
Abstract
Salvia miltiorrhiza Bunge contains various active constituents, some of which have been developed as commercially available medicine. Moreover, some other ingredients in Salvia miltiorrhiza play roles in anti-platelet activity. The aim of the present study was to investigate the effects and the underlying mechanism of miltirone, a lipophilic compound of Salvia miltiorrhiza Bunge. The ability of miltirone to modulate platelet function was investigated by a variety of in vitro and in vivo experiments. Platelet aggregation and dense granule secretion induced by various agonists were measured with platelet aggregometer. Clot retraction and spreading were imaged by digital camera and fluorescence microscope. Ferric chloride-induced carotid injury model and pulmonary thromboembolism model were used to check miltirone antithrombotic effect in vivo. To elucidate the mechanisms of anti-platelet activity of miltirone, flow cytometry and western blotting were performed. Miltirone (2, 4, 8 µM) was shown to suppress platelet aggregation, dense granule, and α granule secretion in a dose-dependent manner. Meanwhile, miltirone inhibited the clot retraction and spreading of washed platelets. It reduced the phosphorylation of PLCγ2, PKC, Akt, GSK3β and ERK1/2 in the downstream signal pathway of collagen receptor. It also reduced the phosphorylation of Src and FAK in the integrin αIIbβ3-mediated “outside-in” signaling, while it did not suppress the phosphorylation of β3. In addition, miltirone prolonged the occlusion time and reduced collagen/epinephrine-induced pulmonary thrombi. Miltirone suppresses platelet “inside-out” and “outside-in” signaling by affecting PLCγ2/PKC/ERK1/2, PI3K/Akt, and Src/FAK signaling. Therefore, miltirone might represent a potential anti-platelet candidate for the prevention of thrombotic disorders.
Similar content being viewed by others


Introduction
Platelets are enucleated cells deriving from megakaryocytes, which play an important role in hemostasis and thrombosis [1]. Platelet activates when the vessel wall is damaged. The formation of thrombus could be accelerated by platelets through the deformation and release of their internal stored granules, such as TxA2 and ADP. Therefore, anti-platelet therapy can effectively inhibit the development of thrombosis [2].
The current anti-platelet therapy mainly interferes the following signaling pathway: the synthesis of TxA2, adenosine diphosphate (ADP)-mediated signal transduction, cAMP, and integrin αIIbβ3-mediated signaling pathways [3]. However, some other potential targets on platelets still need to be found. Because of the critical roles on platelet activation, thrombin and collagen are considered as vital factors on the function of platelets, and their receptors such as glycoprotein VI, α2β1, and protease-activated receptor (PAR) are regarded as superior targets for anti-platelet therapy. The most commonly used medicines are aspirin, clopidogrel, cilostazol, and abciximab. Although the risk of thrombosis and recurrence could be greatly reduced in patients by using these medications, they are associated with many side effects, such as bleeding, rash, neutropenia, and thrombocytopenia [3,4,5]. In addition, these remedies possess some limitations: most of them just have single target; some medicine such as clopidogrel shows a significant delay in the onset of anti-platelet effects; and clopidogrel and asprin resistance [6, 7]. Although some new ADP receptor antagonists show a more rapid onset time and oral αIIbβ3 antagonists are getting some breakthrough, they are still troubled by bleeding and resistance. Thus it remains a pressing priority to find a safer and more effective anti-platelet agent [8, 9].
It is essential and efficient to find new active ingredients from traditional botanicals [10]. Growing evidences have shown that a lot of botanicals possess reliable effects on the treatment of cardiovascular diseases, such as myocardial infarction, stroke, atherosclerosis, and so on [11]. Among these botanicals, Salvia miltiorrhiza Bunge (Danshen in chinese) has been used for thousands of years and officially recognized in China and widely used individually or synergistically with other botanicals. Tanshinone IIA has been shown as a favorable anti-platelet ingredient and been considered as the main active ingredient in Salvia miltiorrhiza, whereas its potency is low and the mechanism is not clear [12, 13]. Miltirone, another active ingredient in Salvia miltiorrhiza, has showed extensive pharmacological activities, such as anti-inflammatory, antileukemic, antioxidant, and reducing the effect of alcohol intake, but whether miltirone has the function of anti-platelet is unknown [14,15,16,17]. We found that miltirone inhibited platelet function and its underlying mechanism involved phosphoinositide phospholipase Cγ2/protein kinase C/extracellular signal–regulated kinase 1/2 (PLCγ2/PKC/ERK1/2), phosphoinositide-3 kinase (PI3K)/Akt, and Src/focal adhesion kinase (FAK) signaling.
Materials and methods
Reagents and chemicals
Miltirone (5, 6, 7, 8-Tetrahydro-8,8-dimethyl-2-isopropyl-3,4-phenanthrenedione, high-performance liquid chromatography grade with ≥98%; Fig. 1) was provided by Chengdu Caoyuankang Bio-tech Co., Ltd. (Sichuan China). It was dissolved in dimethylsulfoxide (DMSO) with a final concentration of 0.4% (v/v) throughout the experiments. ADP, thrombin, prostaglandin E1 (PGE1), EDTA, bovine serum albumin (BSA), arachidonic acid (AA), and phalloidin tetramethylrhodamine isothiocyanate were purchased from Sigma Chemicals (St. Louis, MO). Epinephrine was purchased from Tianjin Pharmaceutial Group Xinzheng Co., Ltd. (Henan, China). Collagen (Type I, from equine tendons suspended in isotonic glucose solution of pH 2.7) and luciferin/luciferase were purchased from Chrono-Log Crop (Havertown, PA). Collagen-related peptide (CRP) was kindly provided by Dr. Newman in Blood Center of Wisconsin, USA. Antibodies against phospho-Akt (Ser473), phospho-GSK3β (Ser9), phospho-p38 (Thr180/Tyr182), phospho-ERK1/2 (Tyr202/204), phoshpo-PLCγ2 (Tyr1217), phospho-PKC substrate and phospho-FAK (Tyr397), phospho-Sr (Y416), total-FAK, total-β3, and total-Src were purchased from Cell Signaling (Beverly, MA). Antibody against phospho-Syk (Tyr525/526) was purchased from EMD Chemicals (Darmstadt, Germany). Antibodies against total-AKT, total-GSK3β, total-Syk, total-p38, total-ERK1/2, and total-PLCγ2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-rabbit immunoglobulin G antibodies were obtained from Life Technologies, UK. Protease and phosphatase inhibitor cocktails were purchased from EMD Millipore Chemicals (Billerica, MA). Fluorescein isothiocyanate (FITC)-conjugated anti-P-selectin (CD62P) antibodies were obtained from BD Biosciences (San Jose, CA). Phycoerythrin (PE)-conjugated anti-αIIbβ3 antibodies (JON/A) were obtained from Emfret Analytics GmbH&Co. KG. The ECL western blotting detection reagent was a product of Pierce Chemical Co. (Rockford, Illinois). All the other chemicals used in the present study were of analytical grade.

Chemical structure of miltirone
Animals
Male Kunming mice (22–25 g, 8–10-week-old, SCXK-2015-0018) were provided by Hubei Experimental Animal Research Center (Wuhan, Hubei, China). All animals were maintained in standard laboratory conditions (specific pathogen free) at a relative humidity of 55–65%, controlled temperature of 22–24 °C, 12 h dark/light cycle, given free access to food and water, and acclimated for at least 1 week before use. All experiments were carried out in accordance with the guidelines and the regulations of Ethics Committee for experimental animals, Tongji Medical College.
Mouse platelets' preparation
Preparations of washed platelets and platelet-rich plasma (PRP) from mouse were described previously [18]. Briefly, mouse whole blood was drawn from the inferior vena cava with a syringe containing 3.8% sodium citrate (9:1, v/v) after the mice were anesthetized with urethane by intraperitoneal injection. PRP was obtained by centrifuging the whole blood for 15 min at 150 × g. The supernatant was diluted with platelet-poor plasma (PPP; the blood samples were centrifuged at 800 × g for 15 min, the supernatant was considered PPP) to different concentrations. Washed platelets were washed twice with Tyrode’s buffer (13.8 mM NaHCO3, 137 mM NaCl, 0.36 mM NaH2PO4, 2.5 mM KCl, 5.5 mM glucose, 20 mM HEPES, pH 7.4) containing 50 ng/ml PGE1 and 1 mM EDTA, and finally resuspended in Tyrode’s buffer not containing PGE1 or EDTA to a final concentration of 3.0 × 108 platelets/ml. The whole process of the platelets' preparation was conducted at room temperature. All washed platelets were placed at room temperature for 30 min before use.
Platelet aggregation and ATP release in vitro
Platelet aggregation and ATP release assays were performed as described previously [19] with an aggregometer (Chrono-Log, Havertown, PA). The concentration of the washed platelets was adjusted to 3.0 × 108/ml with Tyrode’s buffer (PRP were diluted with PPP). CaCl2 (1 mM) was added prior to agonist stimulation. Washed platelets and PRP were preincubated with 0.4% DMSO (vehicle) or different final concentration of miltirone (2 µM, 4 µM, 8 µM) for 5 min under the stirring of 1000 r.p.m. and 37 °C, and then the platelets were stimulated with different agonists. Platelet ATP release was measured by using luciferin–luciferase reagent (Chrono-lume, Chrono-Log). The results of aggregation and ATP release were recorded and analyzed by the aggregolink software (Chrono-Log).
Platelet spreading on fibrinogen
Platelet spreading in vitro was determined according to a modified method previously reported [20]. Washed platelets at a density of 3 × 106 cells/ml were treated with different concentrations of miltirone or vehicle for 5 min and permitted to spread on 24-chamber glasses tissue-culture slides, which were coated with fibrinogen (25 µg/ml) or BSA for 45 min, 37 °C. Then the chambers were washed with phosphate-buffered saline twice after removing platelet suspension. The adhered platelets were fixed with 2% paraformaldehyde, permeabilized with 0.1% triton, and then stained with FITC-labeled phalloidin. Images were obtained using fluorescence microscope (Nikon TE-2000S). The images were analyzed using the Image J software (NIH) to calculate the relative average coverage of spreading platelets and single adherent platelet area.
In the biochemical experiments, the glass tissue culture slides were changed into 6-chambers, and the volume of washed platelet was changed to 1 ml. After incubated with miltirone or vehicle for 45 min at 37 ℃, the platelets were lysed with lysis buffer containing protease and phosphatase inhibitors and then subjected to immunoblot analysis.
Clot retraction
Platelet clot retraction was measured in glass tubes (Chrono-log, Lot 419766) according to a previously described method [18]. Briefly, washed platelet suspension (500 µl, 3 × 108 cells/ml) containing 400 µg/ml fibrinogen and 1 mM CaCl2 was preincubated in the presence and absence of different concentrations of miltirone or vehicle. Clot retraction was initiated by adding thrombin (0.2 U/ml), and then the platelet suspensions were incubated at 37 °C. Images were acquired after 15 min by using a digital camera (Nicon).
Flow cytometric analysis
Platelet membrane antigens were determined by flow cytometric analysis previously reported [21]. Washed platelets (5 × 107 cells/ml) were preincubated with different concentrations of miltirone or vehicle for 5 min prior to activation with collagen (2.0 µg/ml) for 5 min at 37 °C. The platelets were then co-incubated with FITC-conjugated anti-CD62P antibody or PE-conjugated anti-JON/A antibody at room temperature for 15 min in the dark room. The cells were analyzed by counting 10,000 events within the gated population utilizing flow cytometry. Data were analyzed by a BD Bioscience flow cytometer.
Immunoblotting
Washed platelets (3 × 108 cells/ml) were preincubated with miltirone or vehicle for 5 min and stimulated by different agonists or the same volume of Tyrode’s buffer for 3 min. Then the platelets were lysed by the same volume of 2× lysis buffer (300 mM NaCl, 20 mM EGTA, 2% Triton X-100, 30 mM HEPES, 0.2 mM MgCl2, 2× protease and 2× phosphatase inhibitor cocktails). Proteins were quantified by BCA assay and separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and incubated with the corresponding antibodies. Then the membranes were exposed to appropriate secondary antibodies for 1 h at room temperature and visualized using a chemical luminescence image system (DNR bio-imaging systems). The level of fluorescence bands was quantified by Image J.
Ferric chloride-induced carotid injury model
The antithrombotic activity of miltirone was determined by measuring the occlusion time in a ferric chloride-induced carotid injury model with a modified method [19]. Mice were injected with miltirone (10 mg/kg) into the tail vein. After 30 min, the mice were anesthetized with urethane by intraperitoneal injection, and then the carotid artery was exposed by surgical operation. A Doppler flow probe (Transonic, TS420, UK) was proximal to the carotid artery to measure baseline blood flow. The carotid artery adventitial surface was applied by a 2-mm strip of filter paper for 3 min, which was saturated with 10% ferric chloride. After removing the filter paper, blood flow through the carotid artery was monitored for 45 min or until vessel occlusion reached 95%.
Pulmonary thromboembolism model
To investigate the effect of miltirone on thrombosis in vivo, the pulmonary thrombosis mouse model was performed as previously described [22]. Thrombosis was initiated by injecting a mixture of collagen (1 mg/kg) plus epinephrine (10 μg/kg) plus miltirone (10 mg/kg) or vehicle into the tail vein. The mice were euthanized 10 min later, and the lungs were perfused with saline (4 °C) and then fixed in 10% formalin for 24 h. The paraffin-embedded lungs were stained with hematoxylin and eosin (H&E) and observed under a light microscope.
Tail bleeding assay
Mice tail bleeding time was measured according to a method previously reported [23]. Mice were injected with miltirone (10 mg/kg) or vehicle into the tail vein. After 30 min, the distal portion of the tail (3 mm) was amputated with a scalpel, and the transected tail tip was gently touched onto filter paper immediately and after every 30 s interval till the bleeding completely stopped (time to stable cessation of the bleeding was defined as the time where no rebleeding for longer than 1 min was recorded) or until 30 min.
Statistical analysis
All data were analyzed with the Graph Pad Software 6.0 (Graph Pad) and expressed as the means ± standard error of the mean (SEM). Data were assessed by analysis of variance. Differences between each group were analyzed by two-tailed unpaired Student’s t-text. More than two groups were compared using one-way analysis of covariance. P < 0.05 was considered statistically significant.
Results
Effect of miltirone on mouse platelet aggregation induced by various agonists
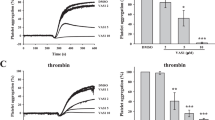
Platelets incubated with miltirone (2, 4, 8 µM) for 5 min exhibited a concentration-dependent inhibitory effect on platelet aggregation induced by collagen (1.0 µg/ml), thrombin (0.06 U/ml), and CRP (0.25 µg/ml). Miltirone showed no inhibitory effects on ADP- and AA-induced platelet aggregation (Fig. 2a, b).

Effects of miltirone on different agonist-induced platelet aggregation of mouse PRP and washed mouse platelets. a PRP (ADP as the agonist, 4 and 10 µM) and washed mouse platelets (3 × 108/ml) were preincubated 5 min with the indicated concentrations of miltirone or vehicle followed by the addition of different kinds of agonists. b The aggregation rates (%) were quantified. Data are presented as mean ± SEM of four independent experiments. *P < 0.05, **P < 0.01, vs. vehicle
Miltirone inhibited collagen- and thrombin-induced platelet granule secretion and integrin αIIbβ3 activation
To assay the effect of miltirone on platelet granule secretion, washed platelets were preincubated with different concentrations of miltirone for 5 min at 37 °C, prior to stimulation by collagen or thrombin. The results showed that miltirone had an inhibitory effect on thrombin and collagen-induced granule release (Fig. 3a). It was able to inhibit P-selectin exposure on the platelet membrane that was caused by collagen stimulation in a concentration-dependent manner. Meantime, the levels of JON/A binding in collagen-stimulated platelets were decreased in the presence of miltirone (Fig. 3b).

The effects of miltirone on ATP release, P-selectin expression, and JON/A binding. a Representative ATP secretion tracing and quantification (%). b Washed mouse platelets suspensions were preincubated with miltirone (2, 4, 8 µM) or vehicle, then FITC-conjugated P-selectin (b1, b2) or PE-conjugated JON/A (b3, b4) were added, followed by collagen (2.0 µg/ml) stimulation. All the values are presented as mean ± SEM of three independent experiments. *P < 0.05, ** P < 0.01, vs. vehicle
Miltirone negatively affects the αIIbβ3-mediated outside-in signaling
After being preincubated with miltirone (4, 8 µM), platelets could not spread on fibrinogen adequately while the control and vehicle group got a full spreading. Most of the platelets treated with miltirone showed a much smaller surface coverage and only a few cells could be observed in filopodia and lamellapodia formation (Fig. 4a, b). Clot retraction was inhibited in the presence of miltrione (2, 4 µM), while the vehicle and control groups showed no change in Fig. 4c. Compared with the vehicle group, clot retraction of platelets treated with miltirone was considerably suppressed.

Miltirone inhibits integrin αIIbβ3-mediated outside-in signaling. a Representative image of platelets spreading on the coated BSA or fibrinogen. b The relative average coverage of spreading platelets in four independent experiments quantified with the control normalized to 100% shown in the left panel. Single adherent platelet area was quantified as shown in the right panel (n = 100). **P < 0.01, ***P < 0.001 compared with the vehicle group. NS means no significance. mil miltirone. c The representative images of clot retraction at 15 min under the intervention of different factors. thr thrombin, fib fibrinogen, mil miltirone. d, e The levels of the phosphorylation of β3/Src/FAK of lysed spreading platelets. All the values are presented as mean ± SEM of three independent experiments. *P < 0.05 compared with the vehicle group
In order to further understand the molecular mechanism of miltirone on the platelets' outside-in signaling, we used miltirone (8 μM) to incubate platelets and investigate the phosphorylation of relative protein in the lysate. First of all, we examined the phosphorylation of β3, which is an indicator that directly reflects the activation of αIIbβ3. Some other molecules, like FAK and Src, are also involved in the outside-in signaling. The results showed that, in the outside-in signaling, the phosphorylation of FAK and Src decreased in the presence of miltirone (Fig. 4d). These data suggest that integrin αIIbβ3-mediated outside-in signaling is remarkably inhibited by miltirone, which plays a vital role in platelet clot retraction and spreading on fibrinogen.
Miltirone inhibited the signaling pathway of platelet GPVI
The abundant subendothelial matrix collagen is an important physiological platelet agonist that initiates the formation of thrombus. In this study, miltirone showed a stronger inhibitory effect on collagen-induced platelet activation [IC50 was 1.21 µM (95% confidence limits 0.798–1.838 µM) vs 2.08 μM (95% confidence limits 1.829–2.369 µM) for collagen and thrombin, respectively] (Fig. 2). To investigate the mechanism of miltirone on inhibiting collagen-induced platelet activation, we tested the phosphorylation of several proteins downstream of GPVI signaling pathway. We found that the phosphorylation of Syk in collagen-induced signaling pathway was not changed, while the phosphorylation of PLCγ2 and PKC substrate (p47) was inhibited by miltirone (Fig. 5).

Miltirone inhibits collagen-induced phosphorylation of signaling molecules. Washed platelets were pretreated with the indicated reagents and stimulated with collagen (1.0 µg/ml) for 3 min and subsequently lysed with lysis buffer followed by immunoblotting using specific antibodies. The blots showed are representative of three independent experiments with similar results. *P< 0.05, **P < 0.01 compared with the control group (n = 3)
The mitogen-activated protein kinase (MAPK) family members, ERK1/2, c-Jun N-terminal kinase (JNK), and p38, in platelets can be activated by a variety of agonists, including collagen. In this study, the phosphorylation of ERK1/2 was inhibited by miltirone; however, the phosphorylation of p38 was enhanced. PI3K is an important part of the downstream of GPVI signaling and plays a crucial role in platelets' activation.
We found that, after preincubation with miltirone, the phosphorylated Akt was inhibited in collagen-activated platelets (Fig. 5). Meanwhile, the phosphorylated glycogen synthase kinase 3β (GSK3β), a substrate of Akt, was restrained by miltirone (Fig. 5). These findings indicate that miltirone might negatively regulate the platelet GPVI signaling.
The antithrombotic activity and the hemostatic parameters of miltirone in vivo
Ferric chloride-induced carotid injury model and pulmonary thromboembolism model were set to establish the influence of miltirone on thrombus formation in vivo. As shown in Fig. 6a, mice treated with miltirone (10 mg/kg) prolonged thrombotic occlusion time in ferric chloride-induced carotid injury model. The occlusion time in mice that received miltirone was 2080 ± 877 s, which was remarkably different from that of the vehicle-treated group (941 ± 450 s). In pulmonary thromboembolism model, mice received miltirone showed a less number of pulmonary vessels associated with thrombi than the mice administered collagen plus epinephrine alone (Fig. 6b). The bleeding time increased slightly when mice were administered with miltirone compared with the vehicle group (Fig. 6c).

Miltirone inhibits thrombus formation in vivo and prolonged bleeding time. a Occlusion time (s) following ferric chloride-induced injury of the carotid artery. Carotid artery occlusion time was measured after ferric chloride-induced injury using doppler flow probe. The occlusion time was compared between the miltirone-treated mice and control group (n = 6). **P < 0.01, compared with model group. b Light microscopic images of H&E-stained lung sections from mice administered mixture of collagen (1000 μg/kg) plus epinephrine (10 μg/kg) plus vehicle and mixture of collagen (1000 μg/kg) plus epinephrine (10 μg/kg) plus miltiorne (10 mg/kg) (n = 4). White arrows point to pulmonary thrombus. c The effect of miltirone on bleeding time. The bleeding time was recorded from the time the distal portion of the tail was amputated to the time when the bleeding was completely stopped. When the bleeding time was >30 min, we recorded that as 30 min (vehicle n = 7; miltirone n = 8). *P < 0.05, compared with the vehicle group
Discussion
Searching for other anti-platelet ingredients in Salvia miltiorrhiza is beneficial for further development of the traditional Chinese medicine and is more conducive to the prevention and treatment of cardiovascular disease. We found that miltirone inhibited collagen-, CRP-, and thrombin-induced platelet aggregation; the release of α granule and dense granule, αIIbβ3 activation, platelet spreading, and clot retraction, as well as attenuated thrombus formation as evaluated by ferric chloride-induced carotid injury model and pulmonary thromboembolism model. Mechanistically, biochemical assay showed that the phosphorylation of molecules involved in PLCγ2-PKC/MAPK and PI3K-Akt-GSK3β signaling pathway downstream of GPVI and αIIbβ3-mediated Src-FAK signaling pathway were interfered by miltirone. Additionally, tail bleeding assay showed that miltiorne had a slightly bleeding risk.
Platelets play a key role in thrombosis, and the aggregation function of platelets is involved in thrombus formation [20]. The main targets of the commercially available anti-platelet medicine include ADP receptors, integrin αIIbβ3, and cyclooxygenase but without GPVI and PAR. Upon GPVI activation, the adjacent Src family kinases will initiate the downstream Syk/PLCγ2/PKC signaling [24,25,26]. Miltirone inhibited collagen- and CRP-induced platelet activation in a concentration-dependent manner. When platelets were stimulated by collagen, 2 µM miltirone got a greater inhibition of aggregation than that in thrombin-stimulated platelet (IC50 was 1.21 μM vs 2.08 μM for collagen and thrombin, respectively). This implies that miltirone possesses a stronger suppression effect on collagen- and thrombin- induced aggregation. However, AA and ADP-induced aggregation could not be inhibited by miltirone. It shows that miltirone has potential specificity for collagen and thrombin receptors, especially for collagen receptors. Because of the favorable role of miltirone on the inhibition of collagen-induced aggregation, this study focused on the GPVI signaling to study the possible mechanisms of miltirone on anti-platelet function.
Platelets contain three kinds of granules: α granules, dense granules, and lysosomes [27]. When platelets are activated, the released granules promote platelet activation in hemostasis, thrombosis, and other pathophysiology process [28]. Platelet α-granules contain abundant proteins, such as P-selectin, fibrinogen, and vWF. Dense granules, however, are rich in ADP, ATP, and 5-HT. The degranulation of platelets makes the release of the above substances to further strengthen the hemostatic effect of platelet. When platelet was activated, the intracellular P-selectin on α-granules membrane was flipped and exposed to the platelet membrane, accompanied by the release of dense granules. The release of platelet dense granules and α-granules were reflected by the releasing of ATP and expression of P-selectin on the cell membrane, respectively. Miltirone inhibited P-selectin expression and ATP release (Fig. 3a). It suggests that miltirone inhibits platelet release function. The αIIbβ3 binding was decreased by miltirone, which suggests that miltirone play an inhibitory role in the inside-out signaling.
After binding to fibrinogen, activated integrin αIIbβ3 are clustered and transforming signaling (outside-in signaling) into platelets, thus initiating platelet structural transformation and the following spreading [29]. The activated αIIbβ3 binding to fibrinogen initiates the outside-in signaling, which synergies together with the inside-out signaling to promote platelet cytoskeletal reorganization and morphological changes, and this process eventually causes platelet spreading and clot retraction [30]. The process will accelerate the formation of stable thrombus. In the early events of outside-in signaling, the β3 subunit of integrin αIIbβ3 can be phosphorylated to result in phosphorylation of Src (Y416), which further leads to phosphorylation of downstream FAK and other proteins [31]. Miltirone inhibited platelet spreading on fibrinogen and clot retraction, which is vital in antithrombosis. Miltirone did not inhibit β3 phosphorylation, but it actually reduced Src and FAK phosphorylation. This indicates that miltirone possesses a significant inhibitory effect on the outside-in signaling.
MAPK is a member of the serine/threonine protein kinase family, which includes ERKs, JNKs, and p38. Among them, ERK1/2, p38 and JNK1 were reported in platelets [32,33,34]. Many kinds of agonists such as collagen, thrombin, and ADP are capable of activating them, whereas their specific roles in platelets are still controversial [34]. ERK1/2 and p38 play crucial roles in platelet activation, while the function of JNK in platelet is poorly defined at present [34]. In consequence, this research mainly studied the effect of miltirone on ERK1/2 and p38 phosphorylation in collagen-induced platelet activation. The result showed that miltirone inhibited ERK1/2 phosphorylation; however it did not inhibit the phosphorylation of p38, even promoted it. But miltirone inhibited platelet aggregation in this study. This may be due to its anti-platelet effects covering the activation role of p38. In addition, as a substrate of p38, cPLA2 phosphorylation increases the production of AA, which is metabolized by cyclooxygenase. AA could be metabolized by cyclooxygenase to generate prostaglandins and its inducing platelet apoptosis function makes platelet could not aggregate effectively [35, 36].
In growing thrombi, the adherent and aggregated platelets play a great role in the process. A suitable arterial thrombus formation model has been considered a classical way to evaluate the antithrombotic activities of active ingredients. The ferric chloride-induced artery injury is a well-established technique to induce the formation of thrombi. Our studies showed that miltirone markedly prolonged the occlusion time. It indicated that milrione has an antithrombotic effect. Due to the great volatility of ferric chloride-induced thrombosis assay, we established a pulmonary thromboembolism model which has been widely used to test the antithrombotic roles of pharmacological ingredients. The results confirmed the same conclusion with ferric chloride-induced artery injury models that milrione has an antithrombotic effect.
In summary, the research showed that miltirone potently inhibited platelet aggregation and release. Besides, it also inhibited platelet adhesion, spreading on fibrinogen, and clot retraction. The results indicated that it suppressed platelet inside-out and outside-in signaling. The underlying mechanism is achieved by inhibiting Syk-PLCγ2-PKC/MAPK and PI3K-Akt-GSK3β signaling downstream of the GPVI signaling cascades. In addition, miltirone has an antithrombotic effect. Thus the research suggests that miltirone might be another potently effective anti-platelet component of Salvia miltiorrhiza and it will be a potential anti-platelet candidate for the prevention of thrombotic disorders.
References
Holinstat M. Normal platelet function. Cancer Metastasis Rev. 2017;36:195–8.
Joshi S, Whiteheart SW. The nuts and bolts of the platelet release reaction. Platelets. 2017;28:129–37.
Yeung J, Holinstat M. Newer agents in antiplatelet therapy: a review. J Blood Med. 2012;3:33–42.
Bryniarski L, Pelc-Nowicka A, Zabojszcz M, Mirek-Bryniarska E. Dual anti-platelet therapy and antithrombotic treatment: recommendations and controversies. Cardiol J. 2009;16:179–89.
Guthrie R. Review and management of side effects associated with antiplatelet therapy for prevention of recurrent cerebrovascular events. Adv Ther. 2011;28:473–82.
Alexopoulos D, Bhatt DL, Hamm CW, Steg PG, Stone GW. Early P2Y12 inhibition in ST-segment elevation myocardial infarction: Bridging the gap. Am Heart J. 2015;170:3–12.
Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet. 2006;367:606–17.
Siddique A, Butt M, Shantsila E, Lip GYH. New antiplatelet drugs: beyond aspirin and clopidogrel. Int J Clin Pract. 2009;63:776–89.
Coccheri S. Antiplatelet drugs-do we need new options? With a reappraisal direct thromboxane inhibitors. Drugs. 2010;70:887–908.
Betz JM, Brown PN, Roman MC. Accuracy, precision, and reliability of chemical measurements in natural products research. Fitoterapia. 2011;82:44–52.
Valli G, Giardina EGV. Benefits, adverse effects and drug interactions of herbal therapies with cardiovascular effects. J Am Coll Cardiol. 2002;39:1083–95.
Maione F, De Feo V, Caiazzo E, De Martino L, Cicala C, Mascolo N. Tanshinone IIA, a major component of Salvia milthorriza Bunge, inhibits platelet activation via Erk-2 signaling pathway. J Ethnopharmacol. 2014;155:1236–42.
Liu J-Q, Lee T-F, Miedzyblocki M, Chan GCF, Bigam DL, Cheung P-Y. Effects oftanshinone IIA, a major component ofSalvia miltiorrhiza, on platelet aggregation in healthy newborn piglets. J Ethnopharmacol. 2011;137:44–9.
Wang H, Gu J, Hou X, Chen J, Yang N, Liu Y, et al. Anti-inflammatory effect of miltirone on inflammatory bowel disease via TLR4/NF-kappaB/I0GAP2 signaling pathway. Biomed Pharm. 2017;85:531–40.
Zhou L, Jiang L, Xu M, Liu Q, Gao N, Li P, et al. Miltirone exhibits antileukemic activity by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction pathways. Sci Rep. 2016;6:20585.
Wang L, Hu T, Shen J, Zhang L, Li LF, Chan RL, et al. Miltirone induced mitochondrial dysfunction and ROS-dependent apoptosis in colon cancer cells. Life Sci. 2016;151:224–34.
Colombo G, Serra S, Vacca G, Orru A, Maccioni P, Morazzoni P, et al. Identification of miltirone as active ingredient of Salvia mil-tiorrhiza responsible for the reducing effect of root extracts on alcohol intake in rats. Alcohol Clin Exp Res. 2006;30:754–62.
Liu G, Xie W, He AD, Da XW, Liang ML, Yao GQ, et al. Antiplatelet activity of chrysin via inhibiting platelet alphaIIbbeta3-mediated signaling pathway. Mol. Nutr. Food Res. 2016;60:1984–93.
Liang ML, Da XW, He AD, Yao GQ, Xie W, Liu G, et al. Pentamethylquercetin (PM0) reduces thrombus formation by inhibiting platelet function. Sci Rep. 2015;5:11142.
Vaiyapuri S, Roweth H, Ali MS, Unsworth AJ, Stainer AR, Flora GD, et al. Pharmacological actions of nobiletin in the modulation of platelet function. BJP. 2015;172:4133–45.
Li W, Tang X, Yi W, Li Q, Ren L, Liu X, et al. Glaucocalyxin A inhibits platelet activation and thrombus formation preferentially via GPVI signaling pathway. PLoS One. 2013;8:e85120.
Sonkar VK, Kulkarni PP, Dash D. Amyloid beta peptide stimulates platelet activation through RhoA-dependent modulation of actomyosin organization. FASEB J. 2014;28:1819–29.
Tyagi T, Ahmad S, Gupta N, Sahu A, Ahmad Y, Nair V, et al. Altered expression ofplatelet proteins and calpain activitymediate hypoxia-induced prothrombotic phenotype. Blood. 2014;123:1250–60.
Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost. 2005;3:1752–62.
Su XL, Su W, Wang Y, Wang YH, Ming X, Kong Y. The pyrrolidinoindoline alkaloid Psm2 inhibits platelet aggregation and thrombus formation by affecting PI3K/Akt signaling. Acta Pharmacol Sin. 2016;37:1208–17.
Watson SP, Herbert JM, Pollitt AY. GPVI and CLEC-2 in hemostasis and vascular integrity. J Thromb Haemost. 2010;8:1456–67.
Eckly A, Rinckel JY, Proamer F, Ulas N, Joshi S, Whiteheart SW, et al. Respective contributions of single and compound granule fusion to secretion byactivated platelets. Blood. 2016;128:2538–49.
Heijnen H, van der Sluijs P. Platelet secretory behaviour: as diverse as the granules.. or not? J Thromb Haemost. 2015;13:2141–51.
Calderwood DA. Integrin activation. J Cell Sci. 2004;117:657–66.
Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. 2012;24:600–6.
Phillips DR, Nannizzi-Alamio L, Prasad KSS. Beta 3 tyrosine phosphoryla-tion in alpha IIb beta 3 (platelet membrane GP IIb-IIIa) outside-in integrin signaling. Thromb Haemost. 2001;86:246–58.
Qi H, Huang Y, Yang Y, Dou G, Wan F, Zhang W, et al. Anti-platelet activity of panaxatriol saponins is mediated by suppression of intracellular calcium mobilization and ERK2/p38 activation. BMC Complement Altern Med. 2016;16:174.
Borst O, Walker B, Munzer P, Russo A, Schmid E, Faggio C, et al. Skepinone-L, a novel potent and highly selective inhibitor of p38 MAP kinase, effectively impairs platelet activation and thrombus formation. Cell Physiol Biochem. 2013;31:914–24.
Adam F, Kauskot A, Rosa JP, Bryckaert M. Mitogen-activated protein kinases in hemostasis and thrombosis. J Thromb Haemost. 2008;6:2007–16.
Kuliopulos A, Mohanlal R, Covic L. Effect of selective inhibition of the p38 MAP kinase pathway on platelet aggregation. Thromb Haemost. 2004;92:1387–93.
Saklatvala J, Rawlinson L, Waller RJ, Sarsfield S, Lee JC, Morton LF, et al. Role for p38 mitogen-activated protein kinase in platelet aggregation caused by collagen or a thromboxane analogue. J Biol Chem. 1996;271:6586–9.
Acknowledgements
This study was supported by grants from The National Natural Science Fundation of China (No. 81273574 to Z.-Y.M.) and Chinese herb key project by Health and Family Planning Commission of Hubei Province (to Z.-Y.M.)
Author information
Authors and Affiliations
Contributions
Z.-Y.M. and W.S. designed the study and wrote the manuscript. Y.-y.M. and S.M. prepared mouse platelets. R.-p.Y. and Y.Z. performed the evaluation of platelet function and analyzed data. All authors reviewed the manuscript. D.S., M.L., and R.M.A. approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Song, W., Ma, Yy., Miao, S. et al. Pharmacological actions of miltirone in the modulation of platelet function. Acta Pharmacol Sin 40, 199–207 (2019). https://doi.org/10.1038/s41401-018-0010-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0010-1
Keywords
This article is cited by
-
Fruitflow inhibits platelet function by suppressing Akt/GSK3β, Syk/PLCγ2 and p38 MAPK phosphorylation in collagen-stimulated platelets
BMC Complementary Medicine and Therapies (2022)
-
Pyroptosis and pyroptosis-inducing cancer drugs
Acta Pharmacologica Sinica (2022)
-
Cytotoxicity, apoptosis inducing activity and Western blot analysis of tanshinone derivatives from Stachys parviflora on prostate and breast cancer cells
Molecular Biology Reports (2022)
