
Abstract
Tissue factor (TF)-dependent coagulation contributes to lung inflammation and the pathogenesis of acute lung injury (ALI). In this study, we explored the roles of targeted endothelial anticoagulation in ALI using two strains of transgenic mice expressing either a membrane-tethered human tissue factor pathway inhibitor (hTFPI) or hirudin fusion protein on CD31+ cells, including vascular endothelial cells (ECs). ALI was induced by intratracheal injection of LPS, and after 24 h the expression of TF and protease-activated receptors (PARs) on EC in lungs were assessed, alongside the extent of inflammation and injury. The expression of TF and PARs on the EC in lungs was upregulated after ALI. In the two strains of transgenic mice, expression of either of hTFPI or hirudin by EC was associated with significant reduction of inflammation, as assessed by the extent of leukocyte infiltration or the levels of proinflammatory cytokines, and promoted survival after LPS-induced ALI. The beneficial outcomes were associated with inhibition of the expression of chemokine CCL2 in lung tissues. The protection observed in the CD31-TFPI-transgenic strain was abolished by injection of an anti-hTFPI antibody, but not by prior engraftment of the transgenic strains with WT bone marrow, confirming that the changes observed were a specific transgenic expression of anticoagulants by EC. These results demonstrate that the inflammation in ALI is TF and thrombin dependent, and that expression of anticoagulants by EC significantly inhibits the development of ALI via repression of leukocyte infiltration, most likely via inhibition of chemokine gradients. These data enhance our understanding of the pathology of ALI and suggest a novel therapeutic strategy for treatment.
Similar content being viewed by others


Introduction
Acute lung injury (ALI) is a clinical syndrome characterized by diffuse interstitial pulmonary and noncardiogenic pulmonary edema that results in severe hypoxemia and dyspnea [1]. Much of the pathogenesis of ALI is due to endothelial cell (EC) injury, which leads to increased vascular permeability and extensive inflammatory pulmonary infiltrates [1]. Because ALI is one of the most frequent causes of morbidity and mortality in critically ill patients, there is a need for novel therapeutic targets.
In ALI or acute respiratory distress syndrome (ARDS), there are mutual amplifying interactions between inflammation and coagulation. Abundant evidence indicates that coagulopathy is a significant event in systemic inflammation, ALI or ARDS. For instance, the infusion of endotoxin or bacteria into the vascular space to activate inflammation is quickly followed by intravascular coagulation that is activated by enhanced tissue factor (TF) activity [2, 3]. In patients with ARDS, evidence of elevated levels of activated coagulation proteins, including thrombin (increased sixfold), soluble TF, activated factor VII (FVIIa), and activated factor X (FXa), can be detected in bronchoalveolar lavage fluid (BALF) [4]. The TF-FVIIa pathway mediates these coagulation disturbances [5]. Procoagulant TF binds FVIIa, and binds and activates FX to FXa, following which downstream coagulation proteases can be activated [6].
These effects are accompanied by changes in the levels of physiological regulators of coagulators. For instance, antithrombin (AT) levels are reduced in the BALF of healthy volunteers after local challenge with either lipoteichoic acid (LTA) (reduced by approximately 75%) [7] or lipopolysaccharide (LPS) (reduced by approximately 83%) [7], as well as in patients with inhalation trauma due to burns (reduced by approximately 66%) [8]. Similarly, the TF inhibitory activity of intra-alveolar tissue factor pathway inhibitor (TFPI) was reduced through truncation and inactivation (in all but 1 of 17 patients with ARDS, the majority of the TFPI was truncated and likely inactive) [9]. All of these observations suggest that coagulation proteases may exacerbate inflammation and thus contribute to ALI.
Coagulation proteases (such as TF-FVIIa complex, FXa, and thrombin) can activate protease-activated receptors (PARs) on the surfaces of EC and monocytes [7], thereby promoting the generation of proinflammatory cytokines, for example, IL-6 and specific chemokines, which can enhance vascular permeability, extravasation of leukocytes, and lung injury [10].
Therapeutic interventions aimed at coagulation blockade have been shown to be useful for ALI/ARDS by dampening the inflammatory response. For instance, in a rat model of Streptococcus pneumoniae pneumonia, AT treatment promoted lung-protective effects (the total histopathology score was improved by approximately 66%) [11]. In another study, TFPI reduced the lung injury due to Escherichia coli septic shock in baboons (the arterial–alveolar oxygen gradient was improved by approximately 50%) [12]. In the current studies, the anticoagulants were administered intravenously, intratracheally, or by nebulization, and no assessment of the mechanism of action was assessed. We have explored the impact of targeted anticoagulant therapy in a mouse model of ALI, using two strains of transgenic mice, CD31-TFPI-Tg and CD31-Hir-Tg, which expressed either a membrane-tethered human tissue factor pathway inhibitor (hTFPI) or a hirudin fusion protein, respectively, on all CD31+ cells, including vascular ECs, monocytes, platelets, polymorphonuclear leukocytes (PMNs), and T cells [13,14,15,16].
Materials and methods
Mice
The CD31-TFPI-Tg and CD31-Hir-Tg mice have been previously described [13]. The transgenic mice express the hTFPI and leech anticoagulant hirudin, which are tethered to the cell surface by fusion with fragments of human CD4 and P-selectin. CD31 [platelet EC adhesion molecule] promoter control of the expression of the transgenes limits the expression to ECs, monocytes, and platelets, PMNs and T cells. In addition, the P-selectin sequence directs expression to secretory granules. Functional cell surface expression only occurs when the cells are activated. Wild-type mice (WT, C57BL/6 background, 18–20 g) were purchased from the Animal Center of Slaccas (Shanghai, China). All mice were maintained in the laboratory animal center of Zhejiang University, which is an accredited animal facility. The mice were housed in a room maintained at 23 ± 2 °C with 50% ± 10% humidity and a 12-h light:12-h dark cycle (lights on from 08:00 a.m. to 08:00 p.m.). In addition, they were allowed free access to water and regular rodent chow (Slaccas, P1101F-25, Shanghai, China). All animal experiments complied with the “Guide for the care and use of laboratory animals” and were approved by the Animal Care and Use Committee at Zhejiang University, China. Genomic DNA from the tails of the CD31-TFPI-Tg and CD31-Hir-Tg mice was extracted for genotyping as described [13].
Bone marrow (BM) transplantation
As described before [17], BM was flushed from the excised ends of long bones from WT mice with CD45.1+ BM with normal saline (NS) and resuspended at 2.5 × 106 cells per mL in NS. CD31-TFPI-Tg or CD31-Hir-Tg and WT mice with CD45.2+ BM were irradiated with 8 Gy (800 rad) before injection of 1 × 106 CD45.1+ BM cells in NS into a tail vein. The CD45.1 and CD45.2 expression in BM of recipient mice before and after BM transplantation was evaluated using flow cytometric analysis (data not shown). CD45.2 mice were purchased from the Animal Center of Slaccas (Shanghai, China). CD45.1 mice were kindly provided by Professor Lie Wang from Zhejiang University School of Medicine. A fluorescein isothiocyanate (FITC)-labeled antibody against CD45.1 (eBioscience, 11-0453-81, San Diego, California, USA) and an allophycocyanin (APC)-labeled anti-CD45.2 (eBioscience, 17-0454-81, San Diego, California, USA) were used.
ALI mouse model
We anesthetized mice with 1% pentobarbital sodium and then injected 50 µL of a suspension of LPS from Pseudomonas aeruginosa (10 mg/kg) (Sigma-Aldrich, L9143, Saint Louis, Missouri, USA) or NS into the trachea with a microsyringe; the mice were kept vertical for 1 min to ensure that the LPS or NS was distributed in lungs. The mice were sacrificed 24 h later.
Clotting time
We incubated 100 µL of mouse plasma in a 5 mL plastic tube, added 50 µL of fresh BALF to the plasma and incubated the tube for 2 min at 37 °C in a water bath. Then, we added 100 µL 25 mM CaCl2 to the tube and mixed it well. We measured the time to clot formation (observed after gently shaking the tube, the formation of the clot observed and the time for the solution to clot was recorded using a stop watch).
Isolated cells from lung
Isolated lung tissues were cut into small pieces before digestion for 1.5 h with collagenase (Sigma-Aldrich, C0130, Saint Louis, Missouri, USA) at 37 °C. The digested tissues and supernatant were ground and then filtered. The resulting cell suspension was centrifuged and resuspended in culture medium [18].
Flow cytometric analysis
Cells were incubated simultaneously with Brilliant Violet (BV) 510-, FITC-, and BV421-labeled antibodies against CD45 (Biolegend, 103138, San Diego, California, USA), CD11b (Biolegend, 101205, San Diego, California, USA), and Gr-1 (Biolegend, 108445, San Diego, California, USA), respectively, for 1 h at 4 °C. The stained cells were prepared for flow cytometric analysis using a flow cytometer (Beckman Cytoflex, Suzhou, Jiangsu, China).
Separate aliquots of cells were sequentially incubated with primary rabbit or goat antibodies directed against mouse TF (Abcam, ab151748, Cambridgeshire, England, UK), PAR1 (Santa Cruz Biotechnology, sc-8203, Dallas, Texas, USA) or PAR2 (Santa Cruz Biotechnology, sc-8205, Dallas, Texas, USA) for 1 h at 4 °C, and then, they were washed and stained with phycoerythrin (PE)-labeled anti-mouse CD144 (VE-Cadherin) (eBioscience, 85-12-1441-80, San Diego, California, USA) and Alexa Fluor 488-labeled goat anti-Rabbit IgG (H + L) secondary antibody (ThermoFisher Scientific, A-11034, Waltham, Massachusetts, USA) or Alexa Fluor 488-labeled donkey anti-goat IgG (H + L) secondary antibody (ThermoFisher Scientific, A-11055, Waltham, Massachusetts, USA) for 30 min to allow for identification of the CD144+ ECs. The stained cells were prepared for flow cytometric analysis using an FC 500 (Cytomics™ FC 500, Beckman, Brea, California, USA).
BALF analysis
The left lungs of the euthanized mice were lavaged three times with 0.4 mL phosphate-buffered saline. Cytospin slides were prepared for Wright-Giemsa staining (BaSO, BA4017, Zhuhai, Guangdong, China). An analysis of the numbers of total and differentiated cell counts of BALF was performed as previously described [19]. Neutrophils contain intracellular granules that stain as a neutral pink and have a nucleus that is divided into 2–5 lobes. Macrophages are larger than neutrophils, stain dark blue and contain a mononucleus of a kidney- or horseshoe-shaped or irregular form.
The total protein levels in the supernatants of BALF were measured using a Pierce BCA protein assay kit (Thermo Scientific, 23225, Rockford, Illinois, USA).
RNA isolation and real-time (RT) PCR assay
We extracted the RNA from lungs with Trizol reagent (Takara Biotechnology, 9109, Kusatsu, Shiga, Japan) according to the manufacturer’s instructions. Using gene expression assays, we performed RT-PCR on a StepOnePlus Real-Time RCR system (Applied Biosystems, Foster City, California, USA). Table 1 shows specific primers for RT-PCR that were used to evaluate the gene expression.
Enzyme-linked immunosorbent assay (ELISA) analysis
The protein expression levels of KC, MIP-2, IL-6, and chemokine (C-C motif) ligand 2 (CCL2) in the lung homogenates were analyzed using Quantikine ELISAs with antibody pairs (R&D Systems, MKC00B, MM200, M6000B, MJE00, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Histological assessment
The right lungs of the euthanized mice were inflated and gravity fixed (20 cm) with 4% paraformaldehyde followed by paraffin embedding. Axial sections (5 μm) were stained with hematoxylin and eosin (H&E). An Olympus BX53 inverted microscope (Olympus, Beijing, China) was used to examine the sections. The histological evaluation was performed using methods previously described [20]: for each mouse, three different lobes were examined for the following features: interstitial edema, hemorrhage, and neutrophil infiltration. Each feature could receive a score of 0 (no injury), 1 (minimal injury), 2 (moderate injury), or 3 (severe injury). These values were totaled for a given lobe’s score, and the results for three lobes were averaged to generate a score for each mouse, giving a minimum score of 0 and a maximum of 9. The histological sections were scored by a pathologist blinded to the treatment or mouse type.
Mouse lung wet/dry ratio assay
The lungs of the euthanized and exsanguinated mice were isolated and weighed to determine the wet weight before being dried at 60 °C for 72 h to obtain the dry weight.
Mouse alveolar-capillary leak assay
Some mice received 20 mg/kg Evans blue (EB) solution (Sigma-Aldrich, E2129, Saint Louis, Missouri, USA) by intravenous injection, 22 h after intratracheal LPS and 2 h prior to euthanasia and exsanguination. The lungs were isolated, dipped into formamide solution (BIOSHARP, BS335A, Hefei, Anhui, China), and incubated at 60 °C for 24 h. Afterwards, the supernatants were measured for the absorbance of EB at 620 nm.
Survival rate
Mice recovered from anesthesia after intratracheal LPS; they were observed daily until 168 h to assess survival of each group.
Statistical analysis
All results were presented as the means ± SEM from each group of three independent experiments, which enrolled 5–11 mice per group. An unpaired t-test was used to compare the two groups. One-way analysis of variance (ANOVA) with the Bonferroni correction was applied to determine the interactions among multiple groups. The log-rank (Mantel–Cox) test was performed for analysis of survival rate. If P < 0.05, the differences were considered statistically significant.
Results
LPS induces lung inflammation and coagulation and activates the TF–thrombin–PARs pathway in mouse lung ECs
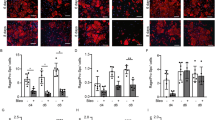
To determine whether LPS induces lung inflammation and coagulation, WT mice were intratracheally instilled with LPS or NS and sacrificed after 24 h. We then evaluated the lung inflammatory cell infiltration using flow cytometric analysis and BALF cell differential counting, and the coagulation using the BALF clotting time assay. The total cell numbers [(4.438 ± 0.7585) × 104 vs (113.9 ± 12.51) × 104, WT-CON vs WT-LPS, mean ± SEM (hereinafter), P < 0.001, Fig. 1a] and the neutrophil numbers [(0.03406 ± 0.008477) × 104 vs (96.83 ± 9.47) × 104, P < 0.001, Fig. 1b)] in the BALF were increased by LPS treatment. The results were consistent with those of the flow cytometric analysis for the percentage of neutrophils in the white blood cells in the suspensions of the lung tissue cells (20.19% ± 2.102% vs 67.96% ± 2.010%, P < 0.001, Figs. 1d-f). The clotting time for BALF (187.8 ± 3.902 vs 71.50 ± 4.890 s, P < 0.001, Fig. 1c) was shortened in the LPS group.

Intratracheal LPS induces lung inflammation (a-b, d-f), coagulation activation (c) and an increased expression of TF (g-i), PAR1 (j–l), and PAR2 (m-o) on CD144+ (the indicative molecule of ECs) cells in lung during ALI. Compared with NS group. n = 5–10 for each group. **P < 0.01; ***P < 0.001; ns no significance. Data are presented as mean ± SEM of three separate experiments. CON control, LPS lipopolysaccharide, ALI acute lung injury, WBC white blood cell, Neut neutrophil, BALF bronchoalveolar lavage fluid, TF tissue factor, PAR protease-activated receptor, ECs endothelial cells, WT wild type
To examine whether the EC-specific TF–thrombin–PARs pathway was involved in the LPS-induced ALI, the expression levels of TF, PAR1, and PAR2 on the ECs from the lung tissue were measured using flow cytometric analysis in the ALI model for WT mouse. Compared with the NS controls, the expression levels of TF (38.12% ± 1.99% vs 74.02% ± 1.65%, P < 0.001, Figs. 1g-i), PAR1 (5.16% ± 0.337% vs 13.44% ± 1.562%, P < 0.001, Figs. 1j-l), and PAR2 (6.933% ± 0.4529% vs 13.86% ± 1.657%, P < 0.01, Figs. 1m-o) in the CD144+ (the indicative molecule of ECs) cells of the WT lung tissue were increased in the LPS group. These data demonstrate that LPS activates the TF–thrombin–PARs pathway in pulmonary ECs.
Both CD31-TFPI-Tg and CD31-Hir-Tg mice protect LPS-induced ALI
To assess the different levels of ALI, the WT, CD31-TFPI-Tg, and CD31-Hir-Tg mice were intratracheally instilled with LPS or NS and sacrificed after 24 h. The total cell numbers [(113.9 ± 12.51) × 104 vs (59.8 ± 5.822) × 104 or (63.88 ± 7.542) × 104, WT-LPS vs CD31-TFPI-Tg-LPS or CD31-Hir-Tg-LPS (hereinafter), P < 0.001, respectively, Fig. 2a], the neutrophil numbers [(96.83 ± 9.47) × 104 vs (54.15 ± 5.411) × 104 or (46.49 ± 9.929) × 104, P < 0.001, respectively, Fig. 2b], and the total protein (1.253 ± 0.1589 vs 0.5767 ± 0.1053 or 0.7108 ± 0.1352 mg/mL, P < 0.001 or P < 0.01, respectively, Fig. 2l) in the BALF were increased by the LPS treatment. Interestingly, compared with WT-LPS group, all of these inflammatory markers were decreased in both CD31-TFPI-Tg-LPS and CD31-Hir-Tg-LPS groups. Consistent with the above results, the increased gene expression of KC (235.5 ± 45.85-fold vs 118.0 ± 27.87-fold or 52.66 ± 9.389-fold, P < 0.01 or P < 0.001, respectively, Fig. 2c), MIP-2 (1124 ± 351.8-fold vs 296.3 ± 77.24-fold or 208.5 ± 41.14-fold, P < 0.01 or P < 0.001, respectively, Fig. 2e), IL-6 (230.3 ± 80.02-fold vs 67.91 ± 15.68-fold or 34.38 ± 4.493-fold, P < 0.01, respectively, Fig. 2g), TNF-α (176.9 ± 41.7-fold vs 15.9 ± 2.308-fold or 20.27 ± 2.147-fold, P < 0.001, respectively, Fig. 2i) and IL-1β (64.77 ± 7.855-fold vs 33.03 ± 4.336-fold or 30.13 ± 4.084-fold, P < 0.001, respectively, Fig. 2j), as well as the lung edema (ratios of 5.079 ± 0.08718 vs 4.51 ± 0.03138 or 4.686 ± 0.03942, P < 0.001, respectively, Fig. 2m), alveolar-capillary leak (28.04 ± 2.845 vs 13.49 ± 2.193 or 16.61 ± 2.319 µg/g, P < 0.001 or P < 0.01, respectively, Fig. 2n) and focal histological injury in the lung tissue (scores of 7.667 ± 0.4216 vs 5.0 ± 0.3651 or 4.667 ± 0.3333, P < 0.001, respectively, Fig. 2p, q) that were seen in the WT-LPS group, were also mitigated in the CD31-TFPI-Tg-LPS and CD31-Hir-Tg-LPS groups. The results for the protein expression levels of KC (431.8 ± 44.08 vs 256.4 ± 23.79 or 187.3 ± 31.95 pg/100 mg, P < 0.001, respectively, Fig. 2d), MIP-2 (361.3 ± 26.04 vs 234.2 ± 28.68 or 215.9 ± 32.17 pg/100 mg, P < 0.01 or P < 0.001, respectively, Fig. 2f), and IL-6 (76.88 ± 9.951 vs 33.06 ± 5.067 or 30.48 ± 2.098 pg/100 mg, P < 0.001, respectively, Fig. 2h) were consistent with those for the RT-PCR for mRNA. Compared with WT-LPS group, the gene expression levels of the anti-inflammatory cytokine IL-10 (42.90 ± 3.482-fold vs 82.62 ± 12.34-fold or 88.59 ± 13.22-fold, P < 0.05 or P < 0.01, respectively, Fig. 2k) were elevated in the CD31-TFPI-Tg-LPS and CD31-Hir-Tg-LPS groups. The survival of the transgenic animals after LPS was higher than that of mice in the WT-LPS group (Fig. 2o) (P < 0.05). These data indicate that targeted expression of hTFPI or hirudin by CD31+ cells can improve ALI in mice.

CD31-TFPI-Tg and CD31-Hir-Tg mice protect against LPS-induced ALI. Total cell numbers (a), neutrophil numbers (b), and total protein (l) in BALF, lung edema (m) and alveolar-capillary leak (n) after intratracheal instillation of LPS for 24 h and the percentage of survival (o). With quantitative real-time PCR, the gene expression levels of KC (c), MIP-2 (e), IL-6 (g), TNF-α (i), IL-1β (j), and IL-10 (k) were detected. With Quantikine ELISA analysis, the protein expression levels of KC (d), MIP-2 (f), and IL-6 (h) were measured. Representative light photomicrographs (100 × ) of H&E staining of lung tissue (p) and histology score (q). Scale bars = 200 μm. Under the heading of “104 cells/mL”, a value of 1 designates 10 000 cells/mL. n = 6–11 for each group. *P < 0.05; **P < 0.01; ***P < 0.001; ns no significance. Data are presented as mean ± SEM of three separate experiments. CON control, LPS lipopolysaccharide, ALI acute lung injury, BALF bronchoalveolar lavage fluid, EB Evans blue, WT wild type
Anti-hTFPI-neutralizing antibody abolishes the protective effect of CD31-TFPI-Tg mice in ALI
To further assess the protective effect of targeted expression of hTFPI in LPS-induced ALI in mice, we administered anti-hTFPI-neutralizing antibody through tail vein injection. In these experiments, the reduced inflammation and injury seen in CD31-TFPI-Tg mice that were administered LPS were again evident, but these effects were abolished by the anti-hTFPI. Therefore, there were no observable differences in total cell numbers [(99.11 ± 6.309) × 104 vs (111.3 ± 6.425) × 104, WT-LPS vs CD31-TFPI-Tg-LPS-anti-TFPI (hereinafter), P > 0.05, Fig. 3a], neutrophil numbers [(93.53 ± 8.318) × 104 vs (105.4 ± 6.203) × 104, P > 0.05, Fig. 3b], and total protein (0.7802 ± 0.07309 vs 0.6839 ± 0.06103 mg/mL, P > 0.05, Fig. 3l) in the BALF, the gene expression levels of KC (96.42 ± 14.02-fold vs 84.97 ± 19.03-fold, P > 0.05, Fig. 3c), MIP-2 (299.9 ± 40.12-fold vs 312.1 ± 33.91-fold, P > 0.05, Fig. 3e), IL-6 (91.01 ± 24.19-fold vs 83.2 ± 12.4-fold, P > 0.05, Fig. 3g), TNF-α (88.32 ± 18.94-fold vs 86.3 ± 13.00-fold, P > 0.05, Fig. 3i), IL-1β (60.62 ± 7.130-fold vs 58.40 ± 8.271-fold, P > 0.05, Fig. 3j), and IL-10 (44.72 ± 4.659-fold vs 46.40 ± 4.877-fold, P > 0.05, Fig. 3k) and focal histological injury in lung tissue (scores of 7.333 ± 0.3333 vs 7.5 ± 0.4282, P > 0.05, Figs. 3m-n) between CD31-TFPI-Tg mice that received the anti-hTFPI-neutralizing antibody and WT mice. The results for the protein expression levels of KC (460.4 ± 41.23 vs 478.3 ± 57.70 pg/100 mg, P > 0.05, Fig. 3d), MIP-2 (398.4 ± 25.02 vs 409.7 ± 23.16 pg/100 mg, P > 0.05, Fig. 3f), and IL-6 (95.09 ± 4.087 vs 79.32 ± 6.602 pg/100 mg, P > 0.05, Fig. 3h) were consistent with those of the RT-PCR for mRNA. These results indicate that the protective effect of CD31-TFPI-Tg mice in ALI is completely dependent on the membrane-expressed hTFPI.

The anti-hTFPI-neutralizing antibody abolishes the protective effect of the hTFPI fusion protein expressed by CD31-TFPI-Tg mice. Total cell numbers (a), neutrophil numbers (b), and total protein (l) in BALF after intratracheal instillation of LPS for 24 h. With quantitative real-time PCR, the gene expression levels of KC (c), MIP-2 (e), IL-6 (g), TNF-α (i), IL-1β (j), and IL-10 (k) were detected. With Quantikine ELISA analysis, the protein expression levels of KC (d), MIP-2 (f), and IL-6 (h) were measured. Representative light photomicrographs (100 × ) of H&E staining of lung tissue (m) and histology score (n). Scale bars = 200 μm. Under the heading of “104 cells/mL”, a value of 1 designates 10 000 cells/mL. n = 6–10 for each group. *P < 0.05; **P < 0.01; ***P < 0.001; ns no significance. Data are presented as mean ± SEM of three separate experiments. CON control, LPS lipopolysaccharide, ALI acute lung injury, BALF bronchoalveolar lavage fluid, WT wild type
CD31-TFPI-Tg and CD31-Hir-Tg mice engrafted with WT BM exert similar protective effects in LPS-induced ALI
As CD31+ cells include ECs, monocytes, platelets, PMNs, and T cells, to exclude the influence of the targeted expression of hTFPI on hemocytes, CD31-TFPI-Tg-LPS and CD31-Hir-Tg mice were engrafted with WT BM for 8 weeks and were then subjected to LPS-induced ALI. Not surprisingly, all of the inflammatory markers, including the total cell numbers [(97.4 ± 8.403) × 104 vs (57.75 ± 6.245) × 104 or (45.9 ± 6.515) × 104, WT/WT-LPS vs WT/CD31-TFPI-Tg-LPS or WT/CD31-Hir-Tg-LPS (hereinafter), P < 0.001, respectively, Fig. 4a], the neutrophil numbers [(93.0 ± 8.104) × 104 vs (52.29 ± 5.881) × 104 or (42.47 ± 11.25) × 104, P < 0.001, respectively, Fig. 4b], and total protein (1.163 ± 0.1521 vs 0.5408 ± 0.05562 or 0.5768 ± 0.1293 mg/mL, P < 0.001 or P < 0.01, respectively, Fig. 4l) in the BALF, as well as the gene expression levels of KC (49.0 ± 7.134-fold vs 23.37 ± 3.612-fold or 25.03 ± 3.473-fold, P < 0.001 or P < 0.01, respectively, Fig. 4c), MIP-2 (105.0 ± 14.57-fold vs 55.28 ± 8.62-fold or 54.72 ± 10.17-fold, P < 0.01, respectively, Fig. 4e), IL-6 (68.58 ± 11.26-fold vs 27.12 ± 10.37-fold or 33.85 ± 6.625-fold, P < 0.01 or P < 0.05, respectively, Fig. 4g), TNF-α (111.4 ± 23.16-fold vs 22.87 ± 2.433-fold or 24.66 ± 2.947-fold, P < 0.001, respectively, Fig. 4i), IL-1β (56.35 ± 6.058-fold vs 35.96 ± 6.525-fold or 32.27 ± 5.388-fold, P < 0.05 or P < 0.01, respectively, Fig. 4j), the lung edema (ratios of 5.118 ± 0.04009 vs 4.781 ± 0.04957 or 4.711 ± 0.09122, P < 0.01 or P < 0.001, respectively, Fig. 4m) and the histological scores (scores of 6.333 ± 0.3333 vs 4.667 ± 0.2108 or 4.5 ± 0.2236, P < 0.01 or P < 0.001, respectively, Figs. 4n, o), were still attenuated in the transgenic mice that had been engrafted with the WT BM. The results of the protein expression levels of KC (760.3 ± 112.2 vs 249.5 ± 48.63 or 244.8 ± 55.93 pg/100 mg, P < 0.001, respectively, Fig. 4d), MIP-2 (700.9 ± 64.25 vs 293.2 ± 50.80 or 317.2 ± 53.50 pg/100 mg, P < 0.001, respectively, Fig. 4f), and IL-6 (72.58 ± 12.09 vs 35.50 ± 3.638 or 34.83 ± 5.188 pg/100 mg, P < 0.001, respectively, Fig. 4h) were consistent with those for the RT-PCR for mRNA. Compared with the WT/WT-LPS group, the gene expression level of the anti-inflammatory cytokine IL-10 (22.71 ± 2.636-fold vs 50.77 ± 4.817-fold or 55.03 ± 8.369-fold, P < 0.001, respectively, Fig. 4k) was elevated in the WT/CD31-TFPI-Tg-LPS and WT/CD31-Hir-Tg-LPS groups. These data indicate that only the expression of the membrane-tethered hTFPI and hirudin anticoagulants on ECs gave similar protection from ALI as was seen in non-engrafted mice of either strain, which suggested that expression by monocytes, platelets, PMNs, and T cells contributed little to the protective phenotype.

CD31-TFPI-Tg and CD31-Hir-Tg mice, engrafted with bone marrow from WT mice (WT/CD31-TFPI-Tg and WT/CD31-Hir-Tg mice respectively) are still protected against LPS-induced ALI. Total cell numbers (a), neutrophil numbers (b), and total protein (l) in BALF, and lung edema (m) after intratracheal instillation of LPS for 24 h. With quantitative real-time PCR, the gene expression levels of KC (c), MIP-2 (e), IL-6 (g), TNF-α (i), IL-1β (j), and IL-10 (k) were detected. With Quantikine ELISA analysis, the protein expression levels of KC (d), MIP-2 (f), and IL-6 (h) were measured. Representative light photomicrographs (100 × ) of H&E staining of lung tissue (o) and histology score (n). Scale bars = 200 μm. Under the heading of “104 cells/mL”, a value of 1 designates 10 000 cells/mL. n = 6–10 for each group. *P < 0.05; **P < 0.01; ***P < 0.001; ns no significance. Data are presented as mean ± SEM of three separate experiments. CON control, LPS lipopolysaccharide, ALI acute lung injury, BALF bronchoalveolar lavage fluid, WT wild type
Significant decrease in the expression level of CCL2 in lung tissue of CD31-TFPI-Tg and CD31-Hir-Tg mice, either engrafted or non-engrafted
To further elucidate the mechanism of the protective effect of the two membrane-tethered anticoagulants, we evaluated the chemokine levels in the lung tissue of mice with ALI and found that the expression of CCL2 was increased in all strains of mice after administration of LPS. However, compared with the WT mice, the expression level of CCL2 was lower in both the CD31-TFPI-Tg-LPS and CD31-Hir-Tg mice. This outcome was true for both the non-engrafted transgenic mice [RT-PCR for mRNA (hereinafter): 228.7 ± 78.01-fold vs 23.73 ± 1.754-fold or 43.76 ± 10.37-fold, P < 0.001 or P < 0.01, respectively; ELISA for protein (hereinafter): 50.48 ± 2.852 vs 34.10 ± 3.454 or 26.04 ± 4.291 pg/100 mg, P < 0.01 or P < 0.001; WT-LPS vs CD31-TFPI-Tg-LPS or CD31-Hir-Tg-LPS, respectively, Fig. 5a, b] and for those engrafted with WT BM (23.85 ± 3.134-fold vs 12.64 ± 1.709-fold or 12.7 ± 1.876-fold, P < 0.01; 68.66 ± 6.677 vs 30.71 ± 4.938 or 17.08 ± 2.553 pg/100 mg, P < 0.001, WT/WT-LPS vs WT/CD31-TFPI-Tg-LPS or WT/CD31-Hir-Tg-LPS, respectively, Figs. 5e, f). However, this reduction in CCL2 was abolished in the CD31-TFPI-Tg mice that received the anti-hTFPI-neutralizing antibody (52.05 ± 9.876-fold vs 52.75 ± 6.513-fold, P > 0.05; 53.40 ± 2.760 vs 54.03 ± 6.856 pg/100 mg, WT-LPS vs CD31-TFPI-Tg-LPS-anti-TFPI, P > 0.05, Fig. 5c, d). These data indicated that the reduced inflammation and injury seen in the two strains of transgenic mice was strongly associated with the reduction in the level of the key chemokine CCL2.

The expression level of CCL2 decreases in lung tissues after LPS-induced ALI in CD31-TFPI-Tg and CD31-Hir-Tg mice, either those engrafted with WT BM or those non-engrafted. Quantitative real-time PCR and ELISA analysis respectively detected the expression level of CCL2 in lung tissues in ALI model of CD31-TFPI-Tg and CD31-Hir-Tg mice (a, b), CD31-TFPI-Tg mice receiving anti-hTFPI-neutralizing antibody (c, d), and WT/CD31-TFPI-Tg and WT/CD31-Hir-Tg mice (e, f). n = 6–10 for each group. *P < 0.05; **P < 0.01; ***P < 0.001; ns no significance. Data are presented as mean ± SEM of three separate experiments. CCL2 chemokine (C-C motif) ligand 2, CON control, LPS lipopolysaccharide, ALI acute lung injury, WT wild type
Rationale for the investigation of the CD31-TFPI-Tg or CD31-Hir-Tg mice
TF and downstream coagulation proteases can exert direct influences on the inflammatory cascade. PAR1 has a key role in transforming coagulation protease activity into inflammatory signals, and thrombin is the main ligand for PAR1. A predominant mechanism by which the fusion proteins hTFPI or hirudin inhibit inflammation is by inhibiting the development of local chemokine gradients, particularly CCL2. In vivo, these gradients are dependent on thrombin signaling through EC-expressed PAR1. The fact that both strains showed the same protective response implies that the predominant effect of the hTFPI fusion protein is to inhibit thrombin generation (Fig. 6).

Rationale for the investigation of the CD31-TFPI-Tg or CD31-Hir-Tg mice. Tissue factor (TF) and downstream coagulation proteases can exert direct influences on the inflammatory cascade. Protease-activated receptor 1 (PAR1) has a cornerstone role in transforming coagulation protease activity into inflammatory signals and thrombin is the main ligand to PAR1. A predominant mechanism by which the fusion proteins, human tissue factor pathway inhibitor (hTFPI) or hirudin, inhibit inflammation is to inhibit the development of local chemokine gradients, particularly CCL2, which in vivo is dependent on thrombin signaling through endothelial cell (EC)-expressed PAR1. The fact that both strains showed the same protective response implies that the predominant effect of the hTFPI fusion protein is to inhibit thrombin generation
Discussion
TF and downstream coagulation proteases can exert direct influences on the inflammatory cascade. All of these coagulation products exhibit extensive crosstalk with inflammatory mediators [21]. Each of these proteins may have independent effects on the lung injury that is associated with ARDS, such as generation of cytokines and migration and activation of cells. PARs have a key role in transforming coagulation protease activity into inflammatory signals [22]. Our study indicates that the expression of TF and PARs was upregulated on the vascular ECs in the lungs of WT mice in LPS-induced ALI.
It is theoretically possible that therapeutic interventions aimed at blocking the coagulation process could influence the inflammation during ALI or ARDS. Several preclinical trials have shown that APC or AT treatment significantly diminished pulmonary inflammation and attenuated histopathologic lung injury [11, 23]. Systemic anticoagulation carries a considerable increase in the risk of bleeding, although for the lung, local (inhaled) anticoagulation could potentially be delivered in the context of ALI. Intratracheal administration of TFPI improved airway obstruction and prevented respiratory failure and death in the rat model of poison gas-induced ALI [24]. Additional preclinical and clinical studies have found that inhaled anticoagulation improved the survival in smoke inhalation-induced ALI [25]. In this study, we have explored several mechanistic elements of the protection afforded by anticoagulation in a model of LPS-induced ALI. We have previously described the generation of two strains of transgenic mice, CD31-TFPI-Tg and CD31-Hir-Tg, which express a membrane-tethered hTFPI or hirudin fusion proteins, respectively, on CD31+ cells, which include vascular ECs, monocytes, platelets, PMNs and T cells [13,14,15,16].
The design of the fusion proteins is such that they are only expressed at the cell surface when the CD31+ cells have been activated by an inflammatory insult. Our previous data suggested that both strains were resistant to the profound coagulation disorder induced by systemic (i.e., intravenous) LPS [13, 26]. Moreover, we have also demonstrated that a predominant mechanism by which the fusion proteins inhibit inflammation is the inhibition of the development of local chemokine gradients, particularly CCL2. In vivo, these gradients are dependent on thrombin signaling through EC-expressed PAR1 [27]. The inhibition of the formation of these gradients prevents the initiation of leukocyte recruitment at sites of inflammation. More recently, using a third strain of mice in which the hTFPI fusion protein expression was targeted to α-smooth muscle actin-positive cells, we showed that specific inhibition of macrophage migration inhibitory factor by these cells prevented development of atherosclerosis, again via inhibition of leukocyte recruitment [28]. This is the first study of ALI with these two strains of transgenic mice.
As reported previously, IL-6 is associated with the inflammation in ARDS [10]. In our study, both transgenic stains showed attenuated expression of inflammatory factors including IL-6, KC, MIP-2, TNF-α, and IL-1β and elevated expression of anti-inflammatory factor, IL-10. In line with previous reports that showed the effects of anticoagulation in ALI or ARDS, in our ALI model, both strains of transgenic mice showed obviously reduced levels of lung inflammation, lung edema, alveolar-capillary leak, and lung injury, and this was associated with a survival rate that was remarkably improved compared with that of the WT mice. The fact that both strains showed the same protective response implies that the predominant effect of the hTFPI fusion protein is to inhibit thrombin generation.
We administered an anti-hTFPI-neutralizing antibody to the CD31-TFPI-Tg mice, and this abolished the protective effect, such that responses to LPS were similar to those seen in WT mice. This confirmed that the protection against LPS-induced ALI was due to the functional impact of the hTFPI expression. Unfortunately, we were unable to obtain an appropriate anti-hirudin-neutralizing antibody, so we were unable to repeat the same maneuver in the second transgenic strain.
To assess the contribution of fusion protein expression by platelets, monocytes, PMNs, and T cells, especially considering that TF can be expressed on the surface of monocytes [29], we made BM reconstitution chimeras by engrafting WT BM into each of the two transgenic strains. These chimeric animals responded to intratracheal LPS in the same way as the parental transgenic mice, which suggested that only the expression of anticoagulant proteins on the EC effectively blocked LPS-induced ALI.
CCL2 is a small cytokine that is part of the CC chemokine family. CCL2 recruits inflammatory cells to the sites of either tissue injury or infection [30, 31]. CCL2 is secreted not only by monocytes [32], macrophages [33], and dendritic cells [34] but also by ECs [35,36,37]. This study focused on the TF–thrombin–PAR1 pathway in the ECs. The fusion proteins, hTFPI or hirudin, inhibited the expression of CCL2, which was dependent on thrombin signaling through EC-expressed PAR1. We speculate that the decrease in the level of expression of CCL2 by the ECs was closely related to the reduction of inflammatory cell recruitment in lung and further led to the decrease in the overall level of inflammation, although the results did not define which cells were the main source of CCL2. A previous study demonstrated that PAR1 signaling impacted the expression of CCL2, which was conducive to the recruitment of neutrophils toward inflamed lung tissue [38]. It is known that PAR1 is the predominant thrombin receptor expressed on ECs [39]. Consistent with this study and our previous work, these new results show that the expression of the chemokine CCL2 is upregulated during ALI in WT mice but was reduced in both transgenic strains. The results from the neutralizing anti-hTFPI and BM chimera experiments were consistent with the concept that inhibition of CCL2 expression within the lung, which results in reduced neutrophil accumulation, is one potential mechanism by which the fusion proteins protect against LPS-induced ALI. Future studies will focus on the identification of the effects of CCL2 in ALI with a CCL2-neutralizing antibody or knockout mice.
All these data are consistent with previous work, mentioned above, which demonstrated that coagulation proteases, by activating specific receptors (PARs), aggravate lung injury. The results further suggest that local delivery of anticoagulants might be a potential therapeutic approach in ALI or ARDS. Specifically, several polypeptides could be developed for targeted anticoagulation aimed at the inflammatory endothelium of the lung in ARDS.
It is worth noting that previous work from Dr. Ware’s research group, using mice with a global absence of mouse TF, has shown that global TF deficiency leads to alveolar hemorrhage and potentiates murine ALI and oxidative stress in a direct lung injury model [40]. Furthermore, using mice with a cell-specific deletion of TF in the lung epithelium, the same investigators have also shown that the lung epithelial TF may be protective in the ALI induced directly by LPS [41]. However, these combined approaches failed to identify the TF-expressing cell type or types that are most critically involved in the lung response to LPS-induced ALI, and the results were consistent with the hypothesis that TF on ECs plays a critical role in LPS-ALI. Moreover, the use of a gene knockout approach increased baseline intra-alveolar hemorrhage, which raised the possibility that the elevated free hemoglobin and lipid peroxidation in lungs of these mice affected the lung inflammation that resulted after LPS. Our work used the same LPS-induced ALI mouse model, but by specifically targeting the membrane-tethered anticoagulants to ECs in a manner in which they were only expressed on the cell membrane upon cell activation, we avoided direct interference with baseline TF expression by any cell type in the lung, thus avoiding any intra-alveolar hemorrhage.
In conclusion, our data indicate that targeting EC-TF/thrombin-dependent inflammation obviously inhibits the development of ALI, and one potential mechanism involves the suppression of chemokine gradients and hence the reduction of leukocyte infiltration. These data remarkably enhance our understanding of the pathology and suggest a novel therapeutic target for the treatment of ALI.
References
Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–49.
van Deventer SJ, Buller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. 1990;76:2520–6.
Biemond BJ, Levi M, ten Cate H, Soule HR, Morris LD, Foster DL, et al. Complete inhibition of endotoxin-induced coagulation activation in chimpanzees with a monoclonal Fab fragment against factor VII/VIIa. Thromb Haemostas. 1995;73:223–30.
Idell S, Koenig KB, Fair DS, Martin TR, McLarty J, Maunder RJ. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am J Physiol. 1991;261:L240–8.
Miller DL, Welty-Wolf K, Carraway MS, Ezban M, Ghio A, Suliman H, et al. Extrinsic coagulation blockade attenuates lung injury and proinflammatory cytokine release after intratracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 2002;26:650–8.
Giesen PL, Rauch U, Bohrmann B, Kling D, Roque M, Fallon JT, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A. 1999;96:2311–5.
Hoogerwerf JJ, de Vos AF, Levi M, Bresser P, van der Zee JS, Draing C, et al. Activation of coagulation and inhibition of fibrinolysis in the human lung on bronchial instillation of lipoteichoic acid and lipopolysaccharide. Crit Care Med. 2009;37:619–25.
Hofstra JJ, Vlaar AP, Knape P, Mackie DP, Determann RM, Choi G, et al. Pulmonary activation of coagulation and inhibition of fibrinolysis after burn injuries and inhalation trauma. J Trauma. 2011;70:1389–97.
Bastarache JA, Wang L, Wang Z, Albertine KH, Matthay MA, Ware LB. Intra-alveolar tissue factor pathway inhibitor is not sufficient to block tissue factor procoagulant activity. Am J Physiol Lung Cell Mol Physiol. 2008;294:L874–81.
Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest. 1995;108:1303–14.
Choi G, Hofstra JJ, Roelofs JJ, Rijneveld AW, Bresser P, van der Zee JS, et al. Antithrombin inhibits bronchoalveolar activation of coagulation and limits lung injury during Streptococcus pneumoniae pneumonia in rats. Crit Care Med. 2008;36:204–10.
Welty-Wolf KE, Carraway MS, Miller DL, Ortel TL, Ezban M, Ghio AJ, et al. Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med. 2001;164:1988–96.
Chen D, Giannopoulos K, Shiels PG, Webster Z, McVey JH, Kemball-Cook G, et al. Inhibition of intravascular thrombosis in murine endotoxemia by targeted expression of hirudin and tissue factor pathway inhibitor analogs to activated endothelium. Blood. 2004;104:1344–9.
Sullivan DP, Watson RL, Muller WA. 4D intravital microscopy uncovers critical strain differences for the roles of PECAM and CD99 in leukocyte diapedesis. Am J Physiol Heart Circ Physiol. 2016;311:H621–32.
Scheible KM, Emo J, Yang H, Holden-Wiltse J, Straw A, Huyck H, et al. Developmentally determined reduction in CD31 during gestation is associated with CD8+ T cell effector differentiation in preterm infants. Clin Immunol. 2015;161:65–74.
Batorov EV, Tikhonova MA, Kryuchkova IV, Sergeevicheva VV, Sizikova SA, Ushakova GY, et al. CD4( + ) memory T cells retain surface expression of CD31 independently of thymic function in patients with lymphoproliferative disorders following autologous hematopoietic stem-cell transplantation. Int J Hematol. 2017;106:108–15.
Liu G, Hu X, Sun B, Yang T, Shi J, Zhang L, et al. Phosphatase Wip1 negatively regulates neutrophil development through p38 MAPK-STAT1. Blood. 2013;121:519–29.
Che L, Jin Y, Zhang C, Lai T, Zhou H, Xia L, et al. Ozone-induced IL-17A and neutrophilic airway inflammation is orchestrated by the caspase-1-IL-1 cascade. Sci Rep. 2016;6:18680.
Ikeda G, Miyahara N, Koga H, Fuchimoto Y, Waseda K, Kurimoto E, et al. Effect of a cysteinyl leukotriene receptor antagonist on experimental emphysema and asthma combined with emphysema. Am J Respir Cell Mol Biol. 2014;50:18–29.
Konter JM, Parker JL, Baez E, Li SZ, Ranscht B, Denzel M, et al. Adiponectin attenuates lipopolysaccharide-induced acute lung injury through suppression of endothelial cell activation. J Immunol. 2012;188:854–63.
Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109:2698–704.
Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–64.
Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, et al. Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood. 1996;87:642–7.
Rancourt RC, Veress LA, Ahmad A, Hendry-Hofer TB, Rioux JS, Garlick RB, et al. Tissue factor pathway inhibitor prevents airway obstruction, respiratory failure and death due to sulfur mustard analog inhalation. Toxicol Appl Pharmacol. 2013;272:86–95.
Miller AC, Elamin EM, Suffredini AF. Inhaled anticoagulation regimens for the treatment of smoke inhalation-associated acute lung injury: a systematic review. Crit Care Med. 2014;42:413–9.
Chen D, McVey JH, Dorling A. Enhanced effect of inhibition of thrombin on endothelium in murine endotoxaemia: specific inhibition of thrombocytopenia. Thromb Res. 2013;132:750–6.
Chen D, Carpenter A, Abrahams J, Chambers RC, Lechler RI, McVey JH, et al. Protease-activated receptor 1 activation is necessary for monocyte chemoattractant protein 1-dependent leukocyte recruitment in vivo. J Exp Med. 2008;205:1739–46.
Chen D, Xia M, Hayford C, Tham el-L Semik V, Hurst S, et al. Expression of human tissue factor pathway inhibitor on vascular smooth muscle cells inhibits secretion of macrophage migration inhibitory factor and attenuates atherosclerosis in ApoE−/− mice. Circulation. 2015;131:1350–60.
Osterud B. Tissue factor expression by monocytes: regulation and pathophysiological roles. Blood Coagul Fibrinolysis. 1998;9(Suppl 1):S9–14.
Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A. 1994;91:3652–6.
Xu LL, Warren MK, Rose WL, Gong W, Wang JM. Human recombinant monocyte chemotactic protein and other C-C chemokines bind and induce directional migration of dendritic cells in vitro. J Leukoc Biol. 1996;60:365–71.
Brigotti M, Carnicelli D, Arfilli V, Porcellini E, Galassi E, Valerii MC, et al. Human monocytes stimulated by Shiga toxin 1a via globotriaosylceramide release proinflammatory molecules associated with hemolytic uremic syndrome. Int J Med Microbiol. 2018;308:940–46.
Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192:3858–67.
Sanseverino I, Rinaldi AO, Purificato C, Cortese A, Millefiorini E, Gessani S, et al. CCL2 induction by 1,25(OH)2D3 in dendritic cells from healthy donors and multiple sclerosis patients. J Steroid Biochem Mol Biol. 2014;144 Pt A:102–5.
Bostrom M, Kalm M. A role for endothelial cells in radiation-induced inflammation. Int J Radiat Biol. 2018;94:259–71.
Shrestha B, Ge S, Pachter JS. Resolution of central nervous system astrocytic and endothelial sources of CCL2 gene expression during evolving neuroinflammation. Fluids Barriers CNS. 2014;11:6.
Ge S, Shrestha B, Paul D, Keating C, Cone R, Guglielmotti A, et al. The CCL2 synthesis inhibitor bindarit targets cells of the neurovascular unit, and suppresses experimental autoimmune encephalomyelitis. J Neuroinflammation. 2012;9:171.
Mercer PF, Williams AE, Scotton CJ, Jose RJ, Sulikowski M, Moffatt JD, et al. Proteinase-activated receptor-1, CCL2, and CCL7 regulate acute neutrophilic lung inflammation. Am J Respir Cell Mol Biol. 2014;50:144–57.
O’Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, et al. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275:13502–9.
Bastarache JA, Sebag SC, Clune JK, Grove BS, Lawson WE, Janz DR, et al. Low levels of tissue factor lead to alveolar haemorrhage, potentiating murine acute lung injury and oxidative stress. Thorax. 2012;67:1032–9.
Shaver CM, Grove BS, Putz ND, Clune JK, Lawson WE, Carnahan RH, et al. Regulation of alveolar procoagulant activity and permeability in direct acute lung injury by lung epithelial tissue factor. Am J Respir Cell Mol Biol. 2015;53:719–27.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81490532 and 81420108001 to H-hS) and the program for Key Site of National Clinical Research Center for Respiratory Disease.
Author contributions
H-hS and AD conceived the project and designed the experiments. JL, YH, M-dW, L-qC, Y-fW, YZ, B-pT, Z-qB, CZ, Y-pW, and L-lH carried out the animal experiments, bone marrow transplantation, and flow cytometric analysis. C-XB and JZ performed critical reading of the manuscript. JL, D-xC, Z-hC, S-mY, and WL analyzed the data. JL, D-xC, and Z-hC prepared the figures and drafted the manuscript. AD and H-hS edited and revised the manuscript. All of the authors participated in data interpretation and approved the final version.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Lou, J., Hu, Y., Wu, Md. et al. Endothelial cell-specific anticoagulation reduces inflammation in a mouse model of acute lung injury. Acta Pharmacol Sin 40, 769–780 (2019). https://doi.org/10.1038/s41401-018-0175-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0175-7
Keywords
This article is cited by
-
Long non-coding RNA OIP5-AS1 aggravates acute lung injury by promoting inflammation and cell apoptosis via regulating the miR-26a-5p/TLR4 axis
BMC Pulmonary Medicine (2021)
-
Lead-induced endothelial cell dysfunction: protective effect of sulfated non-anticoagulant low molecular weight heparin
Toxicology and Environmental Health Sciences (2021)
-
Construction of a 3A system from BioBrick parts for expression of recombinant hirudin variants III in Corynebacterium glutamicum
Applied Microbiology and Biotechnology (2020)
