
Abstract
The chemokine receptor CCR5 is an important anti-HIV (human immunodeficiency virus) drug target owning to its pivotal role in HIV-1 viral entry as a co-receptor. Here, we present a 2.9 Å resolution crystal structure of CCR5 bound to PF-232798, a second-generation oral CCR5 antagonist currently in phase II clinical trials. PF-232798 and the marketed HIV drug maraviroc share a similar tropane scaffold with different amino (N)- and carboxyl (C)- substituents. Comparison of the CCR5–PF-232798 structure with the previously determined structure of CCR5 in complex with maraviroc reveals different binding modes of the two allosteric antagonists and subsequent conformational changes of the receptor. Our results not only offer insights into the phenomenon that PF-232798 has higher affinity and alternative resistance profile to maraviroc, but also will facilitate the design of new anti-HIV drugs.
Similar content being viewed by others

Introduction
Since 1981, when the first case appeared in the USA, acquired immune deficiency syndrome (AIDS) has become a worldwide epidemic disease that cannot be completely cured [1]. The causative agent, HIV-1, is a retrovirus that can infect human immune cells and then destroy the immune system [2]. During the process of HIV-1 entry, the HIV-1 envelope glycoprotein gp120 first binds to the host-cell receptor CD4, followed by the exposure of the third variable loop (V3 loop) of gp120. The V3 loop then interacts with either the CXCR4 or CCR5 coreceptor, and subsequently, HIV-1 glycoprotein gp41 refolds and triggers membrane fusion [3,4,5,6]. With the growing understanding of the HIV virus and the urgent need for effective therapy, a number of drugs that target the virus have been approved in the past years, including the antiretroviral therapy (ART) and highly active antiretroviral therapy (HAART) series of drugs [7]. However, genetic plasticity and the replication rate of the HIV virus have resulted in rapid drug resistance [8]. Coreceptor blockade therapy inhibits virus entry and replication directly and can slow the speed of epitope mutation, making CCR5 an attractive drug target. Through high-throughput compound screening, maraviroc was developed by Pfizer and then approved to be the first CCR5 antagonist as an anti-HIV drug [9,10,11,12,13]. Following this milestone, Pfizer continued to design new compounds based on the structure of maraviroc and discovered a second-generation CCR5 antagonist, N-{(1 S)-1-(3-fluorophenyl)-3-[(3-endo)-3-(5-isobutyryl-2-methyl-4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl]propyl}acetamide (PF-232798), in 2011 [14]. The new candidate went through in Phase II clinical trials for the treatment of HIV-1 infection [14]. PF-232798 shows increased binding affinity and improved oral absorption compared to maraviroc. In addition, it retains activity against a laboratory generated maraviroc-resistant HIV-1 strain, indicating an alternative drug resistance profile [14]. Here, we describe the crystal structure of CCR5 bound to PF-232798 at a resolution of 2.9 Å. The CCR5–PF-232798 structure combined with the previously determined CCR5-maraviroc structure [15] provides insights into the molecular mechanism of allosteric modulator binding modes at CCR5 and will enable lead compound design.
Materials and methods
Plasmid design and protein expression
Codon-optimized human wild type CCR5 DNA was synthesized by Genewiz (Shanghai, China) and cloned into a modified pFastBac1 vector (Invitrogen, Carlsbad, CA, USA) for insect cell expression, using the Asc I and Fse I restriction sites, with a hemagglutinin (HA) signal sequence (MKTIIALSYIFCLVFA) at the N terminus and a FLAG tag (DYKDDDK) followed by a 10 × His tag at the C terminus. A PreScission protease site (LEVLFQGP) was inserted into the vector for removal of the FLAG tag and the 10 × His tag. Residues Met1-Glu54 of rubredoxin [16] were fused into intracellular loop 3 between Arg223 and Glu227 to replace residues Cys224-Asn226. Phe320-Leu352 at the C terminus were truncated to improve protein thermostability. To increase protein homogeneity and stability, four rationally designed mutations (Cys581.60Tyr, Gly1634.60Asn, Ala2336.33Asp, and Lys303Glu) (superscripts indicate Ballesteros–Weinstein nomenclature numbers [17]) were introduced, with the mutation Ala2336.33Asp in particular being able to stabilize the protein inactive state selectively. Spodoptera frugiperda 9 (Sf9) cells at a cell density of 2 × 106–3 × 106 cells ml−1 were infected with baculovirus that were obtained by the Bac-to-Bac Baculovirus Expression System (Invitrogen) at a multiplicity of infection (M.O.I.) of 5. After 48 h infection at 27 °C, cells were harvested by centrifugation and stored at −80 °C until use.
Purification of the CCR5–PF-232798 complex
Frozen cell pellets were thawed in a hypotonic lysis buffer containing 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl and EDTA-free protease inhibitor cocktail (Roche, Basel, Switzerland) (1 tablet per 100 mL buffer). The cell membranes were disrupted and extensively washed by repeated dounce homogenization. Centrifugations were performed in the same hypotonic buffer once and then in a high salt buffer supplemented with 1 M NaCl twice and then again in the hypotonic buffer once to remove the high salt buffer. The final purified membranes were resuspended in the hypotonic buffer supplemented with 30% (v/v) glycerol and flash frozen in liquid nitrogen before storage at −80 °C.
The frozen purified membranes were resuspended by thawing on ice in the presence of 200 μM PF-232798, 1 mg/mL iodoacetamide, and EDTA-free protease inhibitor cocktail (Roche). PF-232798 was synthesized as previously described [14]. After incubation at 4 °C for 1 h, the recombinant CCR5 protein was extracted from the membranes in buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 0.5% (w/v) n-dodecyl-D-maltopyranoside (DDM, Anatrace, Maumee, OH, USA), 0.1% (w/v) cholesteryl hemisuccinate (CHS, Sigma, Duesseldorf, Germany), and 100 μM PF-232798 for 3 h at 4 °C before centrifuging at 40,000 × g for 30 min. The supernatant was incubated in TALON IMAC resin (Clontech, Dalian, China) with 5 mM imidazole overnight. The resin was then washed with 10 column volumes of 25 mM HEPES, pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 30 mM imidazole and 100 μM PF-232798 followed by 10 column volumes of 25 mM HEPES, pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 5 mM ATP (Sigma), 10 mM MgCl2, and 100 μM PF-232798. Excess ATP was washed off with five column volumes of 25 mM HEPES, pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS and 100 μM PF-232798. Finally, the target protein CCR5 was eluted with five column volumes of 25 mM HEPES, pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 300 mM imidazole, and 300 μM PF-232798. The eluted sample was concentrated with a 100 kDa molecular weight cut-off Vivaspin concentrator (Sartorius Stedim Biotech, Gottingen, Germany), imidazole was removed by a PD MiniTrap G-25 column (GE Healthcare, Fairfield, CT, USA), and the protein sample was then supplemented with PF-232798 to a final concentration of 1 mM. The protein was subsequently incubated overnight with His-tagged PreScission protease (custom-made) to remove the C-terminal FLAG tag and 10 × His tag. PreScission protease and the cleaved C-terminal tag fragment were removed from the sample by passing the sample over Ni-NTA superflow resin (Qiagen, Hilden, Germany). The purified, untagged protein was concentrated to 40–50 mg/mL for crystallization. The protein purity and monodispersity were tested by SDS-PAGE and analytical size-exclusion chromatography (aSEC).
Crystallization
Crystallization of the purified protein sample of CCR5 in complex with PF-232798 was performed by the lipidic cubic phase (LCP) crystallization method [18]. The 40 mg/mL protein was mixed with lipid (monoolein and cholesterol 10:1 by mass) in a ratio of 2:3 (protein: lipid, w/w) with a syringe lipid mixer. When the mixture became transparent, it was dispensed onto glass sandwich plates with 45 nL protein sample overlaid with 800 nL precipitant solution per drop using a Mosquito LCP robot (TTP Labtech, Cambridge, UK). The plates were incubated at 20 °C and autoimaged with a Rock imager (Formulatrix, Bedford, MA, USA). The crystals appeared 3 days after crystallization was initiated and reached a maximum size of 100 μm ~2 weeks later. Optimal crystals for X-ray diffraction were obtained in 100 mM HEPES, pH 7.0, 32–38% (v/v) PEG400, 100–200 mM KNO3, and 1 mM PF-232798. Crystals were harvested using 100 μm micromounts (MiTeGen, Ithaca, NY, USA) and flash frozen in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data were collected on the 23ID-D beamline (GM/CA CAT) of the Advanced Photon Source using a 10 μm collimated minibeam at the Argonne National Laboratory. The crystals were exposed for 1 s with 1° oscillation per frame and most crystals diffracted to 3.5–2.5 Å solution. Data from the 5 best-diffracted CCR5–PF-232798 crystals were integrated, scaled and merged at 2.9 Å resolution using HKL2000 [19]. Initial phase information was obtained by molecular replacement (MR) using CCR5-maraviroc (PDB ID:4MBS) [15] and rubredoxin (PDB ID:1IRO) [20] as search models independently with the program Phase [21]. A CCR5–PF-232798 molecule was estimated to exist in an asymmetric unit in the correct solution. All refinements were performed with Refmac5 [22] and Buster [23] followed by manual examination and rebuilding of the refined coordinates in the program Coot [24] using both |2Fo|−|Fc| and |Fo|−|Fc| maps.
Protein stability assay
Protein thermostability was tested using a microscale fluorescent thermal stability assay using the thiol-specific fluorochrome, N-[4-(7-diethylamino-4-methyl-3-coumarinyl) phenyl] maleimide (CPM). The fluorochrome reacted with the native cysteines embedded in the protein interior as a sensor for the overall integrity of the folded state. The stock CPM orange dye (Invitrogen) at 4 mg/mL was diluted (1:20) with buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 10% Glycerol, 0.05% (w/v) DDM and 0.01% (w/v) CHS. The protein to be tested (~ 5 μg) was diluted in the same buffer to a final volume of 120 μL. The diluted dye (1 μL) was added and thoroughly mixed with the protein. The reaction mixture was incubated at room temperature for 20 min, subsequently transferred to a sub-micro quartz fluorometer cuvette (Starna Cells, Inc., Glasgow, UK) and heated in a controlled environment with a ramp rate of 1 °C/min over a temperature range from 10 to 90 °C using a Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). The excitation wavelength was 387 nm, and the emission wavelength was 463 nm.
Results
Ligand screening
More than 30 pharmaceutical ligands of CCR5 were screened with the engineered CCR5 construct described above. Almost all of the compounds improved the thermostability of the CCR5 protein, and the CCR5-ligand complexes had higher melting temperatures (Tm) than CCR5 alone (APO), as shown in the CPM-ramping assay (Fig. 1a). Although most of the ligands were used to set up cocrystallization trials with CCR5, it was hard to obtain high-quality protein-compound crystals except with PF-232798.

Ligand screening and protein quality and crystals of CCR5. a CPM assay of CCR5 in complex with different ligands. b aSEC of the CCR5–PF-232798 crystallization sample. c Crystals of the CCR5–PF-232798 complex
Protein homogeneity and crystallization assay
The large-scale purified CCR5–PF-232798 complex sample exhibited a single peak by aSEC, indicating that the purified protein was highly homogeneous (Fig. 1b). The stick-like crystals reached a maximum size of ~100 × 15 × 14 μm3, and the crystal structure of CCR5 bound to its allosteric antagonist PF-232798 was determined at 2.9 Å resolution (Fig. 1c). The crystals belong to space group P212121 with unit-cell parameters a = 63.5 Å, b = 66.1 Å, c = 134.4 Å, and α = β = γ = 90°. The asymmetric unit contains one binary complex. The Rwork and Rfree are 0.244 and 0.283, respectively (Table 1).
Overall structure of CCR5–PF-232798
The CCR5–PF-232798 structure shares a canonical seven-transmembrane architecture with other solved class A G protein-coupled receptors (GPCRs) (Fig. 2). The overall structure of CCR5 bound to PF-232798 resembles the maraviroc-bound CCR5 structure with a Cα root-mean-square deviation (r.m.s.d.) of 0.708 Å within the helical bundle [15]. The second extracellular loop (ECL2) forms a β-hairpin structure, and the C-terminal amphiphilic helix VIII (Glu302-Phe312) runs parallel to the membrane plane. The disulfide bond formed between Cys1013.25 and Cys178ECL2, which is common in most solved GPCR structures, helps connect ECL2 to helix III and thus stabilizes the conformation of the second extracellular loop. An inactive-state salt bridge interaction, also called an “ionic lock”, between Arg1263.50 and Asp2336.33 and a hydrogen bond between Asp762.50 and Asn2937.49 lock the receptor in an inactive conformation.

Overview of the CCR5–PF-232798 complex structure. a Side view of the CCR5–PF-232798 complex structure. The receptor is shown in cartoon representation in pink. The ligand PF-232798 is shown in stick representation with green carbons. b Top view of the extracellular side of CCR5–PF-232798. The yellow sticks represent disulfide bonds. c Bottom view of the intracellular side of CCR5–PF-232798. Side chains of Arg1263.50, Asp2336.33, Asp762.50, and Gln2937.49 are shown as sticks with pink carbons. Salt bridges and hydrogen bonds are shown as dashed lines colored black and orange, respectively
Comparison of binding modes between PF-232798 and maraviroc
PF-232798 is a second-generation CCR5 antagonist with a higher affinity (0.5 nM) than maraviroc (3 nM). The two compounds maintain the same tropane scaffold, which is necessary for anti-HIV activity (Fig. 3b–e) [14]. The nitrogen of the tropane forms a salt bridge with Glu2837.39 to bury the two drug molecules deep at the bottom of their respective binding pockets. In addition, the conserved carboxamide nitrogen sitting three carbon chains away from the tropane nitrogen [9] forms a hydrogen bond with Tyr2516.51 in both the CCR5-maraviroc and CCR5–PF-232798 structures.

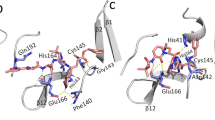
Binding pocket comparison of CCR5-maraviroc (PDG ID: 4MBS) and CCR5–PF-232798. a Alignment of the two structures. CCR5-maraviroc and CCR5–PF-232798 are shown as cartoons colored blue and magenta, respectively. Maraviroc and PF-232798 are shown as sticks with yellow and green carbons, respectively. b Schematic representation of interactions in CCR5-maraviroc. Amino acids interacting only with maraviroc are colored blue. Amino acids interacting with both ligands are colored blue and pink. c Top view of the binding pocket of maraviroc. The backbone of CCR5 is represented as a gray cartoon. Residues involved in maraviroc binding are shown as sticks in blue. d Schematic representation of interactions in CCR5–PF-232798. Amino acids interacting only with PF-232798 are colored pink. Amino acids interacting with both ligands are colored blue and pink. e Top view of the binding pocket of PF-232798. Residues involved in PF-232798 binding are shown as sticks in magenta. Hydrogen bonds, salt bridges and π-π interactions are represented by dashed lines in different colors
The phenyl group between the tropane and carboxamide binds in a deep hydrophobic pocket formed by Phe1123.36, Phe1093.33, Tyr1083.32, Trp2486.48 and Tyr2516.51 in both structures. It is noteworthy that the meta-fluoro modification on the phenyl group in PF-232798 may strengthen the interactions with residues Phe1123.36 and Phe1093.33 in this hydrophobic pocket (Fig. 3d, e).
Consistent with the chemical modifications, the different N- and C- substituents of the two antagonists occupy different subpockets located in the allosteric binding site formed by residues on TM 5/TM 6 and TM 1/TM 2 (Fig. 3a, c, e). The N-fluorine in the cyclohexane ring of maraviroc forms two hydrogen bonds with Thr2596.59 and Thr1955.39 (subpocket 1, Fig. 3b, c) [15]. These hydrophilic interactions are missing in the CCR5–PF-232798 structure, as PF-232798 does not have the N-fluoro-cyclohexane group. On the other hand, the C- substituents (imidazopiperidine and adjacent acetamide groups) in PF-232798 form additional hydrophobic interactions with Tyr892.63, Ala902.64, Ala291.30, and Leu331.34 (subpocket 2, Fig. 3d, e). The important role of the acetamide group was proved by previous structure-activity relationship (SAR) data showing that introduction of an acetyl substituent increased the ability of PF-232798 to inhibit HIV virus attachment to CCR5 by 22-fold [14]. In addition, the π-π stacking interaction between Trp862.60 and the nitro heterocycles in PF-232798 may be stronger than that with maraviroc, as the Trp86Ala mutation decreased the binding affinity of PF-232798 and maraviroc by 629- and 29.8-fold, respectively and Trp86 was predicted to contribute differently to the binding site energy of maraviroc (−2.0 kcal/mol) and PF-232798 (−4.3 kcal/mol) [25]. Moreover, the distance between the amine moiety of imidazopiperidine in PF-232798 and Tyr371.39 (2.7 Å) is shorter than that between the triazole group in maraviroc and Tyr371.39 (3.1 Å), indicating a stronger hydrogen bonding interaction. Those additional or stronger interactions formed between PF-232798 and CCR5 may contribute to the higher affinity of PF-232798.
Structure variation between CCR5–PF-232798 and CCR5–maraviroc
Although the structures of CCR5 bound to each of these two compounds are very similar, the most striking conformational changes in the PF-232798-bound CCR5 structure are movements of helical bundles on the extracellular side, most likely caused by the different N-/C- substituents on the tropane core. The imidazopiperidine in PF-232798 bridges the hydrogen bond network of transmembrane helices 1, 2, and 7 by forming a hydrophobic interaction with Tyr892.63 and a hydrogen bond with Tyr371.39, inducing clockwise movements of TM 2 (~1 Å) and TM 1 (~3 Å) from the central axis of the receptor (Fig. 4). The hydrogen bond between Tyr371.39 and Thr2847.40 further transfers the disturbance to TM 7 and causes an ~2 Å shift toward TM 6 compared to the maraviroc-bound structure. In contrast to the bulkier imidazopiperidine group in the C-substituent, PF-232798 lacks the N-fluoro-cyclohexane group, while in the CCR5-maraviroc structure, this group stretches toward TM 6 and TM 5 and forms strong hydrogen bonds with Thr2596.59 and Thr1955.39. As a result of these modifications, relative to the axis of the 7TM helical bundle in the CCR5–PF-232798 complex structure, TM 6 is pulled inward, and TM 5 is pushed outward.

Comparison of the CCR5-maraviroc and CCR5–PF-232798 complex structures. a Top view of CCR5-maraviroc (blue cylinders) compared with CCR5–PF-232798 (magenta cylinders). Arrows represent the shift of helices. b The residues leading to the shift of TM 5 and TM 6. c The residues leading to the shift of TM 1, TM 2, and TM 7. The colors shown correspond to the colors in Fig. 3d, e
Discussion
Reverse transcriptase inhibitors and protease inhibitors are initial classes of HAART drugs for the treatment of HIV that rely on interactions with viral proteins and have drug resistance problems. Agents that interact with human receptors have a broad spectrum of activity against HIV clades and are attractive alternatives. Among them, the approved drug maraviroc and its imidazopiperidine derivative PF-232798 are allosteric inhibitors that target the HIV-1 coreceptor CCR5. Allosteric ligands confer potential therapeutic advantages in selectivity and safety in targeting GPCRs and have become popular areas of research in the drug development field. To our knowledge, compared with the orthosteric sites, the allosteric sites of GPCRs are less conserved and may be located at deeper sites of the 7TM region (CRF1R) [26], the extracellular surface (M2R) [27], the intracellular surface (CCR2, CCR9, β2AR) [28,29,30] and even outside of the helical bundles (GCGR, P2Y1R, PAR2) [31,32,33,34]. Maraviroc and PF-232798 have been defined as allosteric inhibitors of CCR5 [13] and bind to a pocket deep in the 7 TM region of CCR5, which is similar to the allosteric site of CP-376395 in the CRF1R structure [26].
Structural comparison of the CCR5–PF-232798 and CCR5–maraviroc complexes shows similar allosteric modulation mechanisms. Both maraviroc and PF-232798 sit deep in the 7TM binding pocket, blocking receptor activation through interactions with residues that are important for receptor activation, such as Tyr371.39 and Trp2486.48 (Fig. 3b–e) [13]. However, different N-/C-substituents within the tropane core place the two antagonists into different subpockets and induce large conformational changes, including a 3 Å shift of TM 1 on the extracellular side (Figs. 3 and 4). Previous data showed that the imidazopiperidine-substituted tropane compounds, rather than the triazole- or benzimidazole-substituted tropane compounds, demonstrated inhibition of a laboratory generated maraviroc-resistant (MVCRES) virus [14]. In the CCR5 structures, the interactions between CCR5 and the imidazopiperidine group of PF-232798 display no significant difference from the interactions between CCR5 and the triazole group of maraviroc, indicating that the imidazopiperidine substituent is not the determinant of the different virus resistance profile. Interestingly, the functional imidazopiperidines tested in the anti-MAVRES assay all contain lipophilic amide substituents, which make hydrophobic interactions with residues in subpocket 2 in the CCR5–PF-232798 structure. Overall, it could be proposed that the interactions with residues in subpocket 2 are the leading cause of the alternative drug resistance profile of PF-232798.
Although both PF-232798 and maraviroc are CCR5 antagonists and share a similar tropane scaffold, they have different binding affinities and drug resistance profiles. Comparison of the CCR5–PF-232798 and CCR5-maraviroc structures shows that the modifications on the N-/C- substituents lead to differences in binding modes and trigger conformational changes of the receptor. These findings expand our understanding of CCR5-ligand interaction patterns and will accelerate the development of novel anti-HIV drugs targeting CCR5.
References
Centers for Disease Control. Update on acquired immune deficiency syndrome (AIDS)–United States. MMWR Morb Mortal Wkly Rep. 1982;31:507–8, 513-4.
Acquired immunodeficiency syndrome (AIDS). HIV infections, 1989. Wkly Epidemiol Rec. 1990;65:302–3.
Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700.
Simmons G, Clapham PR, Picard L, Offord RE, Rosenkilde MM, Schwartz TW, et al. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science. 1997;276:276–9.
Murakami T, Nakajima T, Koyanagi Y, Tachibana K, Fujii N, Tamamura H, et al. A small molecule CXCR4 inhibitor that blocks T cell line-tropic HIV-1 infection. J Exp Med. 1997;186:1389–93.
Howard OM, Korte T, Tarasova I, Grimm M, Turpin JA, Rice WG, et al. Small molecule inhibitor of HIV-1 cell fusion blocks chemokine receptor-mediated function. J Leukoc Biol. 1998;64:6–13.
Temesgen Z, Warnke D, Kasten MJ. Current status of antiretroviral therapy. Expert Opin Pharmacother. 2006;7:1541–54.
Gottardo R, Bailer RT, Korber BT, Gnanakaran S, Phillips J, Shen XY, et al. Plasma IgG to linear epitopes in the V2 and V3 regions of HIV-1 gp120 correlate with a reduced risk of infection in the RV144 vaccine efficacy trial. PLoS ONE. 2013;8:e75665.
Palani A, Tagat JR. Discovery and development of small-molecule chemokine coreceptor CCR5 antagonists. J Med Chem. 2006;49:2851–7.
FDA notifications. Maraviroc approved as a CCR5 co-receptor antagonist. AIDS Alert. 2007;22:103.
Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67:1268–82.
Muniz-Medina VM, Jones S, Maglich JM, Galardi C, Hollingsworth RE, Kazmierski WM, et al. The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property? Mol Pharmacol. 2009;75:490–501.
Garcia-Perez J, Rueda P, Alcami J, Rognan D, Arenzana-Seisdedos F, Lagane B, et al. Allosteric model of maraviroc binding to CC chemokine receptor 5 (CCR5). J Biol Chem. 2011;286:33409–21.
Stupple PA, Batchelor DV, Corless M, Dorr PK, Ellis D, Fenwick DR, et al. An imidazopiperidine series of CCR5 antagonists for the treatment of HIV: the discovery of N-{(1S)-1-(3-fluorophenyl)-3-[(3-endo)-3-(5-isobutyryl-2-methyl-4,5,6,7-tetrahydr o-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl]propyl}acetamide (PF-232798). J Med Chem. 2011;54:67–77.
Tan QX, Zhu Y, Li J, Chen ZX, Han GW, Kufareva I, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–90.
Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–76.
Juan A. Ballesteros HW. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Sealfon SC, editor. Methods in Neurosciences; v 25. Receptor Molecular Biology. Amsterdam: Elsevier; 1995;366–428.
Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–31.
Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26.
Dauter Z, Wilson KS, Sieker LC, Moulis JM, Meyer J. Zinc- and iron-rubredoxins from Clostridium pasteurianum at atomic resolution: a high-precision model of a ZnS4 coordination unit in a protein. Proc Natl Acad Sci USA. 1996;93:8836–40.
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74.
Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55.
Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–80.
Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501.
Abrol R, Trzaskowski B, Goddard WA, Nesterov A, Olave I, Irons C. Ligand- and mutation-induced conformational selection in the CCR5 chemokine G protein-coupled receptor. Proc Natl Acad Sci USA. 2014;111:13040–5.
Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–43.
Kruse AC, Ring AM, Manglik A, Hu JX, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–6.
Zheng Y, Qin L, Zacarias NV, de Vries H, Han GW, Gustavsson M, et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature. 2016;540:458–61.
Oswald C, Rappas M, Kean J, Doré AS, Errey JC, Bennett K, et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature. 2016;540:462–5.
Liu XY, Ahn SG, Kahsai AW, Meng KC, Latorraca NR, Pani B, et al. Mechanism of intracellular allosteric β2AR antagonist revealed by X-ray crystal structure. Nature. 2017;548:480–4.
Zhang HN, Qiao AN, Yang DH, Yang LL, Dai AT, de Graaf C, et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature. 2017;546:259–64.
Jazayeri A, Dore AS, Lamb D, Krishnamurthy H, Southall SM, Baig AH, et al. Extra-helical binding site of a glucagon receptor antagonist. Nature. 2016;533:274–7.
Zhang DD, Gao ZG, Zhang KH, Kiselev E, Crane S, Wang J, et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature. 2015;520:317–21.
Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature. 2017;545:112–5.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 31730027). We are grateful to the staff of 23ID-D beamline (GM/CA CAT) of the Advanced Photon Source for support during data collection.
Author contributions
YZ performed the CPM assay, protein purification and crystallization. YZ and Y-LZ analyzed the data and wrote the manuscript. JL synthesized the compounds. HL oversaw compound synthesis. QZ collected the diffraction data and solved the structure. B-LW and Z-LY designed the research and revised the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhu, Y., Zhao, Yl., Li, J. et al. Molecular binding mode of PF-232798, a clinical anti-HIV candidate, at chemokine receptor CCR5. Acta Pharmacol Sin 40, 563–568 (2019). https://doi.org/10.1038/s41401-018-0054-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0054-2
