
Abstract
Inflammatory damage plays an important role in cerebral ischemic pathogenesis and represents a new target for treatment of stroke. Berberine is a natural medicine with multiple beneficial biological activities. In this study, we explored the mechanisms underlying the neuroprotective action of berberine in mice subjected transient middle cerebral artery occlusion (tMCAO). Male mice were administered berberine (25, 50 mg/kg/d, intragastric; i.g.), glycyrrhizin (50 mg/kg/d, intraperitoneal), or berberine (50 mg/kg/d, i.g.) plus glycyrrhizin (50 mg/kg/d, intraperitoneal) for 14 consecutive days before tMCAO. The neurological deficit scores were evaluated at 24 h after tMCAO, and then the mice were killed to obtain the brain samples. We showed that pretreatment with berberine dose-dependently decreased the infarct size, neurological deficits, hispathological changes, brain edema, and inflammatory mediators in serum and ischemic cortical tissue. We revealed that pretreatment with berberine significantly enhanced uptake of 18F-fluorodeoxyglucose of ischemic hemisphere comparing with the vehicle group at 24 h after stroke. Furthermore, pretreatment with berberine dose-dependently suppressed the nuclear-to cytosolic translocation of high-mobility group box1 (HMGB1) protein, the cytosolic-to nuclear translocation of nuclear factor kappa B (NF-κB) and decreased the expression of TLR4 in ischemic cortical tissue. Moreover, co-administration of glycyrrhizin and berberine exerted more potent suppression on the HMGB1/TLR4/NF-κB pathway than berberine or glycyrrhizin administered alone. These results demonstrate that berberine protects the brain from ischemia–reperfusion injury and the mechanism may rely on its anti-inflammatory effects mediated by suppressing the activation of HMGB1/TLR4/NF-κB signaling.
Similar content being viewed by others

Introduction
Ischemic stroke remains one of the most common causes of death and disability throughout the word. Ischemic stroke can be induced by several factors, such as high blood pressure, transient ischemic attack, diabetes, high cholesterol, and atrial fibrillation [1]. As a medical emergency, timely restoration of blood flow and reoxygenation, which reduces the irreversible damage caused by ischemic stroke, is still the only globally approved treatment method [2, 3]. However, the restoration of blood flow following cerebral ischemia initiates an inflammatory cascade that causes secondary neuronal injury [4].
The high-mobility group box 1 (HMGB1) protein is a nuclear nonhistone protein that can be secreted into the extracellular space as a proinflammatory cytokine during injury, infection, and inflammation responses [5]. Elevated serum HMGB1 levels are associated with poor clinical outcome of ischemic stroke and provide accurate prognostic information after ischemic stroke [6]. In the ischemic brain, HMGB1 is released from necrotic and dying neural cells and then binds to myeloid differentiation factor 2, an extracellular adaptor molecule of the Toll-like receptor 4 (TLR4) signalosome, inducing TLR4 signaling and subsequent proinflammatory cytokine synthesis [7]. TLR4, a member of the TLR family, plays a role in immune defense and regulation by recognizing and binding multiple endogenous and exogenous ligands. In cerebral ischemia, TLR4 can modulate the expression of inflammation mediators and activate downstream signaling, including nuclear factor kappa B (NF-κB) [8, 9], which is a transcription factor that can specifically bind promoters and enhancers of many genes and thus participate in multiple cell functions, such as inflammation, immune response, hematogenesis, cell proliferation, and apoptosis [10]. Finally, these signaling pathways and molecules cause neuroinflammatory responses and injury after ischemic stroke [11, 12]. Accordingly, a therapeutic strategy targeting HMGB1/NF-κB signaling may ameliorate cerebral stroke damage by restricting neuroinflammatory processes. Therefore, corresponding anti-inflammatory drug development may be beneficial in treating ischemic stroke-induced brain injury [13, 14].
Berberine, an alkaloid isolated from Chinese herb Coptis chinensis (Huang lian), has been used extensively in China as an antidiabetic, antihyperlipidemic, antidepressant, anti-inflammatory, antibacterial, and anticancer agent [15]. Accumulating evidence demonstrates that berberine possesses potent neuroprotective effects against many neurological disorders, such as anxiety, brain stroke, and Alzheimer’s disease [16, 17]. Based on the involvement of inflammatory stress in the pathogenesis of ischemic brain injury and the proposed activity of berberine as an anti-inflammatory agent, berberine may be used as a new choice for cerebral ischemia pretreatment. With this background, the present study aimed to investigate the neuroprotective role of berberine in ischemic stroke and illustrate the potential mechanism involved in the HMGB1 and NF-κB signaling pathways.
Materials and methods
Animals and groups
Male C57BL/6 J mice (25–30 g) were provided by Vital River Laboratories (Beijing, China). The animals were housed under a 12/12-h dark/light cycle and were allowed free access to food pellets and water. All animal experiments were approved by the Ethics Committee for Animal Experimentation and were carried out in accordance with the National Institutes of Health “Guidelines for Care and Use of the Laboratory Animals”.
Mice were randomly divided into six groups: (1) sham group: mice intragastric (i.g.) administered 0.5% carboxymethylcellulose sodium; (2) vehicle group: mice i.g. administered 0.5% carboxymethylcellulose sodium; (3) berberine-L group: mice i.g. administered berberine (25 mg/kg); berberine-H group: mice i.g. administered berberine (50 mg/kg); Glycyrrhizin (Gly) group: mice interperitonial (i.p.) administered glycyrrhizin (50 mg/kg); berberine-H + Glycyrrhizin group: mice i.g. administered berberine (50 mg/kg), and i.p. administered glycyrrhizin (50 mg/kg). The described treatments were given to all groups once a day for 14 consecutive days before tMCAO.
Animal model of tMCAO
tMCAO surgical procedures were performed on mice by the intraluminal suture method originally described by Longa et al. [18] with some modifications. Mice were anesthetized with chloral hydrate (400 mg/kg, i.p.). In brief, after a midline skin incision, the right common carotid artery, external carotid artery (ECA), and internal carotid artery (ICA) were exposed. A 6-0 suture coated with silicone (0623, Yunshun, Heinan) was introduced into the ECA and advanced from the ICA to the opening of the middle cerebral artery (MCA) until a slight resistance was felt, and the filament was inserted ~ 9–10 mm from the carotid bifurcation, effectively blocking the MCA. The diameter of the tip of the coated suture was between 0.21 and 0.23 mm, and the diameter of the rest of the suture was 0.126 mm. The 6-0 suture was withdrawn after 60 min of occlusion, followed by reperfusion. Animals in the sham-operated group underwent the same surgical procedures except the thread was not inserted. The mice were allowed to recover on a warm blanket with free access to food and water.
Regional CBF measurement
Mice subjected to tMCAO surgeries underwent repeated measurements of regional cerebral blood flow (CBF) before ischemia, 5 min after tMCAO injury and 5 min after reperfusion using the Perfusion speckle image system (FLP12, Gene&I, Beijing).
Neurological score determination
At 24 h after tMCAO, neurological deficit scores were evaluated according to the method described by Longa et al. [18] by an examiner blinded to the experimental groups. A score of 0 indicates no neurologic deficit. A score of 1 indicates that that the mouse fails to extend the left forepaw, 2 reflects the appearance of circling to the left, 3 refers to the failure to turn to the left, and 4 indicates that the mouse cannot walk spontaneously and exhibits a depressed consciousness. The higher the neurological deficit score, the more severe the motor injury.
18F-FDG PET/CT scan
18F-fluorodeoxyglucose (18F-FDG) microPET/CT images of the mouse brain were acquired 24 h after tMCAO surgery. Before scanning, all mice were fasted for 12 h before intravenous (IV) administration of 3.7 MBq 18F-FDG in 0.1 mL saline. Under anesthesia using isoflurane (1%), mice were placed supine near the center of the field of view of the scanner 45 min after IV injection of 18F-FDG. Image acquisition was performed using a small-animal positron emission tomography–computed tomography (PET/CT) scanner (Inveon, Germany SIMENS company) with a microPET acquisition time of 5 min and an energy window of 350–650 keV. MicroCT imaging was performed with a radiographic tube voltage of 80 kV, a current of 500 µA, and an exposure time of 255 ns. The list-mode PET data were post-processed using a three-dimensional ordered subset expectation maximization algorithm (OSEM3D) with CT-based attenuation correction into 256 × 256 × 95 matrices with a voxel size of 1.5 mm3.
Infarct volume evaluation
At 24 h following tMCAO, mice were killed under deep anesthesia for brain tissue collection. The brain was cut into four slices of 2 mm thickness and stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) at 37 °C in a water bath for 20 min to evaluate the infarct volume. Infarct size was determined by digital planimetry of the slices using ImageJ analysis software [19]. Infarct volumes were calculated (in mm3) by multiplying summed section infarct areas by section thickness. The percentage of hemisphere lesion volume was finally calculated. To compensate for the effect of brain edema, the percentage hemisphere lesion volumes were calculated by the following formula: infarct size = (contralateral volume – ipsilateral non-infarct volume)/contralateral volume × 100%.
Measurements of brain edema
Brain tissue was weighed immediately (wet weight) and after dehydration (dry weight) at 100 °C for 24 h. Brain edema was determined using the following formula: brain water content = [(wet weight-dry weight)/wet weight] × 100% [20].
Hematoxylin and eosin staining
After the behavior examination at 24 h, the mice were reanesthetized with chloral hydrate and perfused with normal saline followed by 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS) for fixation. Then, the mouse brain was sliced into 5-µm-thick sections. After hematoxylin and eosin staining, pathological changes in the tissues were analyzed, and all attempts were made to use the same region of cortex as was used for the hematoxylin–eosin evaluation.
ELISA for TNF-α, IL-1β, and IL-6 serum concentrations
Before animals were killed, whole blood was drawn from the orbit of mice and then centrifuged at 4000 r/min for 5 min. The TNF-α, IL-1β, and IL-6 levels in serum were determined using a corresponding ELISA kit (Maijian Biotechnology Center, China).
Real-time reverse transcription-quantitative PCR (RT-qPCR) for mRNA expression
At 24 h after tMCAO, mice were anesthetized, and the ipsilateral cortex tissue was rapidly dissected for RT-qPCR experiments. Total RNA was extracted from the ischemic cortex using an RNA extraction kit and reverse-transcribed to cDNA with a commercially available kit according to the manufacturer’s manual (Abm, China). Real-time PCR was performed using the ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA) with a reaction mixture that consisted of SYBR Green 2 × PCR master Mix (Abm, China), cDNA template, and forward and reverse primers. Thirty-five cycles were conducted as follows: 95 °C for 15 s (denaturation step) and 60 °C for 60 s (to allow extension and amplification of the target sequence). The following primers were used: HMGB1: forward GGATGCTTCTGTC- AACTTCTC, reverse TTCATAACGAGCCTTGTCAG; TLR4: forward GTCAGTGTGATTGTGGTATCC, reverse ACCCAGTCCTCATTCTGACTC; NF-κB p65: forward AGTATTCCTGGCGAGAAAG, reverse CTGTTCCTGGTCCTGTGTAG; TNF-α: forward CAGGCGGTGCCTATGTCTC, reverse CGATCACCCCGAAGTTCAGTA; IL-1β: forward TGACCTGGGCTGTCCTGATG, reverse GGTGCTCATGTCCTCATCCTG; IL-6: forward CCACTTCACAAGTCGGAGGC, reverse GCAAGTGCATCATCGTTGTTCAT; GAPDH: forward GCAGTGGCAAAGTGGAGATTG, reverse TGCAGGATGCATTGCTGACA. Data were analyzed using the ABI 7500 sequence detection system software. The quantity of HMGB1, TLR4, NF-κB, TNF-α, IL-1β, and IL-6 mRNA was normalized to that of GAPDH using the comparative (2-△△Ct) method [21].
Immunohistochemical examination
Brain tissue from each group was fixed in 4% paraformaldehyde for 24 h at 4 °C, dehydrated with alcohol, embedded in paraffin and sliced into 5-µm-thick sections. Sections were incubated with 3% H2O2 and 3% normal goat serum and incubated with the primary antibodies of interest in 0.01 mol/L phosphate-buffered saline overnight. Rabbit monoclonal antibody anti-HMGB1 (1:400, Abcam Biotechnology), mouse monoclonal antibody anti-TLR4 (1:200, Santa Cruz Biotechnology), and rabbit monoclonal antibody anti-NF-κB (1:400, Abcam Biotechnology) were used to detect expression of the corresponding proteins. The secondary antibodies, secondary biotinylated conjugates, and diaminobenzidine were from the Vect ABC kit (zsgb-bio, China). Five visual fields of the ischemic region of the infarct were selected, and slides were viewed and photographed with a × 400 light microscope. The analyses of the results were performed using Image-Pro Plus software.
Western blot analysis
Total protein and cytosolic and nuclear protein in the ischemic cortex tissue were extracted with the corresponding protein extraction kit (Key GEN Biotech, China) following the manufacturer’s protocols, and the protein quantity was assessed with the BCA protein assay kit (Key GEN Biotech, China). Finally, the protein samples were subjected to sulfate polyacrylamide gel electrophoresis (8–12%), separated electrophoretically and transferred to polyvinylidene difluoride membranes (Millipore, USA). After the membranes were blocked for 2 h with 5% milk solution (nonfat dry milk in TBS-Tween 20 (TBST)), they were incubated overnight at 4 °C with rabbit monoclonal antibody anti-HMGB1 (1:2000, Abcam Biotechnology, ab79823), mouse monoclonal antibody anti-TLR4 (1:1000, Santa Cruz Biotechnology, sc-293072), rabbit monoclonal antibody anti-NF-κB (1:1000, Abcam Biotechnology, ab32536), anti-nuclear-β-actin and anti-cytosolic-β-actin (1:1000, Beyotime, China). The membranes were washed three times for 10 min each time, incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (1:5000, zsgb-bio, China) at room temperature for 1 h and washed again three times in TBST buffer. Specific protein signals were visualized on X-ray film using the Super ECL Plus Detection Reagent (Key GEN Biotech, China). The bands were scanned and analyzed quantitatively by densitometry with ImageJ analysis software.
Statistical analyses
All data were expressed as the mean ± SD. Group data in this study were analyzed using SPSS 18.0 software, and a P-value of < 0.05 was considered statistically significant. Statistical analysis was performed by one-way analysis of variance followed by LSD test for intergroup comparisons. An unpaired Student’s t test was used to compare the difference in 18F-FDG uptake between two groups. All statistical figures were performed with Graph Pad Prism Version 5.0 (GraphPad Software, La Jolla, CA).
Results
Regional CBF
CBF occlusion was characterized by a reduction in CBF down to 24.22% ± 4.03% of baseline, and a CBF up to 50.6% ± 5.70% of baseline demonstrated the success of reperfusion (Fig. 1a, b). The results are presented as percent change compared with baseline.

The change in rCBF in the MCAO region. a Representative images of cerebral blood flow for baseline, ischemia, and reperfusion during tMCAO. b Percent change in rCBF compared with baseline during occlusion and reperfusion (n = 5)
Berberine attenuated the neurological deficits
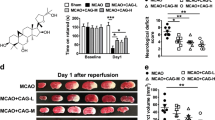
The survival rate was estimated at 24 h after reperfusion. In total, 150 animals were used for experiments, including 21 mice for sham operation. The mortality rate was ~ 30%, and the success rate in compliance with the stroke score standard was ~ 60% in experiments. Neurological deficits were examined and scored on a 5-point scale at 24 h after tMCAO. The scores of the sham group were zero, indicating no neurological deficits, whereas the scores of the vehicle group were significantly higher than those of the sham group. The scores of the berberine-L group and berberine-H group were significantly lower than those of the vehicle group (berberine-L group vs. vehicle group, P < 0.01; berberine-H group vs. vehicle group, P < 0.01, Fig. 2a). There was no significant difference in scores between the berberine-L group and the berberine-H group (P > 0.05, n = 12).

The neuroprotective effect of berberine treatment in ischemic stroke. a Evaluation of the neurological deficit. Pretreatment with berberine significantly decreases neurological deficit scores (n = 12). b Above are the horizontal sections from mice in the vehicle group and berberine-H group. The uptake of 18F-FDG decreases from red to blue. The area indicated by the arrow in the diagram is the area of interest. c Quantitative 18F-FDG uptake in the different groups at 24 h after cerebral reperfusion (n = 3). Data are presented as the mean ± SD. *P < 0.01 versus the vehicle group
Berberine increased 18F-FDG uptake
With the substantial technological advances in small-animal imaging, microPET imaging with 18F-FDG has been successfully used to monitor metabolic changes associated with seizure severity and neuronal activity [22]. To determine whether berberine could improve cerebral ischemic brain injury, 18F-FDG PET/CT scans were performed in mice up to 24 h after tMCAO. As illustrated in Fig. 2b, c, the 18F-FDG uptake ratio in the ischemic hemisphere in the berberine-H group was significantly higher than that in the vehicle group at 24 h after tMCAO (P < 0.05, n = 3). The results indicated that the metabolic condition of the berberine-H group was superior to that of the vehicle group and that berberine treatment alleviated the degree of ischemia in experimental stroke.
Berberine reduced the brain infarction volume
Cerebral infarction was detected by TTC staining and is shown in Fig. 3a. No infarction was observed in the sham group, whereas extensive lesions developed in the ipsilateral cortex in the vehicle group. In the berberine-L group and berberine-H groups, infarct size was significantly reduced compared with that in the vehicle group (P < 0.01, n = 5) (Fig. 3b). There was a significant difference in infarct size between the berberine-L and berberine-H groups (P < 0.05), indicating that the effect of berberine on infarct area was concentration-dependent.

Effects of berberine on infarct volume and brain edema in ischemic stroke (n = 5). a Representative images of TTC-stained brain sections of cerebral ischemia–reperfusion mice with berberine pretreatment, vehicle or sham. b Quantification of the infarct volume in the vehicle or berberine pretreatment groups. c Brain water content. Data are presented as the mean ± SD. #P < 0.01 versus the sham group; *P < 0.01 versus the vehicle group
Berberine decreased the brain water content
The water content of the brain was significantly greater in the vehicle group than in the sham group. The brain water content was significantly reduced by berberine-L and berberine-H treatment in comparison with that in the vehicle group (vehicle group vs. berberine-L group: 82.61% ± 0.40% vs. 81.28% ± 0.35%, P < 0.01; vehicle group vs. berberine-H group: 82.61% ± 0.40% vs. 80.23% ± 0.46%, P < 0.01) (Fig. 3c). There was a significant difference in brain water content between the berberine-L and berberine-H groups (P < 0.05, n = 5).
Berberine alleviated the pathological damage of brain tissue
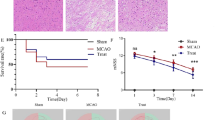
HE staining showed histological changes in the ischemic cortex of vehicle-treated mice compared with that of mice in the sham group. As shown in Fig. 4a–d, the sham group showed normal round intact neurons in the cortex with normal nuclei and integrated tissue structures. HE staining revealed nucleus pyknosis, vacuolation, and shrunken neuronal bodies in the focal point of the infarcted region of the cortex in vehicle-treated mice. After pretreatment with berberine (25 and 50 mg/kg), the focal point of the infarction was significantly shrunken when compared with that in the vehicle control group. Despite partial neuronal loss, many intact cells were still observed. Neurons that had nucleus pyknosis, vacuolation, karyorrhexis, and shrunken cell bodies were defined as ischemically injured cells. The percentages of injured cells in the ischemic cortex region were significantly lower in the berberine-L and berberine-H groups than in the vehicle group (P < 0.01), and there was a significant difference between the berberine-L group and the berberine-H group (P < 0.05, n = 3) (Fig. 4e).

Representative images of ischemic brain tissue with hematoxylin and eosin staining (n = 3). Scale bar, 20 µm. Magnification of the microphotograph, × 400. Arrows indicate injured cells in the ischemic cortex region. a Sham group, b vehicle group, c berberine-L group, d berberine-H group. e Percentage of injured cells in the vehicle, berberine-L, and berberine-H groups in the cerebral cortex. Data are presented as the mean ± SD. *P < 0.01 versus the vehicle group
Berberine decreased serum inflammatory cytokine levels
As shown in Fig. 5a, the serum concentrations of the systemic inflammatory cytokines TNF-α, IL-1β, and IL-6 were significantly increased in the vehicle group relative to those in the sham group at 24 h after the induction of ischemia and reperfusion injury (P < 0.01). Compared with the concentrations in the vehicle group, the serum TNF-α, IL-1β, and IL-6 concentrations were significantly downregulated in the berberine-L and berberine-H groups (P < 0.01, P < 0.05, n = 5). Furthermore, there was a significant difference in serum concentrations between the berberine-L and berberine-H groups (P < 0.05).

Berberine improved brain injury by suppressing the inflammatory response through HMGB1/TLR4/NF-κB inhibition in mice after cerebral ischemia–reperfusion injury (n = 5). a Serum proinflammatory cytokine levels were measured using ELISA kits. b HMGB1, TLR4, and NF-κB mRNA expression in tMCAO-treated mice. c TNF-α, IL-1β, and IL-6 mRNA expression in tMCAO-treated mice. Data are presented as the mean ± SD. #P < 0.01 versus the sham group; ※P < 0.05 versus the vehicle group; *P < 0.01 versus the vehicle group
Berberine downregulated HMGB1, TLR4, NF-κB, and inflammatory cytokine mRNA levels
As shown in Fig. 5b, HMGB1, TLR4, and NF-κB mRNA levels were significantly upregulated in the vehicle group compared with those in the sham group and were suppressed by berberine-L and berberine-H group administration. Finally, the proinflammatory cytokine mRNA levels of TNF-α, IL-1β, and IL-6 were also decreased by berberine-L and berberine-H treatment (P < 0.01, n = 5) (Fig. 5c), and there was a significant difference in mRNA levels between the berberine-L and berberine-H groups (P < 0.05).
Berberine inhibited the HMGB1/ TLR4/ NF-κB signaling pathway
Previous studies have demonstrated that HMGB1, TLR4, and NF-κB are important proinflammatory mediators involved in ischemic brain injury. Therefore, we sought to investigate whether berberine treatment influenced the expression of proinflammatory mediators in the ischemic brain after tMCAO. To evaluate the potential mechanism, the total protein expression of molecules in the HMGB1/TLR4/NF-κB pathway was detected 24 h after tMCAO. As shown in Fig. 6a, the immunohistochemistry results showed that more cells in the cortex of the vehicle group were immunoreactive for HMGB1, TLR4, and NF-κB than in that of the sham group. As expected, the administration of berberine (25 and 50 mg/kg) and glycyrrhizin remarkably decreased the number of cells positive for HMGB1, TLR4, and NF-κB in ischemic brain tissue at 24 h after tMCAO (P < 0.01, n = 3) (Fig. 6b). Notably, the inhibitory effects on HMGB1, TLR4, and NF-κB were weaker in the berberine (25 mg/kg) + glycyrrhizin group than in the berberine (50 mg/kg) + glycyrrhizin group.

a Enlarged images from representative immunohistochemistry photographs of HMGB1, TLR4, and NF-κB in different groups (n = 3). The original images were placed on the top left corner of each photograph. Scale bar, 20 µm. Magnification of the microphotograph, × 400. b Number of cells with positive staining for HMGB1, TLR4, and NF-κB. Data are presented as the mean ± SD. #P < 0.01 versus the sham group; *P < 0.01 versus the vehicle group
Berberine suppressed the protein expression of inflammatory proteins induced by ischemia–reperfusion injury in the brains of mice
We also detected the expression of nuclear and cytoplasmic HMGB1, nuclear and cytoplasmic NF-κB and TLR4 in the ischemic brain tissue. As shown in Fig. 7, the vehicle group exhibited lower nuclear HMGB1 and cytoplasmic NF-κB and higher cytoplasmic HMGB1, nuclear NF-κB, and TLR4 than the sham group. Compared with the vehicle group, all groups given berberine and glycyrrhizin exhibited suppressed expression of cytoplasmic HMGB1, nuclear NF-κB, and TLR4 but upregulated expression of nuclear HMGB1 and cytoplasmic NF-κB (P < 0.01, n = 3 per group). Co-treatment with glycyrrhizin and berberine (50 mg/kg) exerted a greater inhibitory effect on cytoplasmic HMGB1, nuclear NF-κB, and TLR4 expression than treatment with glycyrrhizin or berberine (25 and 50 mg/kg) alone. Finally, there was a significant difference between the berberine-L group and the berberine-H group in the expression of cytoplasmic HMGB1 and TLR4 (P < 0.05).

Effects of berberine on the expression of nHMGB1, cyHMGB1, nNF-κB, cyNF-κB, and TLR4 by western blot analysis at 24 h after cerebral ischemia–reperfusion injury (n = 3). Nuclear and cytosolic protein extracts were prepared and assayed for HMGB1 and NF-κB by western blot analysis. Total protein extracts were prepared for analysis of TLR4 expression. a Representative photographs of western blot for nHMGB1, cyHMGB1, nNF-κB, cyNF-κB, and TLR4. b Ratio of cyHMGB1 to nHMGB1. c Ratio of nNF-κB to cyNF-κB. d Ratio of TLR4 to β-actin. Data are presented as the mean ± SD. #P < 0.01 versus the sham group; *P < 0.01 versus the vehicle group
Discussion
In this study, we found that treatment with berberine improved neurological function and decreased cerebral infarct volume and brain edema after ischemia–reperfusion injury. More importantly, berberine significantly suppressed neuroinflammation via the HMGB1/TLR4/NF-κB signaling pathway. We further explored the possible mechanisms responsible for the anti-inflammatory effect of berberine on cerebral ischemic stroke.
One commonly held view is that inflammation responses contribute to cerebral ischemic stroke injury and secondary damage [23, 24]. HMGB1 regulates transcription to stabilize nucleosomes under normal conditions and is considered a late inflammatory cytokine in many neurodegenerative diseases [25]. A previous study found that the plasma level of HMGB1 is elevated by up to 10-fold in stroke patients and is considered an independent predictor of long-term clinical outcomes in ischemic stroke [26]. HMGB1 translocation and release could activate immunity by inducing proinflammatory cytokine production through its interaction with TLR4, which is one of the main receptors of HMGB1, and the HMGB1-TLR4 pathway contributes to inflammation via multiple mechanisms [27]. Some reports have shown that excessive inflammation resulting from activation of the HMGB1/TLR4 pathway in the brain has been implicated in traumatic brain injury and ischemia–reperfusion injury [12, 28]. Therefore, blocking HMGB1/TLR4 signaling may downregulate the expression of inflammatory pathway genes in cerebral ischemic stroke.
TLR4 has been shown to be one pathway by which HMGB1 can act in a potential feed-forward mechanism, resulting in NF-κB translocation from the cytosol to the nucleus depending on the Myd88 or non-Myd88 pathway, thereby increasing the expression of proinflammatory cytokines [28, 29]. As a signal protein, NF-κB plays a pivotal role in the regulation of immune and inflammatory responses including the expression of p65 and p50 subunits, which are distributed in the cytoplasm. Blocking TLR4 signaling with a specific inhibitor prevented the loss of neurons and suppressed the activation of NF-κB p65 after cardiac arrest and cardiopulmonary resuscitation, suggesting that HMGB1-induced postresuscitation brain damage is probably related to the TLR4-NF-κB pathway [30].
As a transcription factor, NF-κB regulates the expression of many growth factors, immunoreceptors, oxidative stress-related enzymes, and cytokines, such as TNF-α, IL-1β, and IL-6, to initiate inflammatory responses [31]. A previous study reported that the inhibition of NF-κB signaling is neuroprotective because it reduces infarct size, improves neurological deficits, and leads to the inhibition of the expression of inflammatory response genes [32], suggesting that targeting NF-κB activity after acute cerebral infarction may be a potential therapeutic strategy.
Proinflammatory cytokines have long been suggested to contribute to ischemic injury owing to their roles in inducing the infiltration of inflammatory cells and accelerating the pathological process of ischemic stroke. Attenuated levels of proinflammatory cytokines may be associated with poor outcome after stroke [33, 34]. In the present study, HMGB1, TLR4, NF-κB proteins, and inflammatory factors, including TNF-α, IL-1β, and IL-6, were all upregulated in the vehicle group compared with those in the sham group. Berberine interventions obviously reverse these alterations, demonstrating that berberine-inhibiting inflammatory responses were likely to contribute to improving brain function. In addition, the HMGB1 inhibitor glycyrrhizin was also used to further demonstrate that berberine plays a neuroprotective role through the HMGB1/TLR4 signaling pathway.
18F-FDG is an inflammation biomarker for imaging dengue virus infection and assessing treatment response during therapeutic intervention trials, which has been reported in atherosclerosis [35, 36]. A previous study found that imaging with 18F-FDG/PET enabled the detection and quantification of ischemia-induced metabolic deficits and provided a sensitive and reliable means of assessing cerebral ischemic lesions compared with conventional neurological scoring systems [37]. In the present work, we found that 18F-FDG uptake was significantly higher in the frontal cortex in the berberine-H group than in the vehicle group. The correlation between 18F-FDG uptake and inflammation in cerebral stroke suggests a promising role for 18F-FDG PET imaging in the evaluation of neuroinflammation.
Berberine is a well-known natural compound that has various biological functions. Because of its advantages in the treatment and prevention of disease, studies have already discovered that administration of berberine could reduce cerebral ischemia–reperfusion injury by regulating the Akt/GSK signaling pathway and alleviating the inflammatory response [38]. In our study, similar to previous studies, we demonstrated that berberine had a neuroprotective effect on ischemia–reperfusion injury, but we also revealed a novel mechanism of action through the HMGB1/TLR4/NF-κB pathway. Moreover, berberine pretreatment significantly hampered the nuclear-to-cytosolic translocation of HMGB1 and the cytosolic-to-nuclear translocation of NF-κB. Both 25 mg/kg and 50 mg/kg berberine pretreatment relieved nerve defects, reduced brain edema, and decreased infarct size as well as the cortical area affected by ischemic stroke demonstrated by HE staining in vivo. The chosen dose of berberine (25 mg/kg) in the current study was from a previous study, and our study found that a higher dosage (50 mg/kg) yielded more significant neuroprotective effects than the lower dosage. Glycyrrhizin effectively inhibited HMGB1 expression. Moreover, co-administration of glycyrrhizin and berberine exhibited a more potent inhibitory effect on the HMGB1/TLR4/NF-κB pathway than the administration of berberine or glycyrrhizin alone, suggesting that HMGB1 might play a key role in the protective mechanism of berberine on ischemia–reperfusion injury. We found that the different doses of berberine inhibited the HMGB1/TLR4 signaling pathway to different degrees but had the same downstream effect on NF-κB.
In summary, the results of the current study suggested that berberine exhibits significant neuroprotective effects during cerebral ischemia–reperfusion injury, possibly by reducing HMGB1 secretion and nuclear translocation of NF-κB, either directly or indirectly by reducing neuroinflammation. Therefore, we believe that the modulation of interactions among HMGB1, TLR4, and NF-κB by berberine should be considered a useful strategy for pretreatment to protect against ischemia stroke, which features delayed damage after acute and massive neuroinflammation. Of course, further investigations are needed to further elucidate the mechanisms of berberine before its application in patients affected by cerebral ischemic attack.
References
Faralli A, Bigoni M, Mauro A, Rossi F, Carulli D. Noninvasive strategies to promote functional recovery after stroke. Neural Plast. 2013;2013:854597.
Perez-de-Puig I, Miro-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015;129:239–57.
Ma F, Martinez-San Segundo P, Barcelo V, Morancho A, Gabriel-Salazar M, Giralt D, et al. Matrix metalloproteinase-13 participates in neuroprotection and neurorepair after cerebral ischemia in mice. Neurobiol Dis. 2016;91:236–46.
Pundik S, Xu K, Sundararajan S. Reperfusion brain injury focus on cellular bioenergetics. Neurology. 2012;79:S44–S51.
Kang R, Che RC, Zhang QH, Hou W, Wu S, Cao LZ, et al. HMGB1 in health and disease. Mol Asp Med. 2015;40:1–116.
Tsukagawa T, Katsumata R, Fujita M, Yasui K, Akhon C, et al. Elevated serum high-mobility group box-1 protein levels is associated with poor functional outcome in ischemic stroke. J Stroke Cerebrovasc Dis. 2017;26:2404–11.
Yang H, Wang HC, Ju ZL, Ragab AA, Lundback P, Long W, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212:5–14.
Roy A, Srivastava M, Saqib U, Liu DF, Sugathan S, Bishnoi S, et al. Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways. Int Immunopharmacol. 2016;40:79–89.
Zhao H, Chen Z, Xie LJ, Liu GF. Suppression of TLR4/ NF-κB signaling pathway improves cerebral ischemia-reperfusion injury in rats. Mol Neurobiol. 2017;4:1–9.
Van Delft MA, Huitema LF, Tas SW. The contribution of NF-kappaB signalling to immune regulation and tolerance. Eur J Clin Invest. 2015;45:529–39.
Hu J, Liu B, Zhao Q, Jin P, Hua F, Zhang Z, et al. Bone marrow stromal cells inhibits HMGB1-mediated inflammation after stroke in type 2 diabetic rats. Neuroscience. 2016;324:11–9.
Wang J, He GZ, Wang YK, Zhu QK, Chen W, Guo T. TLR4-HMGB1-, MyD88- and TRIF-dependent signaling in mouse intestinal ischemia/reperfusion injury. World J Gastroenterol. 2015;21:8314–25.
LiuT. Y, Zhang TT, Yu HM, Shen HL, Xia WL. Adjudin protects against cerebral ischemia reperfusion injury by inhibition of neuroinflammation and blood-brain barrier disruption. J Neuroinflamm. 2014;11:107.
Burrows F, Haley MJ, Scott E, Coutts G, Lawrence CB, Allan SM, et al. Systemic inflammation affects reperfusion following transient cerebral ischaemia. Exp Neurol. 2016;277:252–60.
Zhang ZJ, Li XH, Li FY, An LJ. Berberine alleviates postperative cognitive dysfunction by suppressing neuroinflammation in aged mice. Int Immunopharmacol. 2016;38:426–33.
Kulkami SK, Dhir A. Berberine: a plant alkaloid with therapeutic potential for central nervous system disorders. Phytother Res. 2010;24:317–24.
Simoes Pires EN, Frozza RL, Hoppe JB, Melo Menezes B, Salbego CG. Berberine was neuroprotective against an in vitro model of brain ischemia: survival and apoptosis pathways involved. Brain Res. 2014;1557:26–33.
Long EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occusion without craniectomy in rats. Stroke. 1989;20:84–91.
Joo SP, Xie W, Xiong X, Xu B, Zhao H. Ischemic postcondition protects against focal cerebral ischemia by inhibiting brain inflammation while attenuating peripheral lymphopenia in mice. Neuroscience. 2013;243:149.
Mdzinarishvili A, Kiewert C, Kumar V, Hillert M, Klein J. Bilobalide prevents ischemia-induced edama formation in vitro and in vivo. Neuroscience. 2007;144:217–22.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)Method. Methods. 2001;25:402–8.
Mirrone MM, Schiffer WK, Siddiq M, Dewey S, Tsirka S. PET imaging of glucose metabolism in a mouse model of temperol lobe epilepsy. Synapse. 2010;59:119–21.
Samson Y, Lapergue B, Hosseini H. Inflammation and ischaemic stroke: current status and future perspectives. Rev Neurol. 2005;161:1177–82.
Zhu Y, Guan YM, Huang HL, Wang QS. Human umbilical cord blood mesenchymal stem cell transplantation suppresses in flammatory responses and neuronal apoptosis during early stage of focal cerebral ischemia in rabbits. Acta Pharmacol Sin. 2014;35:585–91.
Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62.
Huang JM, Hu J, Chen N, Hu ML. Relationship between plasma high-mobility group box-1 levels and clinical outcomes of ischemic stroke. J Crit Care. 2013;28:792–7.
Ojo OO, Ryu MH, Jha A, Unruh H, Halayko AJ. High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1354–66.
Su X, Wang J, Zhao J, Pan H, Mao L. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-κB pathway after traumatic brain injury in the rat. Mediat Inflamm 2011;2011:807142.
Crews FT, Qin L, Sheedy D, Vetrono RP, Zou J. High mobility groupbox1 /Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry. 2013;73:602–12.
Shi X, Li MD, Huang KB, Zhou SM, Hu YF, Pan SY, et al. HMGB1 binding heptamer peptide improves survival and ameliorates brain injury in rats after cardiac arrest and cardiopulmonary resuscitation. Neuroscience. 2017;360:128–38.
Jung HS, Joo JD, Kim DW, Roh M, Jeong JT, Noh SJ, et al. Effect of milrinone on the inflammatory response and NF-κB activation in renal ischemia-reperfusion injury in mice. Korean J Anesthesiol. 2014;66:136–42.
Jiang W, Fu F, Tian J, Zhu H, Hou J. Curculigoside A attenuates experimental cerebral ischemia injury in vitro and vivo. Neuroscience. 2011;192:572–9.
Esenwa CC, Elkind MS. Inflammatory risk factors, biomarkers and associated therapy in ischaemic stroke. Nat Rev Neurol. 2016;12:594–604.
Klimiec E, Kowalska K, Pasinka P, Pera J, Slowik A, Dziedzic T. Reduced release of TNF-α and IP-10 after ex vivo blood stimulation with endotoxin is associated with poor outcome after stroke. Cytokine. 2017;102:51–4.
Chacko AM, Watanabe S, Kalimuddin S, Tham JY, Ong J, Reolo M. 18F-FDG as an inflammation biomarker for imaging dengue virus infection and treatment response. JCI Insight. 2017;93474:1–12.
Montesino Orellana MR, Bentourkia M, Sarrhini O, Fulop T, Paquet N, Lavallee E, et al. Assessment of inflammation in large arteries with 18F-FDG in elderly. Comput Med Imaging Graph. 2013;37:459–65.
Ying KF, Chia JC, Chen KY, Wu KH, Chang KW, Jan ML, et al. Imaging of regional metabolic activity by 18F-FDG/PET in rats with transient cerebral ischemia. Appl Radiat Isot. 2009;67:1743–7.
Zhang XL, Zhang XJ, Wang CH, Li YH, Dong LP, Cui LL, et al. Neuroprotection of early and short-time applying berberine in the acute phase of cerebral ischemia: up-regulated pAkt, pGSK and pCREB, down-regulated NF-κB expression, ameliorated BBB permeability. Brain Res. 2012;1459:61–70.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 81773987).
Author information
Authors and Affiliations
Contributions
Ying-dong Zhang, Chang-qing Yang, and Jian-guo Sun conceived and designed the study. Hai-dan Lu, Yan-li Zhao and Chao Guo performed the experiments. Hong-dong Zhao, Jun-shan Zhou, Feng Wang, and Yun-man Li analyzed the data. Jun-rong Zhu and Wei-rong Fang wrote the paper. Ying-dong Zhang, Chang-qing Yang, and Jian-guo Sun reviewed and edited the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
These are co-first authors: Jun-rong Zhu, Hai-dan Lu, Chao Guo
Rights and permissions
About this article

Cite this article
Zhu, Jr., Lu, Hd., Guo, C. et al. Berberine attenuates ischemia–reperfusion injury through inhibiting HMGB1 release and NF-κB nuclear translocation. Acta Pharmacol Sin 39, 1706–1715 (2018). https://doi.org/10.1038/s41401-018-0160-1
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0160-1
Keywords
This article is cited by
-
Transcriptomics and Metabolomics Unveil the Neuroprotection Mechanism of AnGong NiuHuang (AGNH) Pill Against Ischaemic Stroke Injury
Molecular Neurobiology (2024)
-
Targeting PI3K/Akt in Cerebral Ischemia Reperfusion Injury Alleviation: From Signaling Networks to Targeted Therapy
Molecular Neurobiology (2024)
-
Progesterone improved the behavior of PC12 cells under OGD/R by reducing FABP5 expression and inhibiting TLR4/NF-κB signaling pathway
Journal of Bioenergetics and Biomembranes (2024)
-
Targeting pyroptosis as a preventive and therapeutic approach for stroke
Cell Death Discovery (2023)
-
Matrine exerts its neuroprotective effects by modulating multiple neuronal pathways
Metabolic Brain Disease (2023)
