
Abstract
Brain endogenous cannabinoid (eCB) signaling seems to harmonize appropriate behavioral responses, which are essential for the organism’s long-term viability and homeostasis. Dysregulation of eCB signaling contributes to negative emotional states and increased stress responses. An understanding of the underlying neural cell populations and neural circuit regulation will enable the development of therapeutic strategies to mitigate behavioral maladaptation and provide insight into the influence of eCB on the neural circuits involved in anxiety and depression. This review focuses on recent evidence that has added a new layer of complexity to the idea of targeting the eCB system for therapeutic benefits in neuropsychiatric disease and on the future research direction of neural circuit modulation.
Similar content being viewed by others

Introduction
Cannabis plants have been used medicinally and recreationally for millennia by Chinese, Indian, and Arab pharmacologists [1]. The most commonly reported reason for cannabis use by the majority of users worldwide is its relaxing effect [2, 3], suggesting that cannabinoid signaling could play a role in the control of anxiety and depression. In recent years, a large body of data have demonstrated that the endocannabinoid (endogenous cannabinoid, eCB) system is involved in the regulation of the behavioral domains of anxiety and depression [4, 5].
The eCB system modulates synaptic transmission processes [6, 7], thereby regulating behavioral outputs. Although the eCB system is widely distributed in the central nervous system [8], its activity is highly specific and localized. To understand this specificity in anxiety and depression, one needs an integrated view of the eCB-mediated control of relevant brain regions and their interregional connections. Within distinct cerebral regions, eCB signaling can differentially modulate the activity of multiple cell types (neuronal subtypes [6], astrocytes [9, 10], and microglia [11]) and in turn execute context-related alterations in synaptic transmission. The present review examines the evidence for eCB influence in anxiety- and stress-related dysfunctions, such as depression, and the research on other neural circuits. We evaluate whether the activity of the eCB system is altered in these pathological processes. Potential therapeutic implications of the reviewed literature are also discussed.
The endocannabinoid system
In brief, the eCB system classically consists of cannabinoid receptors (cannabinoid 1 receptors, CB1R (encoded by CNR1) and cannabinoid 2 receptors, CB2R (encoded by CNR2)), endocannabinoids (i.e., anandamide (AEA, also known as N-arachidonoylethanolamine), 2-arachidonoylglycerol (2-AG)), and enzymes that catalyze endocannabinoid synthesis and degradation (Fig. 1). AEA is a high-affinity and low-efficacy CB1R agonist that also activates postsynaptic transient receptor potential cation channel subfamily V member 1 (TRPV1) to induce long-term depression. The level of 2-AG in the brain is 200-fold that of AEA [12] and its affinity for CB1R is lower than that of AEA, but it can excite CB2R in addition to CB1R [13]. Differing from classical neurotransmitters, eCB is not presynthesized and stored in the vesicles of presynaptic membranes, but it is synthesized from cellular membrane lipids following various stimuli in the postsynaptic membrane “on-demand” [14]. This is well documented for 2-AG, whose synthesis was shown to be stimulated after an increased concentration of postsynaptic intracellular Ca2+ or an increased activity of phospholipase Cβ. The on-demand synthesis of AEA is not well characterized yet, it might be synthesized in the presynaptic terminal [15] or synthesized postsynaptically [16], and N-acyl phosphatidyl ethanolamine-phospholipase D is one of the enzymes involved in AEA synthesis. After synthesis, eCB is released into the synaptic cleft, where it combines with CB1R to induce depolarization-induced suppression of inhibition (DSI)or depolarization-induced suppression of excitation (DSE) [17]. For example, isoflurane significantly increased eCB signaling at glutamatergic synapses in both GABAergic and glutamatergic DMH neurons and caused significantly increased DSE in both LTS+ and LTS– neurons [18]. Synaptic eCB signaling is terminated by the presynaptic reuptake and intracellular degradation of eCBs. Fatty acid-binding proteins deliver eCBs to their catabolic enzymes or nuclear receptors [19], and FABP5 is essential for retrograde eCB signaling and may serve as a synaptic carrier of eCBs in central synapses [20]. After presynaptic reuptake, 2-AG and AEA, are hydrolyzed by monoacylglycerol lipase (MAGL) [21] and fatty acid amide hydrolase (FAAH) [22], respectively.

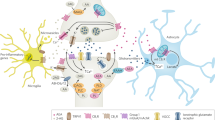
Architecture of eCB system components in neurons and astrocytes. Endocannabinoid (eCB) system shows a distinct anatomical distribution in the central nervous system. On depolarization of the postsynaptic terminal, 2-arachidonoylglycerol (2-AG) is present at the postsynaptic terminal and synthesized “on-demand” by diacylglycerol lipase-α (DAGLα). 2-AG travels to the presynaptic CB1R to inhibit neurotransmitter release, especially when the postsynaptic terminal is strongly activated. The 2-AG-degrading enzyme monoacylglycerol lipase (MAGL) is located at the presynaptic terminal or in astrocytes. The N-arachidonoylethanolamine (AEA)-degrading enzyme fatty acid amide hydrolase (FAAH) is present at the postsynaptic terminal. The cannabinoid receptor type 1 (CB1R) is typically present at the presynaptic terminal. Its stimulation by 2-AG or AEA leads to the inhibition of neurotransmitter release from the presynaptic terminal. CB1R is also present in the astrocytes, and activation of the CB1R leads to an increase in intracellular Ca2+ concentration. AEA can also activate the postsynaptic transient receptor potential cation channel subfamily V member 1 (TRPV1), leading to an increase in the postsynaptic current. CB2R is present on microglial cells and is involved in immune reactions
CB1R, the most abundant G protein-coupled receptor, is the most prominent eCB-binding site in the brain, the main role of which is binding to the endocannabinoid or exogenous cannabinoid and subsequent inhibition of the release of multiple neurotransmitters in the presynaptic membrane [23]. The crystal structure of CB1R and the agonist-bound structure of CB1R help us to understand the atomic framework of the cannabinoid receptor function and will helpaid in the design and optimization of cannabinoid system modulators for therapeutic ends [24,25,26]. The synaptic distribution of CB1R is consistent with its role as a retrograde regulator of synaptic signaling. CB1R expression is high in the presynaptic axon membranes and low in the postsynaptic dendrites and soma [27]. This polarization is driven by the push–pull combination of high MGL levels in the axons and high diacylglycerol lipase expression in the dendrites and soma [28]. CB1R shows particularly dense expression in regions with known involvement in fear, anxiety, and stress, including the hippocampus, prefrontal cortex (PFC), bed nucleus of the stria terminalis, basolateral amygdala (BLA), central amygdala, and various hypothalamic nuclei [6]. It has been demonstrated that CB1R is highly expressed in GABAergic interneurons expressing cholecystokinin (CCK), where it is largely absent from other interneuronal subtypes (for example, calretinin- and parvalbumin-expressing GABAergic interneurons) [29, 30]. CB1R is also present in the cholinergic, serotonergic, and noradrenergic systems [31, 32], suggesting that the eCB system is involved in the inhibition of the release of these neurotransmitters. Much lower levels of CB1R expression are present in glutamatergic neurons in the cortex [8, 29]. However, cortical glutamatergic CB1R has been revealed to have crucial functional roles [33,34,35], including the regulation of excitatory synaptic transmission [6]. Moreover, CB1R is present at extremely low levels in astrocytes [36]. Our previous studies demonstrated that astroglial CB1R plays an important role in the memory formation of cannabis addiction [37], the cannabis-induced impairment of spatial memory [9], the rapid anti-anxiety effects of FAAH inhibition [13] and the JZL195-induced in vivo long-term depression and neuroprotective effect against the ischemic insult [38]. CB1R is also present on the membranes of mouse neuronal mitochondria (mtCB1R), where it directly controls cellular respiration and energy production [39]. Moreover, acute cannabinoid-induced memory impairment in mice requires the activation of hippocampal mtCB1R [40].
CB2R was thought to be primarily expressed in microglial cells, especially activated microglial cells [41]. However, this theory has been challenged by recent studies that have found CB2R expressed in numerous brain areas [42, 43], including ventral tegmental area dopaminergic neurons [44], which also express CB1R [45]. Dopaminergic CB2R plays an important role in modulating dopaminergic neuronal activity and dopamine-related functions [44], inhibiting psychomotor behaviors, and altering anxiety, depression, alcohol dependence [46], and cacaine-induced locomotor-stimulating effects [47]. The recent study proving that CB2R expression in CA2/3 pyramidal neurons of the hippocampus and the novel type of CB2R-mediated neuroplasticity in that region suggest a postsynaptic, cell-intrinsic action for the CB2R that reduces excitability in a cell-specific manner and provide the first evidence for functional coupling between cannabinoid signaling and the sodium-bicarbonate transporter [48, 49]. Several studies have shown that CB2R function is also closely connected to schizophrenia in clinical patients and to schizophrenia-related behaviors in rodent models [50, 51].
Role of the endocannabinoid system in anxiety therapy
Anxiety disorders are a group of mental disorders characterized by significant feelings of anxiety and fear. These feelings may cause physical symptoms, such as a fast heart rate and shakiness [52]. Pathological anxiety is one of the most common types of mental illness, globally ranked as 33rd in its prevalence and the 9th cause of years lived with disability according to the Global Burden of Diseases, Injuries, and Risk Factors Study 2016 [53]. Anxiolytic drugs such as benzodiazepines produce rapid effects, but their long-term use causes severe adverse effects, such as drowsiness, dizziness, and decreased alertness and concentration [54]. Several studies have shown that the eCB system participates in the bidirectional regulation of anxiety circuits and behavior: (1) Exogenous cannabinoids such as ∆9-THC and synthetic cannabinoids influence anxiety‐like behavior in a biphasic manner, with low and high doses exerting anxiolytic and anxiogenic states, respectively [55,56,57]. (2) CB1Rs on cortical glutamatergic and GABAergic neurons exert opposing control on anxiety‐like behaviors [58,59,60]. (3) Global CB1R knockout results in increased anxiety-like behavior [61]. (4) Deletion of the 2-AG MAGL leads to a significant increased anxiety-like behavior [62]. (5) Overexpression of 2-AG hydrolyzyme in the hippocampal glutamatergic neurons also leads to increased anxiety-like behavior [63], whereas pharmacological inhibition of 2-AG hydrolyzyme produces an anxiolytic effect [14]. (6) The genetic knockout or pharmacological inhibition of the AEA hydrolytic enzyme FAAH produces anxiolytic effects [8, 31, 32] without the side effects of marijuana (Table 1). (7) The substrate-selective COX-2 inhibitors may inhibit the oxygenation of 2-AG and AEA and produce anxiolytic effects in preclinical models [64].
FAAH inhibitor PF3845 exerts rapid and long-lasting anxiolytic effects in mice exposed acutely and chronically to the stress hormone corticosterone [10]. This finding expands on the previous findings that the FAAH inhibitor URB597 produces anxiolytic effects 1 day after acute stress exposure [65] and that chronic injection of PF3845 exerts anxiolytic effects in chronic and mild stress-exposed animals [14]. Mice show anxiolytic behavioral responses with increased functional connectivity between the PFC and amygdala (BLA) [66, 67], suggesting a key role for increased FAAH activity and decreased AEA signaling in the development mechanisms of anxiety disorders. Many researchers have recently hypothesized that stress upregulates FAAH activity to decrease AEA signaling, which increases the excitability of BLA pyramidal neurons due to the unavailability of AEA for the suppression of glutamate release, leading to anxiety-like behavior [6, 30]. However, Duan et al. [10] provided evidence that PF3845 induced anxiolytic effects and in vivo long-term depression of synaptic strength at the PFC input into BLA neurons via astroglial CB1R, which strongly supports an alternative mechanism underlying the anti-anxiety effects of FAAH inhibitors [10].
In summary, the eCB system generally exerts anxiolytic functions. However, GABAergic CB1R signaling in anxiety modulation seems to mediate anxiogenic effects under certain conditions.
Role of the endocannabinoid system in the treatment of depression
Major depressive disorder, also known simply as depression, is a mental disorder characterized by at least 2 weeks of low mood that is present across most situations. It is often accompanied by low self-esteem, loss of interest in normally enjoyable activities, low energy, and pain without a clear cause. Major depressive disorder can negatively affect a person’s personal, work, or school life, as well as sleeping and eating habits, and general health [68]. The probability of suffering from depression at some point during one's lifespan is up to 15% for men and 30% for women [69]. The current antidepressant serotonin/noradrenalin reuptake inhibitors have a low remission rate and a delayed onset time of several weeks [70]. It is therefore of great importance to identify new fast-acting antidepressants.
Several findings have shown that the eCB system may be a potential antidepressant candidate [71,72,73,74,75]. Specifically, eCB may help reverse the acute and chronic stress response; it may produce antidepressant physiological changes, including neurogenesis and synaptic plasticity; people with depression have relatively lower eCB levels; and clinical trials in Europe and the United States have shown that one of the side effects of obesity treatment by the selective CB1R antagonist rimonabant is depression. Recent studies have shown that AEA can treat depression induced by acute stress [73, 76]. In addition, although daily injections of MAGL inhibitor JZL184 (8 mg/kg) before stress can prevent the development of depressive behavior [77, 78], JZL184 has no antidepressant effect on chronic stress [77]. In other words, different doses of MAGL inhibitor may produce antidepressant or depressant effects on acute and chronic stress, respectively.
One study found that after the administration of MAGL inhibitor JZL184 2 h before forced swimming, there was a “U-shaped” relationship between the dose and the behavioral indicators of JZL184. That is, a low dose of JZL184 (5 mg/kg) could significantly protect mice against the despair behavior induced by acute stress [79]. However, low doses of JZL184 (5 mg/kg [79] and 8 mg/kg [77]) did not exert antidepressant effects in mice exposed to chronic stress. Interestingly, 20 mg/kg JZL184 produced significant antidepressant behavioral responses in chronic corticosterone-treated mice [79], thus opening a window for the clinical treatment of depression with MAGL inhibitors. In mice with chronic stress, the antidepressant effect may be associated with the activation of GABA neurons via CB1R and the DSI effect [72]. A high dose of MAGL inhibitors produced antidepressant effects in chronically stressed mice because it disinhibited GABAergic synapses via GABAergic CB1R, hence exciting CA1 pyramidal neurons [79].
Collectively, our review provides an overview of the neurobiological interactions between stress and the eCB system. First, after exposure to both acute and chronic stress, it results in a bidirectional regulation of the two eCB ligands AEA and 2-AG, with AEA being reduced by stress and 2-AG being increased by stress. The reduction of AEA appears to occur relatively quickly in response to stress and is mediated by increased AEA hydrolysis by FAAH [80,81,82,83]. With respect to acute stress, the increase in 2-AG is delayed and seems to be mediated by increases in corticosterone due to stress [82, 84,85,86,87]. There seems to be an apparent yin–yang relationship for AEA and 2-AG, with both molecules providing a stress-inhibitory effect, but the reduction of AEA is responsible for the initiation and manifestation of the effects of stress, whereas the increase of 2-AG is responsible for tempering and terminating the stress response [88].
In conclusion, the stress-induced downregulation of CB1R signaling in brain regions is of vital importance for the regulation of emotion processes, such as depression. Stress adaptation, which reduces this effect, is accompanied by enhanced eCB system activity.
Future directions: eCB signaling in neuronal circuit regulation
The eCB system is present in many brain circuits that are well known to regulate anxiety and depression processes. Together with anatomical descriptions and functional evidence, eCB signaling might modulate synaptic activity at many nuclei of these circuits. However, direct evidence of these functions of eCB signaling in freely behaving animals challenged with specific tasks is mostly lacking. Based on the Cre-loxP system to cell-specific modulation of the eCB system, the advent of new technological approaches, such as viral tracing techniques, optogenetics and the DREADD strategy, and calcium imaging techniques will allow the exploration of the direct evidence for CB1R in the regulation of specific circuits and behavior.
How the eCB system regulates anxiety and depression has been studied for a long time. These studies mentioned above raise strong hopes not only for a better understanding of the behavioral processes but also for future therapy to rescue these dysfunctions. In the future, by integrating the cutting-edge research methods, the causal links between the eCB system-meditated synaptic plasticity/electrophysiological modulations and behavioral outcomes will be well revealed.
References
Kmietowicz Z. Cannabis based drug is licensed for spasticity in patients with MS. BMJ. 2010;340:c3363.
Tart CT. Marijuana intoxication common experiences. Nature. 1970;226:701–4.
Volkow ND, Hampson AJ, Baler RD. Don’t worry, be happy: endocannabinoids and cannabis at the intersection of stress and reward. Annu Rev Pharmacol Toxicol. 2017;57:285–308.
Gunduz-Cinar O, Hill MN, McEwen BS, Holmes A. Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends Pharmacol Sci. 2013;34:637–44.
McLaughlin RJ, Hill MN, Gorzalka BB. A critical role for prefrontocortical endocannabinoid signaling in the regulation of stress and emotional behavior. Neurosci Biobehav Rev. 2014;42:116–31.
Lutz B, Marsicano G, Maldonado R, Hillard CJ. The endocannabinoid system in guarding against fear, anxiety and stress. Nat Rev Neurosci. 2015;16:705–18.
Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81.
Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–58.
Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell. 2012;148:1039–50.
Duan T, Gu N, Wang Y, Wang F, Zhu J, Fang Y, et al. Fatty acid amide hydrolase inhibitors produce rapid anti-anxiety responses through amygdala long-term depression in male rodents. J Psychiatry Neurosci. 2016;42:160116.
Mecha M, Feliu A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutierrez S, de Sola RG, et al. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun. 2015;49:233–45.
Gipson CD, Kupchik YM, Kalivas PW. Rapid, transient synaptic plasticity in addiction. Neuropharmacology. 2014;76 Pt B:276–86.
Gonsiorek W, Lunn C, Fan X, Narula S, Lundell D, Hipkin RW. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol Pharmacol. 2000;57:1045–50.
Lomazzo E, Bindila L, Remmers F, Lerner R, Schwitter C, Hoheisel U, et al. Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology. 2015;40:488–501.
Wang J, Ueda N. Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat. 2009;89:112–9.
Huang GZ, Woolley CS. Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR-dependent mechanism. Neuron. 2012;74:801–8.
Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76.
Zhong H, Tong L, Gu N, Gao F, Lu Y, Xie RG, et al. Endocannabinoid signaling in hypothalamic circuits regulates arousal from general anesthesia in mice. J Clin Invest. 2017;127:2295–309.
Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci U S A. 2009;106:6375–80.
Haj-Dahmane S, Shen RY, Elmes MW, Studholme K, Kanjiya MP, Bogdan D et al. Fatty acid-binding protein 5 controls retrograde endocannabinoid signaling at central glutamate synapses. Proc Natl Acad Sci U S A. 2018; 115:3482–87.
Goparaju SK, Ueda N, Taniguchi K, Yamamoto S. Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Biochem Pharmacol. 1999;57:417–23.
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–7.
Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–66.
Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, et al. Crystal structure of the human cannabinoid receptor CB1. Cell. 2016;167:750–62 e14.
Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, et al. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature. 2016; 540: 602-6.
Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 2017;547:468–71.
Thibault K, Carrel D, Bonnard D, Gallatz K, Simon A, Biard M, et al. Activation-dependent subcellular distribution patterns of CB1 cannabinoid receptors in the rat forebrain. Cereb Cortex. 2013;23:2581–91.
Ladarre D, Roland AB, Biedzinski S, Ricobaraza A, Lenkei Z. Polarized cellular patterns of endocannabinoid production and detection shape cannabinoid signaling in neurons. Front Cell Neurosci. 2014;8:426.
Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–25.
Hill MN, Kumar SA, Filipski SB, Iverson M, Stuhr KL, Keith JM, et al. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol Psychiatry. 2013;18:1125–35.
Haring M, Marsicano G, Lutz B, Monory K. Identification of the cannabinoid receptor type 1 in serotonergic cells of raphe nuclei in mice. Neuroscience. 2007;146:1212–9.
Nyiri G, Szabadits E, Cserep C, Mackie K, Shigemoto R, Freund TF. GABAB and CB1 cannabinoid receptor expression identifies two types of septal cholinergic neurons. Eur J Neurosci. 2005;21:3034–42.
Soltesz I, Alger BE, Kano M, Lee SH, Lovinger DM, Ohno-Shosaku T, et al. Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat Rev Neurosci. 2015;16:264–77.
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–8.
Marinelli S, Pacioni S, Cannich A, Marsicano G, Bacci A. Self-modulation of neocortical pyramidal neurons by endocannabinoids. Nat Neurosci. 2009;12:1488–90.
Massi P, Valenti M, Bolognini D, Parolaro D. Expression and function of the endocannabinoid system in glial cells. Curr Pharm Des. 2008;14:2289–98.
Liu Z, Han J, Jia L, Maillet JC, Bai G, Xu L, et al. Synaptic neurotransmission depression in ventral tegmental dopamine neurons and cannabinoid-associated addictive learning. PLoS One. 2010;5:e15634.
Wang F, Han J, Higashimori H, Wang J, Liu J, Tong L, et al. Long-term depression induced by endogenous cannabinoids produces neuroprotection via astroglial CB1R after stroke in rodents. J Cereb Blood Flow Metab. 2018:271678X18755661. [epub ahead of print].
Benard G, Massa F, Puente N, Lourenco J, Bellocchio L, Soria-Gomez E, et al. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat Neurosci. 2012;15:558–64.
Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, et al. A cannabinoid link between mitochondria and memory. Nature. 2016;539:555–9.
Stella N. Endocannabinoid signaling in microglial cells. Neuropharmacology. 2009;56:244–53.
Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol. 2013;64:21–47.
Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–32.
Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang HJ et al. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A. 2014;111:E5007–15.
Avello LM, Pastene NE, Fernandez RP, Cordova MP. [Therapeutic potential of Cannabis sativa]. Rev Med Chil. 2017;145:360–7.
Liu QR, Canseco-Alba A, Zhang HY, Tagliaferro P, Chung M, Dennis E, et al. Cannabinoid type 2 receptors in dopamine neurons inhibits psychomotor behaviors, alters anxiety, depression and alcohol preference. Sci Rep. 2017;7:17410.
Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, et al. Brain cannabinoid CB2 receptors modulate cocaine’s actions in mice. Nat Neurosci. 2011;14:1160–U216.
Stempel AV, Stumpf A, Zhang HY, Ozdogan T, Pannasch U, Theis AK, et al. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the hippocampus. Neuron. 2016;90:795–809.
Quraishi SA, Paladini CA. A central move for CB2 receptors. Neuron. 2016;90:670–1.
Ishiguro H, Horiuchi Y, Ishikawa M, Koga M, Imai K, Suzuki Y, et al. Brain cannabinoid CB2 receptor in schizophrenia. Biol Psychiatry. 2010;67:974–82.
Ortega-Alvaro A, Aracil-Fernandez A, Garcia-Gutierrez MS, Navarrete F, Manzanares J. Deletion of CB2 cannabinoid receptor induces schizophrenia-related behaviors in mice. Neuropsychopharmacology. 2011;36:1489–504.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington DC (USA): American Psychiatric Press; 2013. American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders (DSM-V). p185–95.
Collaborators GBoDS. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800.
Rickels K, Case WG, Downing RW, Winokur A. Long-term diazepam therapy and clinical outcome. JAMA. 1983;250:767–71.
Patel S, Hillard CJ. Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: further evidence for an anxiolytic role for endogenous cannabinoid signaling. J Pharmacol Exp Ther. 2006;318:304–11.
Berrendero F, Maldonado R. Involvement of the opioid system in the anxiolytic-like effects induced by Delta(9)-tetrahydrocannabinol. Psychopharmacol (Berl). 2002;163:111–7.
Rubino T, Guidali C, Vigano D, Realini N, Valenti M, Massi P, et al. CB1 receptor stimulation in specific brain areas differently modulate anxiety-related behaviour. Neuropharmacology. 2008;54:151–60.
Ruehle S, Remmers F, Romo-Parra H, Massa F, Wickert M, Wortge S, et al. Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J Neurosci. 2013;33:10264–77.
Haring M, Kaiser N, Monory K, Lutz B. Circuit specific functions of cannabinoid CB1 receptor in the balance of investigatory drive and exploration. PLoS One. 2011;6:e26617.
Lafenetre P, Chaouloff F, Marsicano G. Bidirectional regulation of novelty-induced behavioral inhibition by the endocannabinoid system. Neuropharmacology. 2009;57:715–21.
Jacob W, Yassouridis A, Marsicano G, Monory K, Lutz B, Wotjak CT. Endocannabinoids render exploratory behaviour largely independent of the test aversiveness: role of glutamatergic transmission. Genes Brain Behav. 2009;8:685–98.
Shonesy BC, Bluett RJ, Ramikie TS, Baldi R, Hermanson DJ, Kingsley PJ, et al. Genetic disruption of 2-arachidonoylglycerol synthesis reveals a key role for endocannabinoid signaling in anxiety modulation. Cell Rep. 2014;9:1644–53.
Guggenhuber S, Romo-Parra H, Bindila L, Leschik J, Lomazzo E, Remmers F, et al. Impaired 2-AG signaling in hippocampal glutamatergic neurons: aggravation of anxiety-like behavior and unaltered seizure susceptibility. Int J Neuropsychopharmacol. 2015;19:2.
Hermanson DJ, Hartley ND, Gamble-George J, Brown N, Shonesy BC, Kingsley PJ, et al. Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci. 2013;16:1291–8.
Bluett RJ, Gamble-George JC, Hermanson DJ, Hartley ND, Marnett LJ, Patel S. Central anandamide deficiency predicts stress-induced anxiety: behavioral reversal through endocannabinoid augmentation. Transl Psychiatry. 2014;4:e408.
Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC, et al. FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun. 2015;6:6395.
Bluett RJ, Baldi R, Haymer A, Gaulden AD, Hartley ND, Parrish WP, et al. Endocannabinoid signalling modulates susceptibility to traumatic stress exposure. Nat Commun. 2017;8:14782.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington DC (USA): American Psychiatric Press; 2013. American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders (DSM-V). p160–8.
Briley M, Moret C. Present and future anxiolytics. IDrugs. 2000;3:695–9.
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40.
Bambico FR, Gobbi G. The cannabinoid CB1 receptor and the endocannabinoid anandamide: possible antidepressant targets. Expert Opin Ther Targets. 2008;12:1347–66.
Hill MN, Patel S. Translational evidence for the involvement of the endocannabinoid system in stress-related psychiatric illnesses. Biol Mood Anxiety Disord. 2013;3:19.
Hillard CJ, Liu QS. Endocannabinoid signaling in the etiology and treatment of major depressive illness. Curr Pharm Des. 2014;20:3795–811.
Leite CE, Mocelin CA, Petersen GO, Leal MB, Thiesen FV. Rimonabant: an antagonist drug of the endocannabinoid system for the treatment of obesity. Pharmacol Rep. 2009;61:217–24.
Griebel G, Stemmelin J, Lopez-Grancha M, Fauchey V, Slowinski F, Pichat P, et al. The selective reversible FAAH inhibitor, SSR411298, restores the development of maladaptive behaviors to acute and chronic stress in rodents. Sci Rep. 2018;8:2416.
Hill MN, Hillard CJ, Bambico FR, Patel S, Gorzalka BB, Gobbi G. The therapeutic potential of the endocannabinoid system for the development of a novel class of antidepressants. Trends Pharmacol Sci. 2009;30:484–93.
Zhong P, Wang W, Pan B, Liu X, Zhang Z, Long JZ, et al. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology. 2014;39:1763–76.
Zhang Z, Wang W, Zhong P, Liu SJ, Long JZ, Zhao L, et al. Blockade of 2-arachidonoylglycerol hydrolysis produces antidepressant-like effects and enhances adult hippocampal neurogenesis and synaptic plasticity. Hippocampus. 2015;25:16–26.
Wang Y, Gu N, Duan T, Kesner P, Blaskovits F, Liu J, et al. Monoacylglycerol lipase inhibitors produce pro- or antidepressant responses via hippocampal CA1 GABAergic synapses. Mol Psychiatry. 2017;22:215–26.
Gray JM, Vecchiarelli HA, Morena M, Lee TT, Hermanson DJ, Kim AB, et al. Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci. 2015;35:3879–92.
Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, et al. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34:2733–45.
Dubreucq S, Matias I, Cardinal P, Haring M, Lutz B, Marsicano G, et al. Genetic dissection of the role of cannabinoid type-1 receptors in the emotional consequences of repeated social stress in mice. Neuropsychopharmacology. 2012;37:1885–900.
Navarria A, Tamburella A, Iannotti FA, Micale V, Camillieri G, Gozzo L, et al. The dual blocker of FAAH/TRPV1 N-arachidonoylserotonin reverses the behavioral despair induced by stress in rats and modulates the HPA-axis. Pharmacol Res. 2014;87:151–9.
McLaughlin RJ, Hill MN, Bambico FR, Stuhr KL, Gobbi G, Hillard CJ, et al. Prefrontal cortical anandamide signaling coordinates coping responses to stress through a serotonergic pathway. Eur Neuropsychopharmacol. 2012;22:664–71.
Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, et al. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci. 2011;31:10506–15.
Atsak P, Hauer D, Campolongo P, Schelling G, McGaugh JL, Roozendaal B. Glucocorticoids interact with the hippocampal endocannabinoid system in impairing retrieval of contextual fear memory. Proc Natl Acad Sci U S A. 2012;109:3504–9.
Bowles NP, Hill MN, Bhagat SM, Karatsoreos IN, Hillard CJ, McEwen BS. Chronic, noninvasive glucocorticoid administration suppresses limbic endocannabinoid signaling in mice. Neuroscience. 2012;204:83–9.
Morena M, Patel S, Bains JS, Hill MN. Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology. 2016;41:80–102.
Acknowledgements
The project was supported by the Shaanxi Province Natural Science Foundation (No. 2016JQ8024) and by the International Cooperation and Exchange of the National Natural Science Foundation of China (No. 81420108013). We thank LetPub for providing linguistic assistance during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Yin, Aq., Wang, F. & Zhang, X. Integrating endocannabinoid signaling in the regulation of anxiety and depression. Acta Pharmacol Sin 40, 336–341 (2019). https://doi.org/10.1038/s41401-018-0051-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0051-5
Keywords
This article is cited by
-
Potential application of endocannabinoid system agents in neuropsychiatric and neurodegenerative diseases—focusing on FAAH/MAGL inhibitors
Acta Pharmacologica Sinica (2020)
-
Cannabis, cannabinoid receptors, and endocannabinoid system: yesterday, today, and tomorrow
Acta Pharmacologica Sinica (2019)
