
Abstract
Astragaloside IV (AsIV) is an active saponin extracted from Astragalus membranaceus, which has shown cardioprotective effects in a number of experimental animals. In this study we investigated the molecular mechanisms by which AsIV attenuated the myocardial ischemia reperfusion (MI/R)-induced injury in vitro and in vivo by focusing on calcium-sensing receptor (CaSR) and extracellular signal-regulated kinase 1/2 (ERK1/2). Rat neonatal cardiac myocytes were subjected to a hypoxia/reoxygenation (H/R) procedure in vitro, which significantly decreased the cell viability, increased lactate dehydrogenase (LDH) release, induced cardiomyocyte apoptosis, and increased [Ca2+]i. H/R also increased the expression of CaSR and decreased ERK1/2 phosphorylation levels in H/R-exposed myocytes. Pretreatment with AsIV (60 μmol/L) significantly improved the cell viability and decreased LDH release, attenuated myocyte apoptosis, decreased [Ca2+]i and CaSR expression, and increased the ERK1/2 phosphorylation levels. The protective effects of AsIV against H/R injury were partially inhibited by co-treatment with a CaSR agonist, gadolinium chloride (GdCl3) or with a specific ERK1/2 inhibitor U0126. For in vivo studies, a rat MI/R model was established. Pre-administration of AsIV (80 mg/kg every day, ig) significantly decreased the myocardium infarct size, creatine kinase-MB (CK-MB) production, serum cardiac troponin (cTnI) levels, and cardiomyocyte apoptosis in the rats with MI/R injury. The therapeutic effects of AsIV were associated with the downregulation of CaSR expression and upregulation of ERK1/2 phosphorylation in myocardial tissues. In summary, astragaloside IV attenuates myocardial I/R injury via inhibition of CaSR/ERK1/2 and the related apoptotic signaling pathways.
Similar content being viewed by others


Introduction
Myocardial ischemia reperfusion (MI/R) injury is a complex pathophysiological process that is associated with various factors and pathways, and the condition remains a major clinical problem in patients who receive thrombolysis, percutaneous coronary intervention, cardiac transplantation, and coronary artery bypass grafting therapies [1, 2]. For ischemic heart disease, reperfusion therapy can preserve myocardial viability and function by reversing myocardial ischemia, but the subsequent ischemia reperfusion injury may reduce the therapeutic benefit, induce cardiomyocyte death, and increase infarct size [3]. Cardiomyocyte apoptosis has been recognized as the main cause of ischemia reperfusion injury, and exploring anti-apoptotic agents represent a novel therapeutic opportunity for ischemia diseases.
Astragaloside IV (AsIV), a purified small molecular saponin, is one of the major and active components of Astragalus membranaceus. Studies on AsIV indicate that it has a wide spectrum of pharmacological effects, including anti-inflammation, antioxidation, regulating energy metabolism, and anti-apoptosis [4,5,6,7]. These beneficial pharmacological and biochemical activities suggest that AsIV has a potential therapeutic role in cardiovascular diseases. According to a previous study, AsIV not only inhibited inward K+ currents to prolong action potential duration (APD) but also suppressed the peak of voltage-dependent Ca2+ channel current to prevent APD over-prolongation, protecting the heart against arrhythmias [8]. Consistent with this study, we previously demonstrated a cardiac protective role of AsIV on cardiac hypertrophy induced by isoprenaline, lipopolysaccharide, and pressure overload [9, 10]. In addition, some reports have indicated that AsIV could attenuate MI/R injury in animal models, and the mechanisms may be related to antioxidative damage, improvement of energy metabolic disturbance, and attenuation of calcium overload [11,12,13]. The present study aimed to further investigate the protective effect of AsIV against MI/R-induced myocardial damage by focusing on possible involvement of the calcium-sensing receptor (CaSR) and its related apoptotic pathways.
CaSR is a member of G protein-coupled receptors (GPCRs) superfamily, which is expressed in hearts and neonatal rat cardiomyocytes [14, 15] and is involved in systemic calcium homeostasis. Moreover, the CaSR in the cardiovascular system plays an important role in cardiac hypertrophy, atherosclerosis, and ischemia/reperfusion injury [16]. Further studies indicated that CaSR contributes to MI/R injury by activating mitogen-activated protein kinases (MAPKs), phospho-protein kinase C δ (p-PKCδ) translocation to the mitochondria, calcium overload, caspase-3, and Fas death receptor pathways [17,18,19]. The extracellular signal-regulated kinase 1/2 (ERK1/2) cascade plays a critical role in cell proliferation and apoptosis and is involved in [Ca2+]i and cell apoptotic gene regulation in MI/R injury [20, 21]. However, the relationship between CaSR and ERK1/2 in MI/R remains unclear. Therefore, the present study was designed to further investigate the mechanism on MI/R by focusing on CaSR and ERK1/2. We tested the hypothesis that AsIV attenuates MI/R injury via inhibiting CaSR and its related apoptotic pathways. We demonstrated for the first time that the CaSR- and ERK1/2-mediated apoptotic pathway contributed to the protective effect of AsIV on MI/R, which further supports the use of AsIV as a promising therapeutic agent for cardiovascular disease.
materials and methods
Materials
AsIV was obtained from Nanjing Jingzhu Biotechnology Company (purity >98%; Nanjing, China). In addition, 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium bromide (MTT), dimethylsulfoxide (DMSO), gadolinium chloride (GdCl3), and U0126 were from Sigma-Aldrich (USA). Nitrotetrazolium blue chloride (NBT) was purchased from Amresco. Antibodies against CaSR, ERK1/2, p-ERK1/2, bcl-2, bax, and cleaved caspase-3 were purchased from Proteintech Biotechnology. In Situ Cell Death Detection, creatine kinase-MB (CK-MB), and cardiac troponin (cTnI) content kits were purchased from Roche Molecular Biochemicals. The kit for determination of lactate dehydrogenase (LDH) content was obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Neonatal cardiac myocyte culture and treatment protocol
The experimental protocols were approved by Committee of Jinzhou Medical University for the Use of Experimental Animals for Research and Teaching. Primary cardiomyocytes were isolated and cultured as previously described [22]. Hypoxia/reoxygenation (H/R) was performed based on the method previously described [23]. Briefly, cardiomyocytes were exposed to anoxia by replacing medium with anoxia buffer (in mM: NaCl 137, KCl 2l, MgCl2 0.49, CaCl2 1.0, HEPES 4, and sodium lactate 20, pH 6.8). Cells were incubated in the hypoxic/ischemic chamber at 37 °C for 2 h in a humidified atmosphere of 5% CO2 and 95% N2. At the onset of reoxygenation, cardiomyocytes were randomly exposed to one of the following treatments: vehicle; AsIV (60 μM); AsIV plus GdCl3 (30 μM), an agonist of CaSR; AsIV plus U0126 (20 μM), a specific inhibitor of ERK1/2; and AsIV plus NPS2143 (1 μM), a specific inhibitor of CaSR. The dosages of these drugs were selected based on previous studies [9, 24,25,26]. Then, cardiomyocytes were incubated again in Dulbecco’s modified Eagle’s medium (DMEM) at 37 °C in 95% air and 5% CO2 (reoxygenation) for 24 h.
Cell viability assay
Briefly, cells were seeded in 96-well plates at a density of 1.0 × 105 cells/mL. After different treatments, MTT solution was added to each well, and cells were incubated for an additional 4 h at 37 °C. Formazan crystals in viable cells were solubilized with 150 μL of DMSO, and the absorbance at 490 nm was measured using a microplate reader (BioTek Instruments, Inc).
Measurement of LDH release in culture supernatant
After reperfusion for 24 h, 200 μL culture supernatant of cardiomyocytes was taken and immediately assayed for LDH release according to the manufacturer’s instruction with a microplate reader.
Mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was determined by JC-1 staining. Cardiomyocytes were cultured in six-well plates (1 × 105/mL cardiomyocytes). After different treatments, cardiomyocytes were incubated 20 min in the presence of 2 μM of JC-1 at 37 °C. Then, the cells were washed twice with JC-1 buffer solution. Thereafter, labeled cells were analyzed and quantified using automatic image analysis software with a fluorescence microscope (Leica, DMI3000B).
Apoptosis assays
Apoptosis was determined using an Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit according to the manufacturer’s instructions. Briefly, cardiomyocytes were seeded in 25-cm2 flasks and incubated with indicated drugs for 24 h. The cells were harvested, washed twice with pre-chilled phosphate-buffered saline (PBS), centrifuged at 2000 r/min for 5 min, and stained with Annexin V-FITC using an assay kit. Data were analyzed using bioconsort software (USA).
Fluo-3/AM measurements of intracellular [Ca2+]i
Cardiomyocytes were cultured in confocal culture dishes at a density of 5 × 104 cells/mL and then exposed to 5 μM Fluo-3/AM for 30 min at 37 °C in the dark. Cardiomyocytes were washed twice with Ca2+-free PBS to remove extracellular Fluo-3/AM and further incubated in DMEM. Changes in [Ca2+]i were measured by the fluorescence intensity induced by Fluo-3 in cardiomyocytes recorded for 5 min using laser confocal scanning microscopy (Leica, TCS SP5II) with excitation and emission at 488 and 530 nm, respectively. Image Pro Plus 6.0 was used for analysis. In this experiment, the loaded cardiomyocytes were divided into control, H/R, GdCl3, H/R+AsIV and GdCl3+AsIV groups.
Animal model of MI/R and experimental protocols
The MI/R model was established as described previously [27]. Briefly, male Sprague Dawley rats (280–300 g) were anesthetized with 20% urethane (0.3 mL/100 g, intraperitoneal). Ischemia was induced by exteriorizing the heart with a left thoracic incision followed by the placement of a slipknot (5-0 silk) around the left anterior descending coronary artery (LAD). After 30 min of ischemia, the slipknot was released, and the animal received 120 min of myocardial reperfusion. Experimental rats were randomly assigned to one of the following three groups: (1) sham group in which the silk was drilled underneath the LAD but the LAD was not ligated; (2) MI/R group in which LAD was ligated for 30 min followed by 120 min of reperfusion; (3) MI/R + 80 mg/kg of AsIV group. The dosage of AsIV was selected according to our previous study [28]. AsIV was dissolved in 0.5% sodium carboxymethylcellulose and was administered daily by gavage 7 days prior to the induction of ischemia until the animals were sacrificed.
Determination of the size of myocardial infarction and TUNEL staining
Myocardial infarction was determined by 0.5% NBT staining. The NBT-stained area (dark blue after staining, non-infarct area) and non-NBT-stained area (white area after staining, infarct area) were analyzed using a digital imaging system and a computer. The TUNEL assay was performed according to the manufacturer’s protocol. The apoptosis index was expressed by the number of positively stained apoptotic cardiomyocytes/total number of cardiomyocytes counted × 100%.
Measurement of CK-MB and cTnI release in serum
Before the heart was moved, blood was collected, and serum was separated by centrifugation for the measurement of CK-MB and cTnT levels according to the manufacturer’s protocol, using a microplate reader (Thermo Scientific, USA).
Western blot analysis
Protein concentrations of the animal heart tissue and cardiomyocytes were determined using the Bradford method. After boiling the samples for 5 min, protein samples were fractionated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were blocked with milk powder at room temperature for 2 h. The membranes were incubated with primary antibodies against CaSR, ERK, p-ERK, bcl-2, bax, and cleaved caspase-3 overnight. On the next day, membranes were incubated with horseradish peroxidase-linked secondary antibodies at room temperature for 1.5 h. Detection was performed with enhanced chemiluminescence reagents. The results were analyzed using Quantity One software (Bio-Rad Laboratories, Hercules).
Statistics
All data are expressed as the mean ± SD. SPSS 17.0 software was used to analyze all the data. Statistical analysis was performed using one-way analysis of variance followed by Bonferroni’s test. P < 0.05 was considered statistically significant.
Results
AsIV reduced cardiomyocyte injury induced by H/R
To study the therapeutic potential of AsIV, cell viability was determined by MTT assay, and cardiomyocytes injury was also assessed by determining the release of LDH in culture medium at the end of reoxygenation. The results revealed that H/R decreased cell viability and increased LDH release. AsIV treatment resulted in a significant increase in cell survival and decrease in LDH release. However, the effects of AsIV were abolished by the pretreatment with the CaSR agonist GdCl3 or the ERK1/2 inhibitor U0126, separately (Fig. 1).

AsIV decreased cardiomyocyte injury induced by H/R. a Cell viability was determined by MTT assay. b The release of LDH in culture medium at the end of reoxygenation was determined. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. Con group, ##P < 0.01 vs. H/R group, &P < 0.05, &&P < 0.01 vs. AsIV group
AsIV attenuated cardiomyocyte apoptosis induced by H/R
In this study, MMP was determined as an index of early myocardial apoptosis by JC-1 staining. In live cells, mitochondria appear red due to the aggregation of accumulated JC-1. In apoptotic and dead cells, the dye remains in its monomeric form, and mitochondria appear green. Therefore, a decreased ratio of red fluorescence to green fluorescence represents MMP dissipation. Annexin V-FITC/propidium iodide double staining was performed to examine the apoptotic rate of cardiomyocytes. Bcl-2, bax, and cleaved caspase-3 expression was also determined as indexes of cardiomyocyte apoptosis. The results showed that H/R induced cardiomyocytes apoptosis. Moreover, treatment with AsIV attenuated cardiomyocyte apoptosis as evidenced by the increased ratio of red fluorescence to green fluorescence and Bcl-2 expression and decreased apoptotic rate and bax and cleaved caspase-3 expression compared with the H/R group. However, the effects of AsIV were partly abolished by treatment with the CaSR agonist GdCl3 or the ERK inhibitor U0126, separately (Figs. 2 and 3).

AsIV attenuated cardiomyocyte apoptosis induced by H/R. a Apoptosis was detected in cardiomyocytes after different treatments by annexin V-FITC/PI double-staining assay. b Bar graphs show that the cell apoptotic rate decreased in cardiomyocytes after treatment with AsIV. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. Con group, ##P < 0.01 vs. H/R group, &P < 0.05, &&P < 0.01 vs. AsIV group

AsIV increased MMP and decreased apoptosis-related proteins induced by H/R. a, b Cells were stained with JC-1 and MMP was assessed by fluorescence microscope. c–f Bcl-2, bax, and caspase-3 expressions were assessed by Western blot. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. Con group, ##P < 0.01 vs. H/R group, &P < 0.05, &&P < 0.01 vs. AsIV group
AsIV inhibited cardiomyocyte [Ca2+]i induced by H/R and GdCl3
To identify the effect of AsIV on [Ca2+]i during H/R, we examined the change of [Ca2+]i upon exposure to H/R or GdCl3 by laser confocal scanning microscopy. The results showed that H/R or GdCl3 increased [Ca2+]i, whereas AsIV pretreatment reduced [Ca2+]i compared with the H/R or GdCl3 group (Fig. 4).

AsIV inhibited cardiomyocytes [Ca2+]i induced by H/R. Changes in the intensity of fluorescence of [Ca2+]i were recorded with a laser scanning confocal microscope under different treatment conditions. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. Con group, ##P < 0.01 vs. H/R group or GdCl3 group
AsIV decreased CaSR protein expression and increased p-ERK1/2 protein expression
As we demonstrated that AsIV inhibited cardiomyocyte apoptosis and enhanced [Ca2+]i induced by H/R, we then investigated the possible mechanisms involved. AsIV significantly decreased the CaSR expression and increased p-ERK1/2 expression in cardiomyocytes compared with the H/R group. Furthermore, the effect of AsIV on CaSR was completely offset by GdCl3. To further investigate the relationship between CaSR and ERK1/2 involvement in the protective effect of AsIV, the CaSR inhibitor NPS2143 was used in this assay. Interestingly, AsIV in combination with the ERK1/2 inhibitor U0126 has no effect on CaSR expression, but AsIV in combination with the CaSR inhibitor NPS2143 further increased p-ERK expression compared with AsIV treatment alone. These results suggested that the CaSR-mediated p-ERK pathway may be involved in the protective effect of AsIV on H/R injury (Fig. 5).

AsIV decreased CaSR expression and increased phosphorylation level of ERK1/2 in cardiomyocytes. a, b CaSR protein expression in cardiomyocytes was determined by western blot. c, d ERK1/2 and p-ERK1/2 expression was determined by western blot. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. Con group, ##P < 0.01 vs. H/R group, &P < 0.05, &&P < 0.01 vs. AsIV group
AsIV reduced myocardial injury in MI/R rats
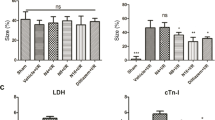
To examine how AsIV influences ischemia/reperfusion-induced myocardial injury, we measured infarct size and the release of CK-MB and cTnI in serum. The area with light white color represented the infarction region, and the area with the dark blue represented normal myocardial tissue. AsIV treatment significantly reduced infarct size and the release of CK-MB and cTnI in serum compared with the MI/R group (Fig. 6).

AsIV decreased myocardial injury in MI/R rats. a Representative photomicrographs of heart sections stained with NBT. b Serum CK-MB activity. c Serum cTnI activity. Data represent the mean ± SD for eight rats in each group. **P < 0.01 vs. Con group, ##P < 0.01 vs. MI/R group
AsIV decreased myocardial cell apoptosis
In the present study, the TUNEL assay was performed using a commercial kit to investigate the anti-apoptotic effect of AsIV. The myocardium in MI/R animals exhibited extensive nuclear changes. In comparison, AsIV treatment resulted in a marked reduction in the number of TUNEL-positive cells. In addition, AsIV treatment also reduced bax and cleaved caspase-3 protein expression and increased bcl-2 protein expression (Fig. 7).

AsIV decreased myocardial cells apoptosis. a Cell apoptosis assayed by TUNEL staining. TUNEL-positive cells were manifested as a marked appearance of dark brown apoptotic cell nuclei. b–d Bax, bcl-2, and cleaved-caspase-3 expressions assayed by western blot. Data represent the mean ± SD of three independent experiments. **P < 0.01 vs. Con group, ##P < 0.01 vs. MI/R group
AsIV reduced CaSR protein expression and increased p-ERK1/2 protein expression in myocardial tissue
To further investigate the role of CaSR and ERK1/2 in the cardioprotective effect of AsIV against MI/R, CaSR and p-ERK1/2 protein expression in myocardial tissue was determined. Consistent with in vitro results, AsIV treatment also reduced CaSR and increased p-ERK1/2 expression in myocardial tissue (Fig. 8).

AsIV decreased CaSR expression and increased ERK1/2 phosphorylation level in myocardial tissue. a–c CaSR and ERK1/2 protein expression in myocardial tissue was determined by Western blot. d–g CaSR and p-ERK1/2 expression in myocardial tissue was determined by immunohistochemistry. Data represent the mean ± SD of eight independent photographs. **P < 0.01 vs. Con group, ##P < 0.01 vs. MI/R group
Discussion
Recently, a number of studies illustrated the benefits of AsIV treatment in MI/R injury. These actions were related to upregulation of hypoxia-inducible factor-1α expression, antioxidation, anti-inflammation, energy metabolism regulation, and upregulation of several KATP channel subunits [29,30,31]. Cardiomyocyte apoptosis is one of the major pathogenic mechanisms underlying MI/R injury, which is initiated shortly after ischemia, accelerated by reperfusion, and partially contributes to overall cardiomyocyte death [32]. Timely and effective inhibition of apoptosis process could prevent the loss of cardiomyocytes, minimize cardiac injury induced by both ischemia and reperfusion, and reduce or even prevent the occurrence of heart failure [33, 34]. In our previous study, we demonstrated that AsIV treatment attenuated MI/R injury via inhibition of Toll-like receptor 4- and nuclear factor-κB-mediated inflammatory responses and subsequent myocardial apoptosis in a rat model [28]. The present study demonstrated that AsIV treatment attenuated myocardial injury, reduced cardiomyocyte apoptosis, decreased [Ca2+]i, inhibited CaSR expression, and increased ERK1/2 phosphorylation levels. These findings not only provided the underlying mechanisms of the cardioprotective effect of AsIV but also further demonstrated the pivotal role of CaSR in MI/R.
CaSR was first cloned in 1993 from bovine parathyroid gland [35] and discovered in rat cardiac tissue by Wang et al. [36]. It was subsequently reported that CaSR is expressed in rat neonatal ventricular cardiomyocytes, regulates systemic calcium homeostasis, and participates in cell proliferation, differentiation, and apoptosis [14]. As a GPCR, CaSR can be activated by extracellular Ca2+ (and Mg2+), polyamines, amino acids, and neomycin, and increase [Ca2+]i via the phospholipase C and inositol 1,4,5 triphosphate pathways. Increased [Ca2+]i by CaSR contributes to cardiac hypertrophy induced by angiotensin II and MI/R, and increases the sensitivity of cardiomyocytes to acute myocardial infarction [24, 37]. In addition, CaSR also arguments mitochondrial Ca2+ via sarcoplasmic reticulum release and induces cardiomyocyte apoptosis through the sarcoplasmic reticulum and mitochondrial apoptotic pathway in heart failure [38]. The translocation of phosphorylated PKCδ to mitochondria was significantly increased when the heart was subject to ischemia reperfusion combined with CaSR agonist GdCl3. In addition, myocardial apoptosis, ERK1/2, protein kinase B (AKT), and glycogen synthase kinase-3β contribute to the effect of CaSR on promoting MI/R [19, 39]. Our present study revealed that myocardial apoptosis was accompanied with increased [Ca2+]i and upregulation of CaSR expression during H/R. These findings were consistent with previous studies. The novel finding of the present study is that AsIV treatment markedly decreased [Ca2+]i and reduced CaSR expression in parallel with reduced cardiomyocyte apoptosis and myocardial injury. Moreover, the downregulation of CaSR and protective effect of AsIV were partly abolished upon pretreatment with the CaSR agonist GdCl3, indicating that downregulation of CaSR contributes to the protective effect of AsIV on MI/R injury.
To gain additional insight into the downstream signaling events following CaSR activation during MI/R, we analyzed the ERK1/2 signaling pathway in vivo and in vitro. As the primary pathway of the MAPK family, the ERK1/2 cascade determines cell proliferation, differentiation, cellular transcriptional activities, and cell survival [40]. The ERK1/2 cascade plays a critical role in myocardial protection against MI/R injury and is involved in ischemic preconditioning and delayed pharmacological preconditioning [41]. Consistent with previous studies, ERK1/2 was inactivated during H/R injury in the present study. Of note, AsIV treatment not only reduced H/R injury but also activated ERK1/2, and the protective effects of AsIV on H/R was partly abolished by the ERK1/2 inhibitor U0126. In addition, AsIV in combination with the CaSR inhibitor NPS2143 could further increase p-ERK expression compared with AsIV treatment alone. These results indicated that activation of ERK1/2 might be involved in the cardioprotective effect of AsIV on MI/R. A previous study demonstrated that CaSR induced hippocampus apoptosis during H/R through ERK1/2 phosphorylation [42]. Enhanced porcine bone marrow mesenchymal stem cell proliferation in response to extracellular calcium is associated with CaSR and ERK1/2 signaling pathway activation [43]. With respect to myocardial tissue, increased CaSR expression promoted cardiomyocyte apoptosis and activated ERK1/2, which contributed to the acceleration of MI/R injury [17]. Treatment with the ERK1/2 inhibitor PD98059 or U0126 caused significant attenuation of ERK1/2 phosphorylation and enhancement of apoptosis in the context of MI/R. However, in the present study, U0126 in combination with AsIV had no effect on CaSR expression compared with AsIV alone, whereas the effect of AsIV on p-ERK1/2 expression was partially offset by the CaSR agonist GdCl3. These differences may due to the complexity and interaction between different signaling pathways, and further studies using signal specific inhibitors or small interfering RNAs are needed.
In summary, we previously demonstrated that AsIV protects myocardium from MI/R injury in vivo. Our present study extended these studies in both rat and cell models and demonstrated for the first time that the protective effect of AsIV against MI/R injury was related with downregulation of CaSR, activation of ERK1/2, and a subsequent reduction in cell apoptosis. These findings might be meaningful to further understand the molecular mechanisms of AsIV cardioprotection.
References
Heusch G. Cardioprotection: chances and challenges of its translation to the clinic. Lancet. 2013;381:166–75.
Levitsky S. Protecting the myocardial cell during coronary revascularization. The William W. L. Glenn Lecture. Circulation. 2006;114:I339–43.
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35.
Zhou X, Sun X, Gong X, Yang Y, Chen C, Shan G, et al. Astragaloside IV from Astragalus membranaceus ameliorates renal interstitial fibrosis by inhibiting inflammation via TLR4/NF-small ka, CyrillicB in vivo and in vitro. Int Immunopharmacol. 2017;42:18–24.
Guo H, Cao A, Chu S, Wang Y, Zang Y, Mao X, et al. Astragaloside IV attenuates podocyte apoptosis mediated by endoplasmic reticulum stress through upregulating sarco/endoplasmic reticulum Ca2+-ATPase 2 expression in diabetic nephropathy. Front Pharmacol. 2016;7:500.
Jiang XG, Sun K, Liu YY, Yan L, Wang MX, Fan JY, et al. Astragaloside IV ameliorates 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis implicating regulation of energy metabolism. Sci Rep. 2017;7:41832.
Xu C, Tang F, Lu M, Yang J, Han R, Mei M, et al. Pretreatment with astragaloside IV protects human umbilical vein endothelial cells from hydrogen peroxide induced oxidative stress and cell dysfunction via inhibiting eNOS uncoupling and NADPH oxidase - ROS - NF-kappaB pathway. Can J Physiol Pharmacol. 2016;94:1–9.
Zhao M, Zhao J, He G, Sun X, Huang X, Hao L. Effects of astragaloside IV on action potentials and ionic currents in guinea-pig ventricular myocytes. Biol Pharm Bull. 2013;36:515–21.
Lu M, Wang H, Wang J, Zhang J, Yang J, Liang L, et al. Astragaloside IV protects against cardiac hypertrophy via inhibiting the Ca2+/CaN signaling pathway. Planta Med. 2014;80:63–9.
Zhang S, Tang F, Yang Y, Lu M, Luan A, Zhang J, et al. Astragaloside IV protects against isoproterenol-induced cardiac hypertrophy by regulating NF-kappaB/PGC-1alpha signaling mediated energy biosynthesis. PLoS One. 2015;10:e0118759.
Zhang WD, Chen H, Zhang C, Liu RH, Li HL, Chen HZ. Astragaloside IV from Astragalus membranaceus shows cardioprotection during myocardial ischemia in vivo and in vitro. Planta Med. 2006;72:4–8.
Tu L, Pan CS, Wei XH, Yan L, Liu YY, Fan JY, et al. Astragaloside IV protects heart from ischemia and reperfusion injury via energy regulation mechanisms. Microcirculation. 2013;20:736–47.
Xu XL, Chen XJ, Ji H, Li P, Bian YY, Yang D, et al. Astragaloside IV improved intracellular calcium handling in hypoxia-reoxygenated cardiomyocytes via the sarcoplasmic reticulum Ca-ATPase. Pharmacology. 2008;81:325–32.
Tfelt-Hansen J, Hansen JL, Smajilovic S, Terwilliger EF, Haunso S, Sheikh SP. Calcium receptor is functionally expressed in rat neonatal ventricular cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;290:H1165–71.
Sun YH, Liu MN, Li H, Shi S, Zhao YJ, Wang R, et al. Calcium-sensing receptor induces rat neonatal ventricular cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;350:942–8.
Li HX, Kong FJ, Bai SZ, He W, Xing WJ, Xi YH, et al. Involvement of calcium-sensing receptor in oxLDL-induced MMP-2 production in vascular smooth muscle cells via PI3K/Akt pathway. Mol Cell Biochem. 2012;362:115–22.
Jiang CM, Han LP, Li HZ, Qu YB, Zhang ZR, Wang R, et al. Calcium-sensing receptors induce apoptosis in cultured neonatal rat ventricular cardiomyocytes during simulated ischemia/reperfusion. Cell Biol Int. 2008;32:792–800.
Lu F, Tian Z, Zhang W, Zhao Y, Bai S, Ren H, et al. Calcium-sensing receptors induce apoptosis in rat cardiomyocytes via the endo(sarco)plasmic reticulum pathway during hypoxia/reoxygenation. Basic Clin Pharmacol Toxicol. 2010;106:396–405.
Zheng H, Liu J, Liu C, Lu F, Zhao Y, Jin Z, et al. Calcium-sensing receptor activating phosphorylation of PKCdelta translocation on mitochondria to induce cardiomyocyte apoptosis during ischemia/reperfusion. Mol Cell Biochem. 2011;358:335–43.
Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3beta pathway after ischemia/reperfusion. PLoS One. 2015;10:e0135988.
Filippone SM, Samidurai A, Roh SK, Cain CK, He J, Salloum FN, et al. Reperfusion therapy with rapamycin attenuates myocardial infarction through activation of AKT and ERK. Oxid Med Cell Longev. 2017;2017:4619720.
Wu G, Wang H, Yang J, Yang Y, Liu C, Jing L, et al. Kappa-Opioid receptor stimulation inhibits augmentation of Ca2+ transient and hypertrophy induced by isoprenaline in neonatal rat ventricular myocytes—role of CaMKIIdelta(B). Eur J Pharmacol. 2008;595:52–7.
Yin Y, Guan Y, Duan J, Wei G, Zhu Y, Quan W, et al. Cardioprotective effect of Danshensu against myocardial ischemia/reperfusion injury and inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2 phosphorylation. Eur J Pharmacol. 2013;699:219–26.
Guo J, Li HZ, Zhang WH, Wang LC, Wang LN, Zhang L, et al. Increased expression of calcium-sensing receptors induced by ox-LDL amplifies apoptosis of cardiomyocytes during simulated ischaemia-reperfusion. Clin Exp Pharmacol Physiol. 2010;37:e128–35.
Greenberg HZE, Jahan KS, Shi J, Vanessa Ho WS, Albert AP. The calcilytics Calhex-231 and NPS 2143 and the calcimimetic Calindol reduce vascular reactivity via inhibition of voltage-gated Ca2+ channels. Eur J Pharmacol. 2016;791:659–68.
Zhang Y, Chen G, Zhong S, Zheng F, Gao F, Chen Y, et al. N-n-butyl haloperidol iodide ameliorates cardiomyocytes hypoxia/reoxygenation injury by extracellular calcium-dependent and -independent mechanisms. Oxid Med Cell Longev. 2013;2013:912310.
Hadi NR, Al-Amran F, Yousif M, Zamil ST. Antiapoptotic effect of simvastatin ameliorates myocardial ischemia/reperfusion injury. ISRN Pharmacol. 2013;2013:815094.
Lu M, Tang F, Zhang J, Luan A, Mei M, Xu C, et al. Astragaloside IV attenuates injury caused by myocardial ischemia/reperfusion in rats via regulation of toll-like receptor 4/nuclear factor-kappaB signaling pathway. Phytother Res. 2015;29:599–606.
Si J, Wang N, Wang H, Xie J, Yang J, Yi H, et al. HIF-1alpha signaling activation by post-ischemia treatment with astragaloside IV attenuates myocardial ischemia-reperfusion injury. PLoS One. 2014;9:e107832.
Han XH, Liu P, Zhang YY, Zhang N, Chen FR, Cai JF. Astragaloside IV regulates expression of ATP-sensitive potassium channel subunits after ischemia-reperfusion in rat ventricular cardiomyocytes. J Tradit Chin Med. 2011;31:321–6.
Li ZP, Cao Q. Effects of astragaloside IV on myocardial calcium transport and cardiac function in ischemic rats. Acta Pharmacol Sin. 2002;23:898–904.
Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–56.
Hu W, Zhang P, Gu J, Yu Q, Zhang D. NEDD4-1 protects against ischaemia/reperfusion-induced cardiomyocyte apoptosis via the PI3K/Akt pathway. Apoptosis. 2017;22:437–48.
Zhang SW, Liu Y, Wang F, Qiang J, Liu P, Zhang J, et al. Ilexsaponin A attenuates ischemia-reperfusion-induced myocardial injury through anti-apoptotic pathway. PLoS One. 2017;12:e0170984.
Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. 1993;366:575–80.
Wang R, Xu C, Zhao W, Zhang J, Cao K, Yang B, et al. Calcium and polyamine regulated calcium-sensing receptors in cardiac tissues. Eur J Biochem. 2003;270:2680–8.
Guo J, Li HZ, Wang LC, Zhang WH, Li GW, Xing WJ, et al. Increased expression of calcium-sensing receptors in atherosclerosis confers hypersensitivity to acute myocardial infarction in rats. Mol Cell Biochem. 2012;366:345–54.
Lu FH, Fu SB, Leng X, Zhang X, Dong S, Zhao YJ, et al. Role of the calcium-sensing receptor in cardiomyocyte apoptosis via the sarcoplasmic reticulum and mitochondrial death pathway in cardiac hypertrophy and heart failure. Cell Physiol Biochem. 2013;31:728–43.
Sun J, Murphy E. Calcium-sensing receptor: a sensor and mediator of ischemic preconditioning in the heart. Am J Physiol Heart Circ Physiol. 2010;299:H1309–17.
Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–47.
Strohm C, Barancik T, Bruhl ML, Kilian SA, Schaper W. Inhibition of the ER-kinase cascade by PD98059 and UO126 counteracts ischemic preconditioning in pig myocardium. J Cardiovasc Pharmacol. 2000;36:218–29.
Wang P, Wang L, Wang S, Li S, Li Y, Zhang L. Effects of calcium-sensing receptors on apoptosis in rat hippocampus during hypoxia/reoxygenation through the ERK1/2 pathway. Int J Clin Exp Pathol. 2015;8:10808–15.
Ye J, Ai W, Zhang F, Zhu X, Shu G, Wang L, et al. Enhanced proliferation of porcine bone marrow mesenchymal stem cells induced by extracellular calcium is associated with the activation of the calcium-sensing receptor and ERK signaling pathway. Stem Cells Int. 2016;2016:6570671.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 81703739), the Natural Science Foundation of Liaoning Province (No 201602284), and the Innovative Training Program for College Students of Liaoning Province (No 201610160000040).
Author contributions
B.Y. and M.-i.L. designed the research. B.Y., X.-w.H. and M.-i.L. performed the research. B.Y. analyzed the data. M.-i.L. wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Yin, B., Hou, Xw. & Lu, Ml. Astragaloside IV attenuates myocardial ischemia/reperfusion injury in rats via inhibition of calcium-sensing receptor-mediated apoptotic signaling pathways. Acta Pharmacol Sin 40, 599–607 (2019). https://doi.org/10.1038/s41401-018-0082-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0082-y
Keywords
This article is cited by
-
Ischemia-reperfusion injury: molecular mechanisms and therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
Solid-phase extraction with the functionalization of calcium-sensing receptors onto magnetic microspheres as an affinity probe can capture ligands selectively from herbal extract
Microchimica Acta (2024)
-
Is telomerase a hidden player? Therapeutic potential of natural telomerase activators against age-related diseases
Phytochemistry Reviews (2023)
-
Astragaloside IV for Heart Failure: Preclinical Evidence and Possible Mechanisms, A Systematic Review and Meta-Analysis
Chinese Journal of Integrative Medicine (2023)
-
Astragaloside IV inhibits adriamycin-induced cardiac ferroptosis by enhancing Nrf2 signaling
Molecular and Cellular Biochemistry (2021)
