant. The concentration of total protein was determined using a BCA protein assay kit (Lot # NH17739, Thermo, Pierce). Aliquots of 60 µg protein were loaded onto 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes (Roche), and subsequently blocked with 5% bovine serum albumin (BSA)-tris-buffered saline, 0.1% Tween 20 solution (BSA powder, GENVIEW, CAS: 9048–46–8) for 1.5 h. The membranes were incubated with primary antibody overnight, and the goat-anti-rabbit/ mouse secondary antibody for 1 h. Protein bands were visualized with ECL (advansta, K-12045-D10) using a chemiluminescence apparatus (GeneGnome5). The protein expression levels were quantified with ImageJ software.
Statistical analysis
All data are presented as the mean ± standard error of mean and were analyzed by Students’ t test, one-way analysis of variance (ANOVA) with Tukey’s post hoc test or two-way ANOVA with Bonferroni’s post hoc test. Statistical significance was considered at P < 0.05.
Results
Huperzine A did not alter the metabolic characteristics of HFD-fed mice or those of ob/ob mice
To generate diet-induced obesity, mice were fed a HFD, and body weight changes were measured weekly. As expected, compared with LFD-fed mice, mice fed a HFD for 18 weeks showed significant body weight gain (increased by 25% vs. LFD, Fig. 1a, b) and impaired glucose tolerance indicated by high glucose levels at 30 min and 60 min after glucose loading and a high area under the curve (Fig. 1c, d).

Peripheral metabolic profiles. a, b Body weight of C56 BL/6 mice before Hup A delivery (LFD and HFD; n = 11 and 12, respectively). c, d Glucose tolerance test of 7-month-old mice (LFD and HFD; n = 5 and 4, respectively). e, g Body weight during Hup A delivery (LFD-Veh, n = 11; HFD-Veh, n = 7; HFD-0.1, n = 9; HFD-0.3, n = 7; ob/ob-Veh, n = 6; ob/ob-0.1, n = 7 and ob/ob-0.3, n = 7). f Glucose tolerance test of 9-month-old mice after two months of Hup A delivery. h Nonfasting blood glucose test during Hup A delivery (n = 4 in each group). Data are presented as the mean ± SEM. Data are compared using Student’s t test b, d and two-way ANOVA with Bonferroni post hoc test a, c, e–g. *P < 0.05; **P < 0.01; ***P < 0.001. There was no significant difference among the Hup A groups
During Hup A treatment, we continued to monitor the body weight of mice in each group. However, Hup A was unable to decrease weight (Fig. 1e, g) or improve glucose intolerance (Fig. 1f) in HFD-fed or ob/ob mice. Hup A treatment was also incapable of reducing hyperglycemia in ob/ob mice (Fig. 1h). These results suggested that Hup A might not improve peripheral glucose metabolism in obese mice.
Hup A enhanced the recognition memory of HFD-fed mice but not ob/ob mice
To investigate whether Hup A reduces cognitive impairment, mice were submitted to the novel object recognition test for an assessment of their short- and long-term memory. In this test, the interaction time used for the recognition index was defined specifically by “sniffing or touching the object with the nose”. Although there was no significant difference in terms of the time spent on the old and novel objects (Fig. 2d), HFD-fed mice treated with 0.1 mg/kg every day Hup A showed a higher recognition index in the short-term memory test than HFD-fed mice treated with vehicle (Fig. 2b), indicating that short-term memory was enhanced. Although there was no statistically significant difference in the long-term memory test, there were increased trends in both the recognition index and time spent on the old and novel objects in HFD-fed mice treated with 0.1 mg/kg every day Hup A (Fig. 2c, e). Most of the ob/ob mice did not exhibit a tendency to explore objects during training; there, we could not obtain relevant data. During the acclimatization period, the movement distance of all the mice was measured in the probe test. According to the results, the obese mice were less active than LFD-fed mice or WT mice, but Hup A increased the activity level of ob/ob mice (Fig. 2f).

Novel object recognition test in 11-month-old mice. a Schematic diagram of the novel object recognition test. b, c The cognition index of short- and long-term memory assessed in 11-month-old mice using a new object recognition test (n = 5 in each group). d, e Time spent on the old and novel objects during short-term and long-term memory tests. f Traveled distance determined during the acclimatization period. Data are presented as the mean ± SEM. Data are compared using two-way ANOVA with Bonferroni post hoc test d, e and one-way ANOVA with Tukey post hoc test a–c, f. *P < 0.05; ***P < 0.001
Hup A enhanced the spatial memory of HFD-fed mice but not ob/ob mice
The Morris water maze was used to confirm whether Hup A improves the spatial learning and memory of obese mice. As shown in Fig. 3a, on the first training day, mice that were treated with Hup A and mice fed a LFD spent less time searching for the hidden platform than the vehicle control mice fed a HFD. However, there was no significant difference on the other training days. There was no difference in escape latency among the ob/ob mice during the training days (Fig. 3b). As expected, in the probe test, mice fed a HFD spent less time in the platform-located quadrant than mice fed a LFD (Fig. 3e). After the HFD-fed mice were treated with 0.1 mg/kg every day Hup A for 3 months, the time spent in the platform-located quadrant was significantly increased to a level almost the same as that of LFD mice (Fig. 3e). Hup A-treated ob/ob mice showed a trend toward an improvement in spatial memory (Fig. 3e). There was no significant difference in the number of hidden platform location crossings between groups (Fig. 3f). Furthermore, ob/ob mice treated with 0.3 mg/kg Hup A showed faster swimming speed on the first 2 training days, and ob/ob mice treated with 0.1 mg/kg Hup A showed faster swimming speed on the fourth training day than those treated with vehicle, whereas the HFD mice treated with either 0.1 mg/kg or 0.3 mg/kg Hup A did not show significant changes (Fig. 3c, d). However, there was no significant difference in swimming speed between the HFD-fed groups or ob/ob groups during the probe test (Fig. 3g).

Morris water maze test in 11-month-old mice. a, b Plots exhibit the average escape latencies of mice for each training day (For day 1, HFD-Veh vs LFD-Veh: P = 0.0286; HFD-Veh vs HFD-0.1: P = 0.0278; HFD-Veh vs HFD-0.3:P = 0.0139). c, d Average speed during the training days. e Time spent in the platform-located quadrant during the probe test (n = 5 in each group). f, g Entries through the platform position and swimming speed during the probe test (n = 5 in each group). h Representative swim paths of mice during the probe test. Data are presented as the mean ± SEM. Data are compared using two-way ANOVA with Bonferroni post hoc test a–d and one-way ANOVA with Tukey post hoc test e–g. *P < 0.05
Hup A enhanced spatial memory involved improvement of cerebral insulin signaling
Insulin signaling has been reported to play an important role in cognitive function [39, 40]. To further investigate the changes in insulin signaling after Hup A treatment, we tested the phosphorylation level of Akt at serine 473 (p-Akt), which is a necessary residue for Akt activity [41]. Compared with obese mice treated with vehicle, obese mice treated with Hup A exhibited remarkably greater Akt-serine 473 phosphorylation in the hippocampus (Fig. 4a, c). Moreover, in the cortex, HFD-fed mice treated with Hup A also showed significantly greater Akt-serine 473 phosphorylation than vehicle-treated HFD-fed mice, whereas Hup A had no significant effect on Akt-serine 473 phosphorylation in ob/ob mice (Fig. 4b, c).

The effects of Hup A on the brain insulin signaling pathway. a Western blot of Akt and phosphorylated Akt (p-Akt) protein levels in the hippocampus of HFD and ob/ob mice. b Western blot of Akt and phosphorylated Akt (p-Akt) protein levels in the cortex of HFD and ob/ob mice. c Quantification of the ratio of p-Akt/Akt protein expression according to A and B (n = 5 in each group). d The insulin levels in the cortex by ELISA (n = 4 in each group). Data are presented as the mean ± SEM. Data are compared using one-way ANOVA with Tukey’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001
Because the insulin concentration in the brain decreases with aging [20] and the high density of insulin in the brain enhances spatial learning and memory [42], the insulin level in the cortex was tested. The HFD-fed and ob/ob mice treated with Hup A showed notably higher insulin concentrations in the cortex than those mice treated with vehicle (Fig. 4d). Therefore, Hup A may improve spatial memory in obese mice by enhancing insulin signaling and increasing cortical insulin levels.
High-dose Hup A inhibited cerebral BACE1 expression and reduced Aβ levels
Studies have shown that BACE1 has a pivotal role in dementia [43, 44] and correlates with metabolic disorders caused by obesity or T2D [6, 9, 45]. Moreover, Hup A prevents increases in the membrane distribution of BACE1 in the cortex [34]. The ob/ob mice treated with 0.3 mg/kg every day Hup A showed significantly lower hippocampal BACE1 expression than vehicle-treated ob/ob mice, whereas HFD mice did not exhibit a significant difference with Hup A treatment (Fig. 5a, c). Obese mice treated with a high dose of Hup A showed remarkably lower cortical BACE1 expression than those treated with vehicle (Fig. 5b, c). HFD mice treated with the low dose of Hup A showed a trend toward a decrease in cortical BACE1 expression compared with vehicle-treated HFD mice (Fig. 5b, c).

The effects of Hup A on BACE1 and Aβ42 levels in the hippocampus or cortex. a Western blot of BACE1 protein expression in the hippocampus of HFD and ob/ob mice. b Western blot of BACE1 protein expression in the cortex of HFD and ob/ob mice. c Quantification of BACE1/β-actin protein expression according to A and B (n = 5 in each group). d The level of Aβ42 in the cortex by ELISA (n = 4 in each group). Data are presented as the mean ± SEM. Data are compared using one-way ANOVA with Tukey’s post hoc test. *P < 0.05; **P < 0.01
To investigate whether decreased BACE1 expression affects the Aβ42 concentration, we examined the concentration of Aβ42 in the cortex. Here, ob/ob mice administered 0.3 mg/kg every day Hup A had a lower concentration of Aβ42 than vehicle-treated ob/ob mice (Fig. 5d). These results suggested that decreased expression of BACE1 may contribute to the protection of Hup A against cognitive dysfunctions caused by obesity.
Discussion
A substantial number of studies have shown the classic effects of Hup A in inhibiting acetylcholinesterase, increasing acetylcholine, and activating cholinergic receptors, eventually improving the cognitive decline for AD patients and animals [46,47,48]. In this study, we observed increased cortical insulin levels in the brains of obese (HFD or ob/ob) mice after Hup A treatment. Cortical insulin levels in the vehicle HFD and ob/ob group were higher than those in the vehicle LFD and WT group, which together with the GTT results indicated that the obese mice exhibited insulin resistance. Hup A further increased the insulin concentration accordingly to meet the needs of the brain for neuroprotection and to help to maintain higher cognitive processes, including learning, memory, executive function, and attention [21, 49]. Brain insulin levels have been reported to be slightly higher in ob/ob mice at 8–10 weeks of age than in their lean littermates [50]. Similarly, our data showed that brain cortical insulin levels were higher in HFD-fed and ob/ob mice at 44–45 weeks of age than in their respective controls. Treatment with Hup A further elevates insulin levels in the brain in obese mice, but how higher insulin levels are beneficial to the memory deficits in these animals is still unclear. Excessive insulin likely facilitates the insulin signaling pathway despite insulin resistance, allowing insulin to be effective in controlling glucose homeostasis under conditions of hyperinsulinemia. In addition, intranasally administered insulin has been reported to penetrate directly from the nose to the brain to improve the functions of the central nervous system [51, 52], and more specifically, intracerebroventricular (ICV) STZ-induced memory impairments in rats were prevented by treatment with intranasal insulin [52, 53]. Previous studies have reported that impairments in insulin or insulin-like growth factor signaling are observed in the brains of AD patients, and these abnormalities are involved in the decreased levels of phospho-Akt, the insulin receptor substrate (IRS), and the IRS-associated phosphatidylinositol 3-kinase and the increased levels of glycogen synthase kinase-3β activity and APP [54]. Insulin is well known to promote neurite outgrowth, modulate activity-dependent synaptic plasticity via PI3K-AKT signaling, and have a key role in the development and maintenance of excitatory synapses [54]. Although epidemiologic studies have indicated that long-term hyperinsulinemia is a risk factor for dementia, insulin administered to the AD brain maintains glucose homeostasis and improves memory function [55, 56]. In addition, HFD has been shown to impair brain insulin signaling and synaptic plasticity [57, 58]. In our study, HFD or ob/ob mice treated with Hup A for 3 months exhibited an increase in the insulin concentration in the cortex, resulting in improved cognitive function.
In our current study, obese mice exhibited lower levels of phospho-Akt in the brain than WT mice, which suggests impaired neuronal insulin signaling. After Hup A treatment, the expression of p-Akt was fully restored. Hup A has also been shown to ameliorate oxidative glutamate toxicity in immortalized hippocampal HT22 cells, and this protection might be involved in regulating the BDNF/TrkB-dependent PI3K/Akt/mTOR signaling pathway [32]. Therefore, we speculated that the improvement in HFD-induced obesity-related cognitive dysfunction by a low dose of Hup A (0.1 mg/kg) is attributable to the increased insulin concentration and expression of p-Akt in the brain.
In addition, a previous study demonstrated that Hup A could reduce body weight and improve blood glucose levels in STZ-induced diabetic rats [33]. However, our study did not show similar results in HFD-fed or ob/ob mice. Neither body weight nor glucose homeostasis was changed by Hup A treatment. This discrepancy may result from the difference between the animal species and diabetic models. Mao et al. [33] used Wistar rats treated with STZ, which are generally recognized as a model of type 1 diabetes. In contrast, the HFD and ob/ob mice used in our study, which showed obvious obesity and insulin resistance, are accepted as models of T2D. HFD-fed rodents gradually develop significant weight gain, obesity and insulin resistance. These features are more relevant to human T2D. However, ob/ob mice are severely obese owing to a genetic leptin deficiency, which is an important anorexigenic hormone that controls food intake. The metabolic and behavioral differences in these two models strongly influence the degree of obesity.
We also demonstrated for the first time that Hup A decreased BACE1 expression in the hippocampus and cortex of ob/ob mice and in the cortex of HFD-fed mice. BACE1, a proamyloidogenic enzyme, is known to cleave APP, which has a critical role in the generation of the amyloid-β peptides implicated in the pathogenesis of AD. There is a positive correlation between BACE1 levels and plaque numbers in AD brains [59, 60]. Although highly expressed in the brain, BACE1 is also expressed in nonneuronal tissues, including the pancreas, skeletal muscle, and liver. BACE1 activity in these tissues may be increased under stress conditions [61, 62]. Deletion or reduction of BACE1 decreases mouse body weight and enhances insulin sensitivity when fed a regular chow diet but also partially prevents HFD-induced obesity. These actions may be involved in the increase in the uncoupling of proteins in skeletal muscle and brown fat tissues. Therefore, decreased BACE1 activity and its level may ameliorate metabolic abnormalities caused by HFD [9]. Moreover, a recent study showed that neuronal BACE1-overexpressing mice exhibit systemic diabetes. Essentially, BACE1 knock-in causes systemic glucose intolerance, fatty liver and impaired hepatic glycogen storage in mice. These phenomena are also associated with hypothalamic pathology, such as a dysregulated melanocortin system and advanced endoplasmic reticulum stress [6]. Studies have shown that the expression of both hippocampal and cortical BACE1 increases considerably in obese and diabetic mice, and inhibiting BACE1 may serve as a new strategy to intervene in T2D and AD [63,64,65]. Consequently, neuronal BACE1 is a key factor for metabolic homeostasis, thus affording a potential mechanism for metabolic disturbance in the AD brain. Apart from its reversible, potent, and selective inhibitory effect on AChE, Hup A treatment regulates APP processing by activating ADAM 10 and modulating extracellular signal-regulated kinase 1/2 pathways and protein kinase C, resulting in elevated sAPPα expression and decreased Aβ production in HEK293swe cells [34, 66].
We used two doses of Hup A, 0.1 and 0.3 mg/kg every day, according to previous reports [22, 28, 33, 66, 67]. Obviously, low-dose Hup A is most commonly used and shows the best effect, which is reflected in the enhanced short-term memory and spatial learning and memory and corresponds to the increase in insulin in the cortex and phospho-Akt expression in the hippocampus and cortex. Although low-dose Hup A did not affect the levels of BACE1 and Aβ42, studies have indicated that Aβ itself is not the primary neurotoxin causing AD but is the initiator of the pathologic changes in the brain that finally cause neurodegeneration years later [68]. In any case, cognitive performance can be improved independent of Aβ [69, 70]. We also gave mice 0.3 mg/kg Hup A to determine whether the higher dose would have stronger effects than 0.1 mg/kg in our mouse models. Nevertheless, 0.3 mg/kg Hup A did not further improve cognitive function.
In summary, our results have demonstrated for the first time that Hup A protects against the obesity-associated cognitive impairment in HFD-fed mice but not in ob/ob mice. Although Hup A had no effect on body weight change and peripheral glucose metabolism regulation, it significantly enhanced the activity of the neuronal insulin signaling pathway, thereby contributing to its improvement in cognitive dysfunctions associated with T2DM. The novel findings in this study provide important information for a potential new application of Hup A in addition to its classical treatment of AD.
References
Hu C, Yu D, Sun X, Zhang M, Wang L, Qin H. The prevalence and progression of mild cognitive impairment among clinic and community populations: a systematic review and meta-analysis. Int Psychogeriatr. 2017;29:1595–608.
Sugimoto T, Sakurai T, Ono R, Kimura A, Saji N, Niida S, et al. Epidemiological and clinical significance of cognitive frailty: a mini review. Ageing Res Rev. 2018;44:1–7.
Gujral UP, Weber MB, Staimez LR, Narayan KMV. Diabetes among non-overweight individuals: an Emerging Public Health Challenge. Curr Diab Rep. 2018;18:60.
Caselli RJ, Beach TG, Yaari R, Reiman EM. Alzheimer’s disease a century later. J Clin Psychiatry. 2006;67:1784–800.
Miyoshi K. What is ‘early onset dementia’? Psychogeriatrics. 2009;9:67–72.
Plucinska K, Dekeryte R, Koss D, Shearer K, Mody N, Whitfield PD, et al. Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia. 2016;59:1513–23.
Zou L, Yang R, Zhang P, Dai Y. The enhancement of amyloid precursor protein and beta-site amyloid cleavage enzyme 1 interaction: amyloid-beta production with aging. Int J Mol Med. 2010;25:401–7.
Ham S, Kim TK, Lee S, Tang YP, Im HI. MicroRNA profiling in aging brain of PSEN1/PSEN2 double knockout mice. Mol Neurobiol. 2018;55:5232–42.
Meakin PJ, Harper AJ, Hamilton DL, Gallagher J, McNeilly AD, Burgess LA, et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem J. 2012;441:285–96.
Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280:30797–806.
Kravitz E, Schmeidler J, Schnaider Beeri M. Type 2 diabetes and cognitive compromise: potential roles of diabetes-related therapies. Endocrinol Metab Clin North Am. 2013;42:489–501.
Adam AP. A potential new mechanism linking type II diabetes mellitus and Alzheimer’s disease. Bioessays. 2018;40:e1800061.
Riederer P, Korczyn AD, Ali SS, Bajenaru O, Choi MS, Chopp M, et al. The diabetic brain and cognition. J Neural Transm (Vienna). 2017;124:1431–54.
Hassing LB, Dahl AK, Thorvaldsson V, Berg S, Gatz M, Pedersen NL, et al. Overweight in midlife and risk of dementia: a 40-year follow-up study. Int J Obes (Lond). 2009;33:893–8.
Alford S, Patel D, Perakakis N, Mantzoros CS. Obesity as a risk factor for Alzheimer’s disease: weighing the evidence. Obes Rev. 2018;19:269–80.
Okereke OI, Kang JH, Cook NR, Gaziano JM, Manson JE, Buring JE, et al. Type 2 diabetes mellitus and cognitive decline in two large cohorts of community-dwelling older adults. J Am Geriatr Soc. 2008;56:1028–36.
Chen F, Dong RR, Zhong KL, Ghosh A, Tang SS, Long Y, et al. Antidiabetic drugs restore abnormal transport of amyloid-beta across the blood-brain barrier and memory impairment in db/db mice. Neuropharmacology. 2016;101:123–36.
Morris JK, Vidoni ED, Mahnken JD, Montgomery RN, Johnson DK, Thyfault JP, et al. Cognitively impaired elderly exhibit insulin resistance and no memory improvement with infused insulin. Neurobiol Aging. 2016;39:19–24.
Abolhassani N, Leon J, Sheng Z, Oka S, Hamasaki H, Iwaki T, et al. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech Ageing Dev. 2017;161:95–104.
Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (Vienna). 1998;105:423–38.
Baranowska-Bik A, Bik W. Insulin and brain aging. Prz Menopauzalny. 2017;16:44–6.
Wang R, Yan H, Tang XC. Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol Sin. 2006;27:1–26.
Zhu HF, Yan PW, Wang LJ, Liu YT, Wen J, Zhang Q, et al. Protective properties of Huperzine A through activation Nrf2/ARE-mediated transcriptional response in X-rays radiation-induced NIH3T3 cells. J Cell Biochem. 2018;119:8359–67.
Xu SS, Cai ZY, Qu ZW, Yang RM, Cai YL, Wang GQ, et al. Huperzine-A in capsules and tablets for treating patients with Alzheimer disease. Zhongguo Yao Li Xue Bao. 1999;20:486–90.
Ye JW, Cai JX, Wang LM, Tang XC. Improving effects of huperzine A on spatial working memory in aged monkeys and young adult monkeys with experimental cognitive impairment. J Pharm Exp Ther. 1999;288:814–9.
Howes MJ, Perry E. The role of phytochemicals in the treatment and prevention of dementia. Drugs Aging. 2011;28:439–68.
Sheng L, Qu Y, Yan J, Liu GY, Wang WL, Wang YJ, et al. Population pharmacokinetic modeling and simulation of huperzine A in elderly Chinese subjects. Acta Pharmacol Sin. 2016;37:994–1001.
Mei Z, Zheng P, Tan X, Wang Y, Situ B. Huperzine A alleviates neuroinflammation, oxidative stress and improves cognitive function after repetitive traumatic brain injury. Metab Brain Dis. 2017;32:1861–9.
Zhu N, Lin J, Wang K, Wei M, Chen Q, Wang Y. Huperzine A protects neural stem cells against Abeta-induced apoptosis in a neural stem cells and microglia co-culture system. Int J Clin Exp Pathol. 2015;8:6425–33.
Tao LX, Huang XT, Chen YT, Tang XC, Zhang HY. Acetylcholinesterase-independent protective effects of huperzine A against iron overload-induced oxidative damage and aberrant iron metabolism signaling in rat cortical neurons. Acta Pharmacol Sin. 2016;37:1391–400.
Wang R, Tang XC. Neuroprotective effects of huperzine A. A natural cholinesterase inhibitor for the treatment of Alzheimer’s disease. Neurosignals. 2005;14:71–82.
Mao XY, Zhou HH, Li X, Liu ZQ. Huperzine A alleviates oxidative glutamate toxicity in hippocampal HT22 cells via activating BDNF/TrkB-dependent PI3K/Akt/mTOR signaling pathway. Cell Mol Neurobiol. 2016;36:915–25.
Mao XY, Cao DF, Li X, Yin JY, Wang ZB, Zhang Y, et al. Huperzine A ameliorates cognitive deficits in streptozotocin-induced diabetic rats. Int J Mol Sci. 2014;15:7667–83.
Wang Y, Tang XC, Zhang HY. Huperzine A alleviates synaptic deficits and modulates amyloidogenic and nonamyloidogenic pathways in APPswe/PS1dE9 transgenic mice. J Neurosci Res. 2012;90:508–17.
Molz P, Ellwanger JH, Zenkner FF, Campos D, Pra D, Putzke MT, et al. Recognition memory and DNA damage in undernourished young rats. Acad Bras Cienc. 2016;88:1863–73.
Lueptow LM. Novel object recognition test for the investigation of learning and memory in mice. J Vis Exp. 2017;126:e55718.
Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, et al. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest. 2015;125:234–47.
Bromley-Brits K, Deng Y, Song W. Morris water maze test for learning and memory deficits in Alzheimer’s disease model mice. J Vis Exp. 2011;53:2920.
Gupta S, Yadav K, Mantri SS, Singhal NK, Ganesh S, Sandhir R. Evidence for compromised insulin signaling and neuronal vulnerability in experimental model of sporadic Alzheimer’s disease. Mol Neurobiol. 2018;55:8916–35.
Kasper JM, Milton AJ, Smith AE, Laezza F, Taglialatela G, Hommel JD, et al. Cognitive deficits associated with a high-fat diet and insulin resistance are potentiated by overexpression of ecto-nucleotide pyrophosphatase phosphodiesterase-1. Int J Dev Neurosci. 2018;64:48–53.
Lucic I, Rathinaswamy MK, Truebestein L, Hamelin DJ, Burke JE, Leonard TA. Conformational sampling of membranes by Akt controls its activation and inactivation. Proc Natl Acad Sci U S A. 2018;115:e940–e9.
Rajasekar N, Dwivedi S, Nath C, Hanif K, Shukla R. Protection of streptozotocin induced insulin receptor dysfunction, neuroinflammation and amyloidogenesis in astrocytes by insulin. Neuropharmacology. 2014;86:337–52.
Kamikubo Y, Takasugi N, Niisato K, Hashimoto Y, Sakurai T. Consecutive analysis of BACE1 function on developing and developed neuronal cells. J Alzheimers Dis. 2016;56:641–53.
Crunkhorn S. Alzheimer disease: BACE1 inhibitor reduces beta-amyloid production in humans. Nat Rev Drug Discov. 2016;16:18.
Devi L, Alldred MJ, Ginsberg SD, Ohno M. Mechanisms underlying insulin deficiency-induced acceleration of beta-amyloidosis in a mouse model of Alzheimer’s disease. PLoS ONE. 2012;7:e32792.
Wang J, Zhang HY, Tang XC. Huperzine a improves chronic inflammation and cognitive decline in rats with cerebral hypoperfusion. J Neurosci Res. 2010;88:807–15.
Wang ZF, Tang XC. Huperzine A protects C6 rat glioma cells against oxygen-glucose deprivation-induced injury. FEBS Lett. 2007;581:596–602.
Wang J, Chen F, Zheng P, Deng W, Yuan J, Peng B, et al. Huperzine A ameliorates experimental autoimmune encephalomyelitis via the suppression of T cell-mediated neuronal inflammation in mice. Exp Neurol. 2012;236:79–87.
Akintola AA, van Heemst D. Insulin, aging, and the brain: mechanisms and implications. Front Endocrinol (Lausanne). 2015;6:13.
Havrankova J, Roth J, Brownstein MJ. Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J Clin Invest. 1979;64:636–42.
Plum L, Schubert M, Bruning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab. 2005;16:59–65.
Kern W, Born J, Schreiber H, Fehm HL. Central nervous system effects of intranasally administered insulin during euglycemia in men. Diabetes. 1999;48:557–63.
Rajasekar N, Nath C, Hanif K, Shukla R. Intranasal insulin administration ameliorates streptozotocin (ICV)-induced insulin receptor dysfunction, neuroinflammation, amyloidogenesis, and memory impairment in rats. Mol Neurobiol. 2017;54:6507–22.
Nguyen TTL, Chan LC, Borreginne K, Kale RP, Hu C, Tye SJ. A review of brain insulin signaling in mood disorders: from biomarker to clinical target. Neurosci Biobehav Rev. 2018;92:7–15.
Salameh TS, Bullock KM, Hujoel IA, Niehoff ML, Wolden-Hanson T, Kim J, et al. Central nervous system delivery of intranasal insulin: mechanisms of uptake and effects on cognition. J Alzheimers Dis. 2015;47:715–28.
Maimaiti S, Anderson KL, DeMoll C, Brewer LD, Rauh BA, Gant JC, et al. Intranasal insulin improves age-related cognitive deficits and reverses electrophysiological correlates of brain aging. J Gerontol A Biol Sci Med Sci. 2016;71:30–9.
Liu Z, Patil IY, Jiang T, Sancheti H, Walsh JP, Stiles BL, et al. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS ONE. 2015;10:e0128274.
Gralle M. The neuronal insulin receptor in its environment. J Neurochem. 2017;140:359–67.
Cheng X, He P, Lee T, Yao H, Li R, Shen Y. High activities of BACE1 in brains with mild cognitive impairment. Am J Pathol. 2014;184:141–7.
Sadleir KR, Eimer WA, Cole SL, Vassar R. Abeta reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Mol Neurodegener. 2015;10:1. https://doi.org/10.1186/1750-1326-10-1.
Huse JT, Byant D, Yang Y, Pijak DS, D’Souza I, Lah JJ, et al. Endoproteolysis of beta-secretase (beta-site amyloid precursor protein-cleaving enzyme) within its catalytic domain. A potential mechanism for regulation. J Biol Chem. 2003;278:17141–9.
Vattemi G, Engel WK, McFerrin J, Pastorino L, Buxbaum JD, Askanas V. BACE1 and BACE2 in pathologic and normal human muscle. Exp Neurol. 2003;179:150–8.
Zhang T, Pan BS, Zhao B, Zhang LM, Huang YL, Sun FY. Exacerbation of poststroke dementia by type 2 diabetes is associated with synergistic increases of beta-secretase activation and beta-amyloid generation in rat brains. Neuroscience. 2009;161:1045–56.
MacPherson RE, Baumeister P, Peppler WT, Wright DC, Little JP. Reduced cortical BACE1 content with one bout of exercise is accompanied by declines in AMPK, Akt, and MAPK signaling in obese, glucose-intolerant mice. J Appl Physiol. 2015;119:1097–104.
Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–24.
Zhang HY, Yan H, Tang XC. Huperzine A enhances the level of secretory amyloid precursor protein and protein kinase C-alpha in intracerebroventricular beta-amyloid-(1-40) infused rats and human embryonic kidney 293 Swedish mutant cells. Neurosci Lett. 2004;360:21–4.
Shi Q, Fu J, Ge D, He Y, Ran J, Liu Z, et al. Huperzine A ameliorates cognitive deficits and oxidative stress in the hippocampus of rats exposed to acute hypobaric hypoxia. Neurochem Res. 2012;37:2042–52.
Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci. 2015;18:800–6.
Chen G, Chen KS, Kobayashi D, Barbour R, Motter R, Games D, et al. Active beta-amyloid immunization restores spatial learning in PDAPP mice displaying very low levels of beta-amyloid. J Neurosci. 2007;27:2654–62.
Denver P, McClean PL. Distinguishing normal brain aging from the development of Alzheimer’s disease: inflammation, insulin signaling and cognition. Neural Regen Res. 2018;13:1719–30.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81871222, 81570763, and 81270947 to XQX), the Fundamental Science and Advanced Technology Research of Chongqing (Major Project, CSTC2015jcyjB0146), and the National Program on Key Basic Research Project of China (973 Program, 2012CB517505, to XQX).
Author contributions
HYW and XQX designed the research; HYW, MW, and JLD performed the research; HYW and MW analyzed the data; JBL and YXS contributed the materials; and HYW and XQX wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Wang, Hy., Wu, M., Diao, Jl. et al. Huperzine A ameliorates obesity-related cognitive performance impairments involving neuronal insulin signaling pathway in mice. Acta Pharmacol Sin 41, 145–153 (2020). https://doi.org/10.1038/s41401-019-0257-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0257-1
