
Abstract
Schizophrenia is a psychiatric disorder with significant impact on individuals and society. The current pharmacologic treatment, which principally alleviates psychosis, is focused on neurotransmitters modulation, relying on drugs with severe side effects and ineffectiveness in a significant percentage of cases. Therefore, and due to difficulties inherent to diagnosis and treatment, it is vital to reassess alternative cellular and molecular drug targets. Distinct risk factors – genetic, developmental, epigenetic, and environmental – have been associated with disease onset and progression, giving rise to the proposal of different pathophysiological mechanisms and putative pharmacological targets. Immunity is involved and, particularly microglia – innate immune cells of the central nervous system, critically involved in brain development – have captured attention as cellular players. Microglia undergo marked morphologic and functional alterations in the human disease, as well as in animal models of schizophrenia, as reported in several original papers. We cluster the main findings of clinical studies by groups of patients: (1) at ultra-high risk of psychosis, (2) with a first episode of psychosis or recent-onset schizophrenia, and (3) with chronic schizophrenia; in translational studies, we highlight the time window of appearance of particular microglia alterations in the most well studied animal model in the field (maternal immune activation). The organization of clinical and translational findings based on schizophrenia-associated microglia changes in different phases of the disease course may help defining a temporal pattern of microglia changes and may drive the design of novel therapeutic strategies.

Similar content being viewed by others

Introduction
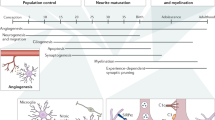
Schizophrenia is a chronic multifactorial mental disorder, presenting variable clinical manifestations. Patients manifest different symptoms, classified as positive (e.g. delusions, hallucinations), negative (e.g. social withdrawal, self-neglect, avolition, and loss of motivation) and/or cognitive (e.g. working memory and attention deficits) [1]. The first symptoms usually manifest during late adolescence and early adulthood and are, in the majority of cases, depressive and negative symptoms (in a minority of cases, psychotic symptoms are the first to appear) [2]. Due to the scarce symptomatology until the occurrence of the first episode of psychosis, if we consider the chronopathology of schizophrenia, the diagnosis is late and patients already present substantial brain changes, in terms of structure, neurochemistry, and connectivity [3]. Based on schizophrenia onset and progression, three stages are established: (1) latent, pre-morbid – implicates biological processes intrinsic to brain development and maturation and extends from the prenatal period to childhood; (2) high-risk, prodromal – usually initiates in adolescence, hampering brain reorganization (e.g. sexual maturation) and (3) chronic, fluctuating – occurs at adulthood (for more details see ref. [3]; Fig. 1).

(1) latent, pre-morbid (from the prenatal period until childhood) implicates biological processes intrinsic to brain development and maturation; (2) high-risk, prodromal (usually initiates in adolescence) - impacts on brain reorganization (e.g. sexual maturation); and (3) chronic, fluctuating (adulthood).
In addition to the diversity in clinical presentation, the identified risk factors (from development and genetic to environmental and epigenetic) also reflect the involvement of several biological systems in the genesis and progression of the disease [4, 5]. In consequence, several theories (focused on environmental and genetic factors, neurochemical and neuroanatomical abnormalities [6,7,8]) have emerged in an attempt to clarify the biological basis of schizophrenia. Objectively, if we consider the drugs currently used in the clinical practice (different generations of modulators of the subtype D2 of dopamine receptors, modified over the years in order to attenuate severe side effects associated with D2 receptor blockade [9]), dopamine hypothesis is the most well accepted theory. It postulates an (1) excess of dopamine transmission in subcortical limbic areas, such as the nucleus accumbens (NAc), amygdala (AMY), and hippocampus (HIP) (mesolimbic pathway) – responsible for the positive symptoms [10, 11] - and a (2) dopamine deficiency in the prefrontal cortex (PFC; mesocortical pathway) – responsible for the negative and cognitive symptoms [12] (Fig. 2). Despite the unquestionable beneficial effects, antipsychotics still present important limitations, including behavioral, neurological (namely extrapyramidal effects), hematological, cardiovascular, metabolic, and endocrine (weight gain, hyperlipidemia, and diabetes mellitus) adverse effects [7, 13,14,15,16]. The identification of alternative cellular and molecular drug targets would fill the gap that still remains in schizophrenia treatment.

Schematic representation of hypofunctional (decreased dopamine transmission to the prefrontal cortex (PFC)) and hyperfunctional (increased dopamine transmission to nucleus accumbens (NAc), amygdala (AMY) and hippocampus (HIP).
Over the past years, immunity has been implicated in the genesis, progression and clinical manifestations of psychiatric disorders, including schizophrenia (for review [17,18,19,20]). Although out of the scope of this review, we cannot afford to mention the immunomodulatory properties of antipsychotic drugs [21, 22], namely the ability to normalize altered levels of inflammatory mediators, reported as a mechanism underlying the remission of psychotic symptoms [23].
Innate immunity involves different cells able to mount an inflammatory response to insults, including microglia, which colonize the central nervous system during early neurodevelopment. Throughout life and from the very beginning of brain development, these innate immune cells perform an essential function – synapse pruning, which involves the phagocytosis of synapses [24,25,26]. In the past, it has been proposed that an aberrant synaptic pruning occurs in schizophrenia [4, 27,28,29], raising the idea that microglia could be involved in its genesis and progression. Notably, the effect of antipsychotics on microglia (for a review see ref. [30]) is not well established, a nuclear question to clarify if drug efficacy is related with the modulation of disease-associated microglia sequelae. The aim of this review is not to summarize the inflammatory-immune hallmarks of schizophrenia, but rather focused on disease-associated changes of this specific cellular candidate – microglia. These cells are in tight communication with the first newborn neurons, being sensitive and responsive to neurochemical signs (reviewed in refs. [31, 32]), exerting a crucial modulatory role in the formation/maturation of neurons and neuronal circuits in critical periods of brain development, and later, contributing to brain homeostasis. We here gather clinical and translational findings related with microglial changes (sequelae) in schizophrenia.
Microglia in brain development and homeostasis: kickoff to schizophrenia
Microglia are immune cells derived from embryonic yolk-sac myeloid progenitors. After the colonization of the brain (early in the embryonic period, before the completion of blood-brain barriers) [33], microglia migrate towards different brain regions, proliferate and mature at different paces from species to species. After the initial colonization, self-renewal by proliferation is accepted as the only mechanism of repopulation throughout life [34].
These heterogeneous cells are categorized in subpopulations, according to the expression of particular gene clusters, and present distinct transcriptomic profiles with brain region specificity, that change over time to functionally respond to particular demands of different developmental phases [34,35,36]. Microglia also present a panoply of morphologic phenotypes, ranging from amoeboid to varied degrees of cellular processes ramification [37]. In response to different stimuli, a notable ability to change from amoeboid to ramified phenotypes (and vice-versa) is granted by cellular processes well adapted to the surveillance of brain parenchyma, through dynamic extension and retraction mechanisms [38, 39]. Importantly, microglia do not transit only between a “healthy” ramified and a “pathological” amoeboid phenotype, as initially believed. Instead, microglia undergo morphological adaptions, depending on their brain location and stimuli, a topic that our group has been studying in the context of mental diseases [40,41,42].
Due to their phagocytic ability and capacity to produce and release inflammatory mediators and growth factors, microglia actively contribute to the proper establishment and maturation of neural circuits. Microglia can stimulate neurogenesis by supporting survival, proliferation, and maturation of neuronal progenitor cells (NPCs) and neurons [43]) or, instead, eliminate neurons, NPCs or apoptotic cells by phagocytosis (reviewed in refs. [32, 44]).
The settlement between elimination or strengthening depends on microglia-neuron communication, that occurs through the release of soluble factors [45] or physical contact [31, 46]. Distinct signaling pathways, involving fractalkine/fractalkine receptor (CX3CL1/CX3CR1), OX-2 membrane glycoprotein/OX-2 membrane glycoprotein receptor (CD200/CD200R), Toll-like receptors (TLRs), cytokines, complement pathways (complement component 1q/complement component 1q receptor – C1q/C1qR, complement component 3/complement component 3 receptor – C3/C3R), extracellular vesicles, among others [47,48,49], mediate neuron-microglia interactions.
Microglia is able to eliminate or strengthen cells and subcellular domains, namely synapses [24], a function of high relevance in the onset of schizophrenia, proposed to be coincident with an aberrant process of synapse pruning [50, 51], as firstly reported by Feinberg [52]. In schizophrenia, the complement system is a key intervenient in synapse elimination by microglia, in particular the components C4 and C3. The former, which is increased in the hippocampus of patients (in neurons and synapses) [53], activates C3, that binds to a receptor exclusively expressed by myeloid cells, including microglia [26, 54, 55]. Other authors demonstrated that C4 variants associated with schizophrenia, increase C3 deposition in neurons [51], synaptic engulfment by microglia [51, 56] and reduce the connectivity in the PFC of prepubertal rats [56].
This group of findings clearly points to the involvement of the complement pathway, further supporting the proposed theory of excessive synaptic pruning in the genesis of schizophrenia [50]. If this represents one of the triggering events of the disease, the pharmacologic modulation of the complement system emerges as a potential therapeutic approach. However, as previously mentioned, diagnosis is usually subsequent to the first episode of psychosis, when substantial changes already occurred in the brain, including cellular plastic events of microglia, eventually contributing to disease progression. To clearly define microglia as a cellular target in the disease, one must know the nature and the time of appearance of specific microglial changes with disease progression. In the following sections, we reunite data from clinical and non-clinical studies focused on microglia sequelae.
Microglia sequelae: clinical findings in SCZ patients
Several epidemiological studies have shown that changes in the immune system are linked to psychosis and, therefore, to schizophrenia pathophysiology [57,58,59,60]. Most studies describe alterations in cellular and/or molecular mediators assessed in patient blood, rather than immune changes in the brain. The detection of peripheral immune changes is a more feasible and low-cost approach, that may help finding novel biomarkers of disease, improving diagnosis and progression monitoring. For instance, a correlation was established between increased levels of interleukine-6 in serum (measured at 9 years old) with a twofold increase risk of developing psychotic symptoms at 18 years [61]. Although aware of the importance of peripheral immune changes that may have the contribution of factors produced and released by microglia (properly reviewed elsewhere, e.g. [17,18,19,20]), the main goal of the present article is to review changes exclusively related with this particular type of immune cells of the central nervous system.
Here, we organized the clinical studies in two subsections, neuroimaging techniques, that allow in vivo monitorization and longitudinal analyses, and post-mortem assessments, a complementary approach that helps finding molecular and cellular pathophysiological traits of the disease.
Neuroimaging approaches as a readout for neuroinflammation mediated by microglia
In the last years, new techniques and methodologies granted significative advances in neuroimaging. Even though, there is a lack of reliable markers to properly evaluate microglia. The available methods mainly target a mitochondrial protein (18-kDa translocator protein, TSPO) highly expressed in activated microglia, that is, involved in an inflammatory response to pathological conditions, such as psychiatric diseases [62]. Thus, the measurement of TSPO levels through the use of radiotracers has been commonly performed to investigate microglia activation in SCZ patients [63], although we would like to emphasize that TSPO is not exclusively expressed in microglial cells [64] and its expression does not correlate with the expression of canonical activation markers [65]. Anyway, we consider of importance to reunite and carefully analyze clinical findings based on TSPO evaluation to finally take objective conclusions about the validity of considering TSPO as a readout of microglia-mediated neuroinflammation (see Table 1).
To better understand the potential conflicting results between studies, it is important to consider some bias related to TSPO radiotracers, starting from the binding affinity of the radioligands, that ranges from low (for first generation radiotracers) to high (for second-generation radiotracers), which potentially influences the analysis. Besides, the binding of some radiotracers is conditioned by a common genetic polymorphism in exon 4 of the TSPO gene (rs6971), resulting in distinct TSPO binding profiles (for more details see ref. [66]). The methods applied to calculate total/regional TSPO binding also vary among studies: simplified reference tissue model (method mostly used for 11C-PK11195 radiotracer) [67,68,69,70]; two-tissue component model (2TCM) with metabolite-corrected arterial plasma curve as input function (11C-PK11195 [71, 72] or second-generation radiotracer [64, 71,72,73,74,75,76,77,78]) and/or two-tissue compartmental model accounting for endothelial vascular TSPO binding (2TCM-1K) [74,75,76, 79]. Finally, schizophrenia itself accounts for important confounding factors, such as cohorts heterogeneity, medication, stage of disease progression, to mention a few (see review [80]).
For the sake of clarity, we divided neuroimaging studies in three groups of patients in different stages of the disease: (1) ultra-high risk of psychosis, (2) first episode of psychosis (FEP) or recent-onset schizophrenia and (3) chronic schizophrenia.
Patients at ultra-high risk of psychosis
In the literature, we only found two studies exploring TSPO binding in patients at ultra-high risk of psychosis. Curiously, normal values of TSPO tracing were described when a low binding affinity radiotracer (11C-PK11195) was used (the low affinity may hypothetically limit the detection of subtle changes) [67]. When a second-generation radiotracer (11C-PBR28) was used, the authors established a correlation between increased TSPO tracing and the severity of symptoms [75]. It is important to emphasize that, in addition to differences in the affinity of radiotracers and methodologic approaches, the conclusions were supported by observations in different brain regions.
Patients with FEP or recent-onset schizophrenia
As described in patients at risk of psychosis, in this group of patients with FEP, the results are conflicting, with three main studies describing an increase [76], a decrease [74] or the absence of changes [73] in TSPO tracing. Again, besides differences in radiotracer affinities, the heterogeneity of the individuals involved in the studies may influence the results, as occurs with the study reporting an increase in TSPO [76], that includes patients with an acute relapse, that likely present exacerbated microglia-mediated neuroinflammation. The complexity of the analysis increases if we segregate patients under medication and not treated, since pharmacotherapy is an important confounding effect, mainly considering the immune modulatory effect of antipsychotics, as previously mentioned. Normal TSPO was observed in a cohort under antipsychotic medication [73], whereas decreased TSPO levels were found in non-treated FEP patients [74] or recent-onset schizophrenia [69].
In patients with recent-onset schizophrenia under antipsychotic treatment, most studies indicate the absence of neuroinflammation, as assessed by TSPO evaluation [67, 69, 70, 77]. Even though, we were able to find two studies reporting abnormal TSPO levels [64, 71].
Patients with chronic schizophrenia
Studies including patients with chronic schizophrenia, that are usually under antipsychotic treatment, report increases [68, 72, 75] or normal levels of TSPO [67, 69, 78]. In this case, one cannot justify the absence of TSPO changes with the use of low affinity radiotracers, since the use of the low affinity 11C-PK11195 radiotracer did not allow the detection of changes in two studies [67, 69], but detected increased TSPO expression in another study/cohort [72].
In summary, the available published studies applying TSPO tracing, are not sufficient to correlate disease progression and/or severity of symptoms with microglia-mediated inflammation. One cannot exclude the existence of a correlation, simply because the studies were not designed taking into account the influence of strong bias, from the stage of the disease, the severity of symptoms or the influence of medication (a problem even more important if we consider that different classes of drugs with potential different immune modulatory effects exist). Efforts have been made to circumvent this heterogeneity, increasing the level of patient stratification; however, stratification based on highly selective criteria implies the inclusion of higher number of individuals, which constitutes a serious limitation in clinical practice.
Schizophrenia-associated microglia alterations revealed by post-mortem studies
Post-mortem analysis is an important tool to identify and characterize changes in microglia that are not possible to detect in live patients, although one must be aware of important confounding factors, namely the cause of death, co-morbidities, medication, lack of knowledge of disease stage, and/or symptoms predominance, to mention a few. Some studies do not mention the stage or the predominant symptomatology and only refer the involvement of patients with a diagnosis [65, 81,82,83] or chronic schizophrenia [84,85,86,87,88]. The lack of information about disease staging led us to organize post-mortem data by brain region (as a summary, please see Table 2).
Glia plasticity, in general, and microglia plasticity, in particular, have been a matter of debate in the fields of basic neurosciences and psychiatry/neurology. The concept refers to the ability of microglia to undergo adaptative changes at different levels, namely in cell density, morphology and/or function, in response to physiological or pathological stimuli. For instance, alterations in the number of microglial cells may reflect a proliferative status, that typically occurs in non-physiological conditions (with the exception of normal brain development, characterized in certain phases by intense microglia proliferation). The morphological remodeling of microglia is also a biologic finding of several diseases and, interestingly, pre-clinic studies point towards a correlation between symptoms amelioration and the recovery of the healthy, normal morphology of microglia by pharmacologic interventions [40, 89]. One of the advantages of post-mortem analyses is precisely the possibility of performing a characterization of the so-called disease-associated microglia (DAM), as described in the context of neurodegenerative diseases [90]. Typically, the presence of amoeboid microglia was taken for decades as an index of activation, usually implicated in the response to an injury or brain disease. So, several studies in the literature report microglia changes in schizophrenia patients, based on the dual analysis of two phenotypes, ramified (resting) or amoeboid (activated) cells. This approach has been gradually replaced by more sophisticated morphometric methodologies, that include the determination of morphologic parameters considered relevant for microglia function, such as the degree of ramification or the brain area occupied per cell (hypothetically related with the efficacy of immune surveillance). We have also contributed to the finding that microglia present distinct regional morphologic phenotypes in the brain and that the morphologic remodeling in pathologic conditions is also different according to the brain region under analysis [40,41,42, 89, 91].
Besides the characterization of microglia morphology, post-mortem studies allow the quantitative analysis of parameters that may reflect the functional state of microglia, namely the local levels of inflammatory mediators (although other cells in the brain are able to produce and release these substances), enzymes involved in the inflammatory response operated by microglia, among others, that will be presented throughout this and the next sections. The data obtained deserve careful interpretation, as the readouts of cellular physiology are, as expected, seriously affected by the process of cell death and the underlying lesion.
Considering the regional segregation of microglia, we will then review the main findings per brain region, in particular because schizophrenia-associated microglia phenotypes were recently described in schizophrenia patients (increase of microglia soma size and decrease of arborization) [86].
Prefrontal region
Starting with the analysis of data collected in the frontal cortex, several studies report an increase in the density of microglia [84,85,86, 92, 93]. We found one study where, in general, no changes were detected, but reporting an increase in the number of activated microglia in three patients (without convincing justification/discussion) [81]. One of the referred studies also demonstrates the increase of cytokine levels [93] and other the integrity of the phagocytic ability of microglia [87]. In opposition, a recent meta-analysis [82] stands for the absence of changes in microglia density in the frontal cortex from schizophrenia post-mortem tissue. Of note, this meta-analysis included all studies with patients with a diagnosis, without considering the disease stage, symptoms predominance and other bias, that must be taken in consideration, as suggested by a study claiming for a correlation between the increased number of amoeboid microglia and the predominance of positive symptoms [92]. Other factor that may feed the controversy is the effect of lateralization (differences between hemispheres); it was recently observed, although specifically in the cingulated cortex, a higher density of microglia in the right hemisphere, as compared with the left one [83]. Finally, some authors evaluate microglia transcripts in the frontal cortex (particularly in the middle frontal gyrus), but no changes were detected, namely in TSPO labeling or in genes associated with microglia in physiologic (Allograft inflammatory factor 1 - AIF1, Integrin alpha M - ITGAM) or activated state (Cluster of Differentiation 163 - CD163, HLA-DRA, interleukin (IL)-1β) [65]. Nevertheless, a down-regulation of CX3CR1 ligand (CX3CL1) [94], mainly released by neurons, and its receptor (CX3CR1), present in microglia [95], was found in this brain region, suggesting a compromise of neuron-microglia communication.
Temporal region
In the temporal region, the evidences gathered so far are consistent, converging for an increase of microglia density [82, 84, 86] accompanied by a decrease in several transcripts (e.g. CX3CR1, Transmembrane Protein 119 - TMEM 119, Purinergic Receptor P2Y12 - P2RY12, HLA class II histocompatibility antigen, DR alpha chain - HLA-DRA, Colony stimulating factor 1 receptor - CSFR1) [82].
Subcortical regions
In subcortical regions, we found two reports about microglia transcripts in schizophrenia patients, one claiming for no changes in the corpus callosum [65] and the other showing an increase of several transcripts in the substantia nigra [88]. In the hippocampus, we found a very interesting finding on the relation between symptoms and microglia changes: the authors observe higher microglia numbers in patients with predominant psychotic/positive symptoms, as compared with patients with predominant negative symptoms [96]. This very relevant observation is clearly related with other finding by Purves-Tyson in 2019, that failed to find transcriptomic changes in schizophrenia patients until their stratification, in the case, by splitting the group in high and low immune biotypes [88]. The increased levels of AIF-1, CD68, and TSPO transcripts was only detectable in the subgroup of high immune biotype [88], whereas increased numbers of T and B lymphocytes were detected in schizophrenia with a different clinical presentation, categorized as paranoid [96], reinforcing the contribution of the peripheral immune system in particular contexts of the disease [17, 18]. In line with this concept, it was reported the activation of microglial cells in culture by exposure to serum samples from recent-onset schizophrenia patients [97], proving the responsiveness of microglia to peripheral signals and strengthening the interest of these cells as targets in the context of schizophrenia pharmacologic treatment.
Microglia sequelae: findings from SCZ animal models
Animal models, although unable to replicate human diseases, are valuable tools in the study of neurobiological basis, mainly in the case of brain, that is of particularly difficult access. Despite the obvious limitations of animal models, such as the absence of symptoms of human schizophrenia (e.g., hallucinations, delusions), the fact is that several neurochemical changes, behavioral alterations, and biomarkers accomplish validity criteria to address pathophysiological mechanisms and to test novel therapeutic tools [98].
Although the etiology of schizophrenia is not fully understood, it is known that gestation is a period particularly sensitive, namely to genetic and environmental risk factors. In the literature, studies on microglia alterations in schizophrenia are mainly described in a model of maternal immune activation, the reason why this model is the most mentioned along this section. This animal model consists in the administration of an agonist of the Toll-like receptor 3, poly I:C (polyinosinic:polycytidylic acid) to pregnant rodents. Several protocols are found in the literature, namely concerning the gestational time window of administration (early gestation: 8–9.5 days; mild gestation: 12.5–13.5; mild-late gestation: 14.5–15 days; late gestation: 16.5–18.5 days). These differences in the protocol have a tremendous impact on brain development and require cautious interpretation of results. Adult descendants develop neuroanatomical, neurochemical and behavioral changes, some replicating disease traits found in schizophrenia patients, such as dopamine hyperactivity, ventricles enlargement, social and cognitive deficits and repetitive behaviors, that are clearly dependent of the gestational period of administration (for more details see [99]). In this article, we organize data related with microglia changes, mainly in poly I:C model (poly I:C - Table 3; others - Table 4), covering different periods of life (prenatal period, postnatal period, adolescence and adulthood; the main findings are summarized in Fig. 3). This chronological organization of microglia alterations (even aware of the importance of the time of exposure to poly I:C) may help identifying precise period(s) of microglia changes and, thus, of therapeutic modulation, hopefully as precociously as possible in the trajectory of the disease.

The alterations are organized in four periods of development: prenatal, neonatal, adolescence and adulthood. Titles in capital letters evidence the main alteration(s) observed at a particular period of life. ED embryonic day, PND postnatal day, MG microglia, C cortex, HIP hippocampus, SCC supraventricular cortex, CC corpus callosum, PFC prefrontal cortex.?imprecise time window of appearance of microglia alterations. 1evidences from other SCZ animal models.
Prenatal period
In the prenatal period, microglia are actively proliferating and migrating to the appropriate regions of the brain [100]. After this period of brain colonization, microglia are engaged in the active regulation of the number of newborn neurons and/or synapses, according to their functional state [100]. This function of microglia is mainly supported by their phagocytic ability and by the production and secretion of molecular mediators, able to influence neuronal/synapse fate [43, 101, 102].
As previously mentioned, in the studies found in the literature, maternal immune activation model is the main experimental option to model schizophrenia when the main aim is to evaluate microglia. Although we found different protocols of poly I:C administration (different gestational days, GD, single shot or double injection), a factor that certainly interferes with brain development, the fact is that the reported microglia changes are consistent between studies.
When the administration of poly I:C is performed early in gestation (GD 9.5), the offspring (embryonic day, ED, 17) presents transcriptomic alterations in microglia, particularly in genes associated with cell motility structures (protrusions) and neuritogenesis [103]. The administration some days later (GD 12.5 or 14.5) accelerates the establishment of a transcriptional profile (assessed in ED 14) characteristic of later developmental phases [34]. Poly I:C administration at mild/mild-late gestation period (at GD 12 or 15), triggers an increase in microglia dynamics (total distance moved over time), without interfering with morphology (evaluated at ED 18) [104]. A single or double injection of poly I:C (at GD 11.5 and/or GD 15.5) did not alter microglia density or the levels of pro-inflammatory cytokines at ED 17.5 [105].
In summary, apart differences in the maternal immune stimulation plan, this prenatal stimulus produces congruent changes of microglia transcriptome, mainly associated with the early acquisition of a profile typical from a later stage (one study report alterations in motility that, however, do not impact on colonization or cell density).
Postnatal period
Maternal immune activation during gestation, besides the alterations already observed in utero, is also associated with changes in microglia density after birth. However, the published works analyze different brain regions upon exposure to poly I:C at different gestational periods, protocol details that hinder concluding about increases or decreases in density. Microglia density per brain region may be expressed as the total number of cells or as the number of specific cellular phenotypes, namely amoeboid (in perinatal phases indicates greater immaturity) or already equipped with protrusions or cellular processes and ramifications.
The administration of poly I:C close to the delivery day (GD 20) is associated to an increase in the total density and in the density of amoeboid microglia in the hippocampus, immediately after birth, that is, at postnatal day (PND) 1 [106]. Earlier administration of poly I:C (mild-late administration, GD15) increased the number of amoeboid microglia in supraventricular corpus callosum (region where primitive microglia accumulate and then migrate tangentially across corpus callosum, acquiring a ramified phenotype), while associated with a decrease in corpus callosum, striatum, somatosensory cortex and hippocampus [107], suggesting an effect upon density dependent on the brain region under analysis. It is important to note that changes in microglia density, in this period of development, can result from changes in proliferation, death or migration; to disentangle which specific mechanism drives microglia changes may guide assertive therapeutic options.
In addition to changes in microglia density, alterations in the systems CX3CR1-CX3CL1 and CD200R-CD200 (evaluation of mRNA and protein levels of the ligands and respective receptors) were observed in the cortex and in the hippocampus of the offspring of mothers exposed to poly I:C (GD 15) or to a different immune stimulus such as lipopolysaccharide (LPS), at GD7 [108]. These two systems are important hubs of communication between microglia and neurons, and the described alterations may suggest an impairment in the crosstalk between different types of cells, that is fundamental to adequate circuit wiring and development.
Adolescence
Adolescence, a period of intense plasticity and neuronal circuit rearrangements, is also a critical time window of susceptibility to brain disorders, including schizophrenia [109]. Moreover, schizophrenia diagnosis is often associated with adolescence, the driver for pre-clinical studies in this phase of life using animal models of the disease.
In the period of rodent weaning (PND 21) no changes were observed in the total number or in the number of phagocytic microglia [110]. However, a different study reports an increase in the number of amoeboid cells in the hippocampus of male descendants of rodent mothers subjected to poly I:C administration at GD 15 [111]. Again, the moment of poly I:C administration is different (GD 9 when no changes were observed) between studies.
A few days later (PND 28), LPS offspring (injections from GD 17 until delivery) still not present changes in microglia density in the hippocampus [112]. Nevertheless, 2 days later, poly I:C offspring (PND 30) present a higher number of microglial cells in the hippocampus and striatum, but not in the frontal cortex. This increase was accompanied by morphologic alterations (e.g. reduced number of processes and branches) only in the hippocampus [113].
More recently, it was observed that morphologic changes parallel changes of functional markers in the poly I:C model, and described enlarged microglial cell bodies and a retraction of processes (alterations often taken as indicators of a reactive state, typically associated to disease conditions) in the hippocampus, corpus callosum, striatum, and PFC, accompanied by an increase in the expression of inducible nitric oxide synthase (iNOS), a key enzyme involved in inflammatory processes [114].
In line with the present knowledge about microglia heterogeneity throughout the healthy brain, but also in psychiatric conditions [40,41,42, 89], differences found in microglia at adolescence are likely dependent on the brain region under study. Interestingly, at this age, sex-dependent changes in microglia immune profile were also observed in poly I:C offspring (only females present alterations in genes related to a pro-inflammatory state, namely an up-regulation of interleukin 4 receptor (CD124) and macrophage mannose receptor (CD206) and a down-regulation of Cluster of Differentiation 54 (CD54), C-C chemokine receptor type 2 (CCR2, CX3CR1) [115]. This observation is particularly meaningful and aligned with the work developed in our lab, which is focused in sex-specific morphologic remodeling processes in psychiatric diseases [40,41,42, 89, 91].
The results gathered so far, highly suggestive of a pro-inflammatory profile of microglia in the period of adolescence, are corroborated by studies with different designs. For instance, if the administration of poly I:C is performed after birth (e.g. at PND 5-7), a pattern of microglia reactivity by a pro-inflammatory response is still observed in the hippocampus, striatum and PFC, at PND 35 [116]. Concordantly, others showed that the increase of pro-inflammatory cytokines in the hippocampus and PFC of poly I:C offspring (early administration, GD 9) at PND 40 is paralleled by the increase of microglia numbers in these brain regions, thus suggesting that microglia are probably a major player in this immune response [117].
Despite the consistency of the presented studies, one study reported the absence of microglia changes (in terms of number, morphology and phagocytosis) in the hippocampus of poly I:C offspring at PND 40 [110]. Controversial evidences were found in the number of microglial cells in the hippocampus of poly I:C offspring at PND 40 (even though the coincident administration of poly I:C at GD 9), but the studies were conducted in different animal species, presenting temporal differences in the appearance of some phenotypic characteristics (in fact, rat development lags ∼1.5 days behind mice [99]). In addition, the markers used to stain microglia in these studies were also different (IBA-1 versus CD68) and stain different microglia populations (IBA-1 – total microglia; CD68 – phagocytic microglia).
Finally, to further support the contribution of microglia for the pathophysiology of schizophrenia in rodent models and, in particular, during adolescence (PND 21-42), the depletion of these cells and the subsequent repopulation ameliorates several phenotypic traits of the disease. Particularly, poly I:C offspring presented changes in microglia transcriptome early in development (E17 and PND 7) that were no longer observed after microglia depletion and repopulation during adolescence, therefore contributing for the recovery of microglia-neuron communication and behavioral changes in this animal model at adulthood [103]. Additionally, the administration of minocycline (antibiotic with anti-inflammatory effects, usually used as a microglia modulator) during adolescence (PND 21-35) was also able to normalize the hyper-ramification of microglia in DG, as well as behavior alterations observed in adult mice prenatally exposed to poly I:C [118].
In summary, the data gathered so far at adolescence point to a general increase in microglia density, accompanied by a shift to a pro-inflammatory phenotype, typically associated to pathological conditions. The contribution of these alterations to the presentation of schizophrenia in the animal model is strongly supported by the elimination/repopulation experiment and is clearly suggestive of the relevance of microglia at adolescence to the pathophysiology of schizophrenia.
Adulthood
Similarly to what happens in other time windows of rodent life, at adulthood, the majority of the studies available report alterations in the density, as well as morphologic remodeling of microglia and a deviation to a pro-inflammatory state, commonly referred as activation state and often related with pathologic conditions or threats to homeostasis.
At adulthood (PND 56), higher levels of a TSPO radiotracer were detected in the PFC and hippocampus of rats prenatally exposed to poly I:C (early gestational period, GD 9) [119]. Conversely, others describe a decrease in TSPO staining in the PFC (but not in the hippocampus) in the poly I:C model [64]. As previously explained, caution must be taken when considering TSPO levels as an index of neuroinflammation mediated by microglia or even microglia activation, since other cells, including astrocytes and vascular endothelial cells, also express TSPO [64] and it was recently demonstrated that its expression does not correlate with the expression of microglia activation markers [65].
Other studies support a pro-inflammatory profile in the adult brain of schizophrenia models, gathering evidences for the increase in microglia density in different brain regions without [120] or with the presence of phagocytic structures compatible with the so-called activated state [121, 122]. Two studies performed later (PND 60–69) with rats prenatally [123] or postnatally [124] exposed to poly I:C further support the general idea of higher density and the presence of activated microglia. Curiously, one of these studies refers a reduction in the phagocytic ability of microglia [123] and other a pro-inflammatory environment in the PFC of rats subjected to prenatal poly I:C [117, 125], thus suggesting that microglia are predominantly in a pro-inflammatory state.
Interestingly, at PND 80–90, sex-specific changes in the density (more microglia clusters in the hippocampus of males), ramification level (lower ramification in the hippocampus of males), phagocytic activity (lower phagocytic activity in males) and interaction with synapses (more contact points with neurons in the case of females) have been described in the poly I:C model [126]. Recently, the same authors demonstrated that these phenotypic alterations are associated to sex-specific changes in excitatory and inhibitory synapse density. Poly I:C males present more synapses and inhibitory inputs, probably due to a deficient synaptic elimination, whereas females present a reduction in the number and activity of excitatory synapses [127]. These studies are highly relevant in the sense they describe a functional disturbance of the neuronal network, possibly correlated with poly I:C-induced microglia phenotypic alterations. These observations are in line with already described failures in the communication between microglia and neurons, which depends on the ability of microglia to sense their local environment. In fact, Mattei and co-workers described changes in a cluster of genes involved in microglia sensing abilities (sensome) [123]. Further suggesting a deficiency in microglia-neuron communication in schizophrenia models, a study demonstrated a disruption in the CX3CL1 (fractalkine released by neurons)-CX3CR1 (fractalkine receptor expressed by microglia) axis in poly I:C animals [128]. Finally, Manitz and co-workers proposed an impairment of the proper surveillance of brain parenchyma by microglia, based on the detection of alterations in microglia markers, namely a decrease of CD11b (integrin of the complement receptor 3) levels in poly I:C offspring at PND 100 (at both sexes) and a decrease of CD45 (negative regulator of microglia activation), exclusive to males. As described in other models of psychiatric disorders, these observations suggest that microglia sequelae are dependent, not only on the brain region under analysis, but also on sex [129], a question that deserves further investigation, considering differences in the incidence and clinical presentation found in clinics.
In summary, at adulthood, the majority of studies point to increased microglia density and activation state associated with a pro-inflammatory environment, a pattern of alterations apparently starting during adolescence. Some evidences also demonstrate a functional compromise of microglia, associated with their phagocytic ability, an alteration potentially implicated in the incorrect selection of synapses for pruning and behavior abnormalities. Future studies should clarify this issue.
Main conclusions and implications for schizophrenia diagnosis and therapeutics
Innate immunity and, in particular, its cellular elements (typified by microglia in the central nervous system), have been implicated in several psychiatric diseases with genesis during brain development. In early development, the fine regulation of synapse formation and selection for elimination is assisted by microglia-mediated immune functions, subsequent to the proper detection of neuronal/synaptic molecular signs. Thus, any brain or peripheral alteration detected by microglia may elicit a response with potential to interfere with the normal course of development, with impact in the number, degree of maturation and function of synapses/neurons and, ultimately, in behavior and health.
We find scattered studies exploring schizophrenia-associated changes in microglia cells, both in animal models of the disease (mainly obtained by gestational immune activation) and in patients. However, we were not able to find a review organizing and integrating pre-clinical and clinical information, to clearly characterize microglia changes associated to schizophrenia. The main aim of this work is to fill this gap, that we consider mandatory to rethink therapeutic targets and strategies for schizophrenia.
As main conclusion, schizophrenia-associated microglia sequelae are consistently observed in patients and animal models of disease, even considering the lack of consistency in the design of clinical and pre-clinical studies, that generates apparently conflicting data.
Clinical findings, mainly obtained by neuroimaging (PET analysis), are biased by a serious of confounding factors that must be considered in future studies. These factors include, mainly but not exclusively, individual and inter-individual variability associated to: sex and age of onset, predominance of positive or negative symptoms, type and duration of pharmacological treatment of schizophrenia and/or comorbidities. It is important to have in mind that any immune-inflammatory concomitant condition may affect microglia and this is a critical aspect, often undervalued in the literature. In addition, there are important technical limitations in assessing microglia or microglia correlates in live patients. For instance, a variety of different tracers (with variable affinities for endogenous ligands and erroneously taken as microglia markers) are used in PET, and a clear definition of the outcomes to be analyzed must be rethought and implemented. The clear meaning of normal, increased or decreased PET tracing must be reinterpreted in a consensus evaluation of its pathophysiological implications and correlation with the clinical presentation of schizophrenia (including the correlation suggested by some studies with the predominance of positive or negative symptoms).
The post-mortem study of microglia from schizophrenia patients globally demonstrates: (1) transcriptomic changes; (2) density variations – increase density in frontal and temporal cortices and a lateralization effect, that deserves further investigation; (3) the presence of amoeboid microglia in frontal and temporal cortices and in the hippocampus (Table 2), apparently more evident in the case of positive symptoms predominance (this finding reinforces the importance of establishing rigorous criteria in patient’s stratification). As claimed for in vivo studies, microglia markers used in post-mortem studies present some limitations, namely in selectivity (an overestimation of microglia alterations may result from the contribution of other cells also stained by the same markers, including perivascular macrophages and/or infiltrating peripheral macrophages). Finally, one cannot ignore the contribution of the cause of death to microglia changes, as well as the influence of time of post-mortem, aspects to be considered in the design of future studies.
Animal models of schizophrenia, namely based on the immune stimulation during gestation, are very important sources of information, otherwise impossible to get from clinical studies. It is important to note that the main data from animal studies presented and discussed in this article were obtained modeling schizophrenia by maternal immune stimulation. As previously mentioned, this animal model presents neurochemical changes (e.g. dopamine dysfunction from early development – for review see ref. [130]) and behavioral traits characteristic of schizophrenia (as well as of other developmental brain diseases, including autism, with similar symptoms and risk factors; for detail see refs. [131, 132]). So, it is of importance to further study microglia in alternative models to establish a universal mechanism of disease and disease manifestations. From different pre-clinical studies, it is unquestionable that microglia undergo alterations associated with schizophrenia modeling. Overall, microglia alterations will vary according to the period of life under analysis and can include changes in: transcriptomic profile, motility, migration/colonization, activation state (including morphologic alterations and cytokine release) and function (phagocytosis, crosstalk with neurons; Table 3 and Fig. 3). Prenatally, most changes involve the transcriptome, with microglia acquiring an adult-like phenotype earlier than in basal conditions. The main alterations described in microglia at postnatal period rely on density (increase) and migration (delay). At adolescence, the increased density is still present and is accompanied by a pro-inflammatory environment (characteristics typically associated with the so-called activated microglia). Notably, adolescence seems to be a critical period for intervention, since microglia elimination/depletion or modulation (by the control of the pro-inflammatory environment through minocycline treatment) rescue microglia transcriptomic and morphologic alterations, as well as microglia-neuron communication, in parallel with the recovery of behavior traits typically present in schizophrenia models. At adulthood, microglia still present an activation state and some functional issues emerge, namely the impairment of phagocytic ability and of microglia-neuron communication.
According to the information organized in the present work, it is clear that microglia is involved in the pathophysiology of schizophrenia. If, as suggested by pre-clinical research, the normalization of disease-associated changes of microglia, is also able to ameliorate human symptoms of the disease, it is important to design future studies including immune modulation strategies able to limit microglia numbers, inflammatory phenotype and/or phagocytosis. In this regard, considering the lack of efficacy of anti-inflammatory compounds in symptoms remission (corticosteroids or non-steroid anti-inflammatory drugs, already tested in clinical trials (https://clinicaltrials.gov/), it would be desirable to improve the selectivity towards microglia. In parallel, it is also mandatory to clarify if and how the immune modulatory effects of antipsychotics impact on microglia and if these eventual effects contribute to drug efficacy. The limitations associated to neuroimaging data highlights the need of more adequate tools to evaluate schizophrenia-associated microglia sequelae, that may include data from human iPSC-derived glia (as reviewed in refs. [133, 134]).
Far behind symptoms manifestation and diagnosis, we consider it is also important to better understand schizophrenia genesis during development. If, as suggested in the literature, an excessive synaptic pruning may be the kickoff for neurobiological deviations from normality, we think alternative mechanisms of disease need to be explored in order to explain the described hypodopaminergic function, for instance, deficits in synaptic pruning in these particular pathways. Synaptic pruning is a cellular response to local cues in the brain, thus, synaptic pruning may be differently affected in different brain areas. We hypothesize that the main pathways implicated in the disease, which are dually hypo- or hyperfunctional, may result from the excessive (as already described) or from the compromised (a hypothesis not tested so far) synaptic pruning (Fig. 4).

Schematic representation of novel putative mechanisms by which microglia may oppositely influence dual dopaminergic pathways: imbalance between the ability to eliminate and strengthen synapses.
In conclusion, very early in development, a brain-region determined microglia defect could trigger different mechanisms of disease (kickoff). On the other hand, subsequent microglia changes (sequelae) likely persist during disease progression and it is apparently sufficient to target these sequelae to ameliorate psychotic symptoms. The second approach is relatively easy to test and implement in patients, but preventing kickoff mechanisms would require advanced knowledge about microglia mechanisms of pruning at the molecular level and in utero immune or genetic modulation of those mechanisms. Recent discoveries on the involvement of specific components of the complement system in microglia-mediated synaptic pruning clearly helps defining new therapeutic targets.
Data availability
Data sharing is not applicable to this article.
References
Picchioni MM, Murray RM. Schizophrenia. BMJ 2007;335:91–5.
Hafner H, Heiden WAD. Clinical Handbook Of Schizophrenia - Course And Outcome. The Guilford Press; 2008.
Millan MJ, Andrieux A, Bartzokis G, Cadenhead K, Dazzan P, Fusar-Poli P, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discov. 2016;15:485–515.
Insel TR. Rethinking schizophrenia. Nature 2010;468:187–93.
Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet 2016;388:86–97.
Downar J, Kapur S. Clinical Handbook Of Schizophrenia - Biological theories. The Guilford Press; 2008.
Patel KR, Cherian J, Gohil K, Atkinson D. Schizophrenia: overview and treatment options. P T 2014;39:638–45.
McCutcheon RA, Reis Marques T, Howes OD. Schizophrenia-an overview. JAMA Psychiatry. 2019;1–10.
Johnsen E, Kroken RA. Drug treatment developments in schizophrenia and bipolar mania: latest evidence and clinical usefulness. Ther Adv Chronic Dis. 2012;3:287–300.
Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III−the final common pathway. Schizophr Bull. 2009;35:549–62.
Walter H, Kammerer H, Frasch K, Spitzer M, Abler B. Altered reward functions in patients on atypical antipsychotic medication in line with the revised dopamine hypothesis of schizophrenia. Psychopharmacology. 2009;206:121–32.
Fusar-Poli P, Howes OD, Allen P, Broome M, Valli I, Asselin MC, et al. Abnormal prefrontal activation directly related to pre-synaptic striatal dopamine dysfunction in people at clinical high risk for psychosis. Mol Psychiatry. 2011;16:67–75.
Allison DB, Mentore JL, Heo M, Chandler LP, Cappelleri JC, Infante MC, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. 1999;156:1686–96.
Saarni SE, Saarni SI, Fogelholm M, Heliovaara M, Perala J, Suvisaari J, et al. Body composition in psychotic disorders: a general population survey. Psychol Med. 2009;39:801–10.
Monteleone P, Martiadis V, Maj M. Management of schizophrenia with obesity, metabolic, and endocrinological disorders. Psychiatr Clin North Am. 2009;32:775–94.
Maroney M. An update on current treatment strategies and emerging agents for the management of schizophrenia. Am J Manag Care. 2020;26:S55–S61. 3 Suppl
Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry. 2015;2:258–70.
Kroken RA, Sommer IE, Steen VM, Dieset I, Johnsen E. Constructing the immune signature of schizophrenia for clinical use and research; an integrative review translating descriptives into diagnostics. Front Psychiatry. 2018;9:753.
Strous RD, Shoenfeld Y. Schizophrenia, autoimmunity and immune system dysregulation: a comprehensive model updated and revisited. J Autoimmun. 2006;27:71–80.
Hughes HK, Ashwood P. Overlapping evidence of innate immune dysfunction in psychotic and affective disorders. Brain Behav Immun Health. 2020;2:100038.
Pollmacher T, Haack M, Schuld A, Kraus T, Hinze-Selch D. Effects of antipsychotic drugs on cytokine networks. J Psychiatr Res. 2000;34:369–82.
Drzyzga L, Obuchowicz E, Marcinowska A, Herman ZS. Cytokines in schizophrenia and the effects of antipsychotic drugs. Brain Behav Immun. 2006;20:532–45.
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70:663–71.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011;333:1456–8.
Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron 2013;77:10–8.
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012;74:691–705.
Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–93.
Parellada E, Gasso P. Glutamate and microglia activation as a driver of dendritic apoptosis: a core pathophysiological mechanism to understand schizophrenia. Transl Psychiatry. 2021;11:271.
Germann M, Brederoo SG, Sommer IEC. Abnormal synaptic pruning during adolescence underlying the development of psychotic disorders. Curr Opin Psychiatry. 2021;34:222–7.
Dinesh AA, Islam J, Khan J, Turkheimer F, Vernon AC. Effects of antipsychotic drugs: cross talk between the nervous and innate immune system. CNS Drugs. 2020;34:1229–51.
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–80.
Miyamoto A, Wake H, Moorhouse AJ, Nabekura J. Microglia and synapse interactions: fine tuning neural circuits and candidate molecules. Front Cell Neurosci. 2013;7:70.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841–5.
Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016;353:aad8670.
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019;50:253–71. e6
Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell 2018;172:500–16. e16.
VanRyzin JW, Pickett LA, McCarthy MM. Microglia: driving critical periods and sexual differentiation of the brain. Dev Neurobiol. 2018;78:580–92.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314–8.
Caetano L, Pinheiro H, Patricio P, Mateus-Pinheiro A, Alves ND, Coimbra B, et al. Adenosine A2A receptor regulation of microglia morphological remodeling-gender bias in physiology and in a model of chronic anxiety. Mol Psychiatry. 2017;22:1035–43.
Simoes-Henriques C, Mateus-Pinheiro M, Gaspar R, Pinheiro H, Mendes Duarte J, Baptista FI, et al. Microglia cytoarchitecture in the brain of adenosine A2A receptor knockout mice: brain region and sex specificities. Eur J Neurosci. 2019;51:1377–87.
Gaspar R, Soares-Cunha C, Domingues AV, Coimbra B, Baptista FI, Pinto L, et al. Resilience to stress and sex-specific remodeling of microglia and neuronal morphology in a rat model of anxiety and anhedonia. Neurobiol Stress. 2021;14:100302.
Prinz M, Jung S, Priller J. Microglia biology: one century of evolving concepts. Cell 2019;179:292–311.
Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013;36:209–17.
Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T. Bidirectional microglia-neuron communication in health and disease. Front Cell Neurosci. 2018;12:323.
Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–24.
Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89.
Paolicelli RC, Bergamini G, Rajendran L. Cell-to-cell communication by extracellular vesicles: focus on microglia. Neuroscience 2019;405:148–57.
Wang M, Zhang L, Gage FH. Microglia, complement and schizophrenia. Nat Neurosci. 2019;22:333–4.
Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci. 2019;22:374–85.
Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 1982;17:319–34.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016;530:177–83.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007;131:1164–78.
Bordeleau M, Carrier M, Luheshi GN, Tremblay ME. Microglia along sex lines: from brain colonization, maturation and function, to implication in neurodevelopmental disorders. Semin Cell Dev Biol. 2019;94:152–63.
Comer AL, Jinadasa T, Sriram B, Phadke RA, Kretsge LN, Nguyen TPH, et al. Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biol. 2020;18:e3000604.
Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry. 2011;168:1303–10.
Benros ME, Eaton WW, Mortensen PB. The epidemiologic evidence linking autoimmune diseases and psychosis. Biol Psychiatry. 2014;75:300–6.
Khandaker GM, Zimbron J, Dalman C, Lewis G, Jones PB. Childhood infection and adult schizophrenia: a meta-analysis of population-based studies. Schizophr Res. 2012;139:161–8.
Khandaker GM, Zimbron J, Lewis G, Jones PB. Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studies. Psychol Med. 2013;43:239–57.
Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry. 2014;71:1121–8.
Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306–28.
Marques TR, Ashok AH, Pillinger T, Veronese M, Turkheimer FE, Dazzan P, et al. Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. Psychol Med. 2019;49:2186–96.
Notter T, Coughlin JM, Gschwind T, Weber-Stadlbauer U, Wang Y, Kassiou M, et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol Psychiatry. 2018;23:323–34.
Sneeboer MAM, van der Doef T, Litjens M, Psy NBB, Melief J, Hol EM, et al. Microglial activation in schizophrenia: is translocator 18 kDa protein (TSPO) the right marker? Schizophr Res. 2019;215:167–72.
Turkheimer FE, Rizzo G, Bloomfield PS, Howes O, Zanotti-Fregonara P, Bertoldo A, et al. The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans. 2015;43:586–92.
Di Biase MA, Zalesky A, O’Keefe G, Laskaris L, Baune BT, Weickert CS, et al. PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Transl Psychiatry. 2017;7:e1225.
Holmes SE, Hinz R, Drake RJ, Gregory CJ, Conen S, Matthews JC, et al. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: a [(11)C](R)-PK11195 positron emission tomography study. Mol Psychiatry. 2016;21:1672–9.
Conen S, Gregory CJ, Hinz R, Smallman R, Corsi-Zuelli F, Deakin B, et al. Neuroinflammation as measured by positron emission tomography in patients with recent onset and established schizophrenia: implications for immune pathogenesis. Mol Psychiatry. 2020;26:5398–406.
van der Doef TF, de Witte LD, Sutterland AL, Jobse E, Yaqub M, Boellaard R, et al. In vivo (R)-[(11)C]PK11195 PET imaging of 18 kDa translocator protein in recent onset psychosis. NPJ Schizophr. 2016;2:16031.
van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11 C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–2.
Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med. 2009;50:1801–7.
Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, et al. Imaging microglial activation in untreated first-episode psychosis: a PET study with [(18)F]FEPPA. Am J Psychiatry. 2017;174:118–24.
Collste K, Plaven-Sigray P, Fatouros-Bergman H, Victorsson P, Schain M, Forsberg A, et al. Lower levels of the glial cell marker TSPO in drug-naive first-episode psychosis patients as measured using PET and [(11)C]PBR28. Mol Psychiatry. 2017;22:850–6.
Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [(11)C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173:44–52.
Ottoy J, De Picker L, Verhaeghe J, Deleye S, Wyffels L, Kosten L, et al. (18)F-PBR111 PET imaging in healthy controls and schizophrenia: test-retest reproducibility and quantification of neuroinflammation. J Nucl Med. 2018;59:1267–74.
Coughlin JM, Wang Y, Ambinder EB, Ward RE, Minn I, Vranesic M, et al. In vivo markers of inflammatory response in recent-onset schizophrenia: a combined study using [(11)C]DPA-713 PET and analysis of CSF and plasma. Transl Psychiatry. 2016;6:e777.
Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G, et al. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18 F]-FEPPA. Schizophr Bull. 2015;41:85–93.
Selvaraj S, Bloomfield PS, Cao B, Veronese M, Turkheimer F, Howes OD. Brain TSPO imaging and gray matter volume in schizophrenia patients and in people at ultra high risk of psychosis: An [(11)C]PBR28 study. Schizophr Res. 2018;195:206–14.
De Picker L, Morrens M. Perspective: solving the heterogeneity conundrum of TSPO PET imaging in psychosis. Front Psychiatry. 2020;11:362.
Hercher C, Chopra V, Beasley CL. Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J Psychiatry Neurosci. 2014;39:376–85.
Snijders G, van Zuiden W, Sneeboer MAM, Berdenis van Berlekom A, van der Geest AT, Schnieder T, et al. A loss of mature microglial markers without immune activation in schizophrenia. Glia 2021;69:1251–67.
Petrasch-Parwez E, Schobel A, Benali A, Moinfar Z, Forster E, Brune M, et al. Lateralization of increased density of Iba1-immunopositive microglial cells in the anterior midcingulate cortex of schizophrenia and bipolar disorder. Eur Arch Psychiatry Clin Neurosci. 2020;270:819–28.
Wierzba-Bobrowicz T, Lewandowska E, Lechowicz W, Stepien T, Pasennik E. Quantitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenics. Folia Neuropathol. 2005;43:81–9.
Uranova NA, Vikhreva OV, Rakhmanova VI. Abnormal microglial reactivity in gray matter of the prefrontal cortex in schizophrenia. Asian J Psychiatr. 2021;63:102752.
Gober R, Ardalan M, Shiadeh SMJ, Duque L, Garamszegi SP, Ascona M, et al. Microglia activation in postmortem brains with schizophrenia demonstrates distinct morphological changes between brain regions. Brain Pathol. 2021:e13003;32:e13003.
Tzioras M, Stevenson AJ, Boche D, Spires-Jones TL. Microglial contribution to synaptic uptake in the prefrontal cortex in schizophrenia. Neuropathol Appl Neurobiol. 2021;47:346–51.
Purves-Tyson TD, Weber-Stadlbauer U, Richetto J, Rothmond DA, Labouesse MA, Polesel M, et al. Increased levels of midbrain immune-related transcripts in schizophrenia and in murine offspring after maternal immune activation. Mol Psychiatry. 2019;26:849–63.
Duarte JM, Gaspar R, Caetano L, Patricio P, Soares-Cunha C, Mateus-Pinheiro A, et al. Region-specific control of microglia by adenosine A2A receptors: uncoupling anxiety and associated cognitive deficits in female rats. Glia 2019;67:182–92.
Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 2018;173:1073–81.
Gaspar R, Soares-Cunha C, Domingues AV, Coimbra B, Baptista FI, Pinto L, et al. The duration of stress determines sex specificities in the vulnerability to depression and in the morphologic remodeling of neurons and microglia. Front Behav Neurosci. 2022;16:834821.
Uranova NA, Vikhreva OV, Rakhmanova VI, Orlovskaya DD. Ultrastructural pathology of oligodendrocytes adjacent to microglia in prefrontal white matter in schizophrenia. NPJ Schizophr. 2018;4:26.
Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 2013;18:206–14.
Hill SL, Shao L, Beasley CL. Diminished levels of the chemokine fractalkine in post-mortem prefrontal cortex in schizophrenia but not bipolar disorder. World J Biol Psychiatry. 2020;22:94–103.
Bergon A, Belzeaux R, Comte M, Pelletier F, Herve M, Gardiner EJ, et al. CX3CR1 is dysregulated in blood and brain from schizophrenia patients. Schizophr Res. 2015;168:434–43.
Busse S, Busse M, Schiltz K, Bielau H, Gos T, Brisch R, et al. Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: further evidence for disease course-related immune alterations? Brain Behav Immun. 2012;26:1273–9.
van Rees GF, Lago SG, Cox DA, Tomasik J, Rustogi N, Weigelt K, et al. Evidence of microglial activation following exposure to serum from first-onset drug-naive schizophrenia patients. Brain Behav Immun. 2018;67:364–73.
Feifel D, Shilling PD. Promise and pitfalls of animal models of schizophrenia. Curr Psychiatry Rep. 2010;12:327–34.
Haddad FL, Patel SV, Schmid S. Maternal immune activation by poly I:C as a preclinical model for neurodevelopmental disorders: a focus on autism and schizophrenia. Neurosci Biobehav Rev. 2020;113:546–67.
Harry GJ, Kraft AD. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology 2012;33:191–206.
Thion MS, Garel S. On place and time: microglia in embryonic and perinatal brain development. Curr Opin Neurobiol. 2017;47:121–30.
Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19:622–35.
Ikezu S, Yeh H, Delpech JC, Woodbury ME, Van Enoo AA, Ruan Z, et al. Inhibition of colony stimulating factor 1 receptor corrects maternal inflammation-induced microglial and synaptic dysfunction and behavioral abnormalities. Mol Psychiatry. 2020;26:1808–31.
Ozaki K, Kato D, Ikegami A, Hashimoto A, Sugio S, Guo Z, et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci Rep. 2020;10:21378.
Smolders S, Smolders SM, Swinnen N, Gartner A, Rigo JM, Legendre P, et al. Maternal immune activation evoked by polyinosinic:polycytidylic acid does not evoke microglial cell activation in the embryo. Front Cell Neurosci. 2015;9:301.
Ratnayake U, Quinn TA, Castillo-Melendez M, Dickinson H, Walker DW. Behaviour and hippocampus-specific changes in spiny mouse neonates after treatment of the mother with the viral-mimetic Poly I:C at mid-pregnancy. Brain Behav Immun. 2012;26:1288–99.
Zhang J, Jing Y, Zhang H, Bilkey DK, Liu P. Maternal immune activation altered microglial immunoreactivity in the brain of postnatal day 2 rat offspring. Synapse. 2018:e22072.
Chamera K, Kotarska K, Szuster-Gluszczak M, Trojan E, Skorkowska A, Pomierny B, et al. The prenatal challenge with lipopolysaccharide and polyinosinic:polycytidylic acid disrupts CX3CL1-CX3CR1 and CD200-CD200R signalling in the brains of male rat offspring: a link to schizophrenia-like behaviours. J Neuroinflammation. 2020;17:247.
Thion MS, Ginhoux F, Garel S. Microglia and early brain development: an intimate journey. Science 2018;362:185–9.
Giovanoli S, Weber-Stadlbauer U, Schedlowski M, Meyer U, Engler H. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav Immun. 2016;55:25–38.
Murray KN, Edye ME, Manca M, Vernon AC, Oladipo JM, Fasolino V, et al. Evolution of a maternal immune activation (mIA) model in rats: early developmental effects. Brain Behav Immun. 2019;75:48–59.
Vojtechova I, Maleninska K, Kutna V, Klovrza O, Tuckova K, Petrasek T, et al. Behavioral alterations and decreased number of parvalbumin-positive interneurons in wistar rats after maternal immune activation by lipopolysaccharide: sex matters. Int J Mol Sci. 2021;22:3274.
Juckel G, Manitz MP, Brune M, Friebe A, Heneka MT, Wolf RJ. Microglial activation in a neuroinflammational animal model of schizophrenia−a pilot study. Schizophr Res. 2011;131:96–100.
Esshili A, Manitz MP, Freund N, Juckel G. Induction of inducible nitric oxide synthase expression in activated microglia and astrocytes following pre- and postnatal immune challenge in an animal model of schizophrenia. Eur Neuropsychopharmacol. 2020;35:100–10.
Esslinger M, Wachholz S, Manitz MP, Plumper J, Sommer R, Juckel G, et al. Schizophrenia associated sensory gating deficits develop after adolescent microglia activation. Brain Behav Immun. 2016;58:99–106.
Ribeiro BM, do Carmo MR, Freire RS, Rocha NF, Borella VC, de Menezes AT, et al. Evidences for a progressive microglial activation and increase in iNOS expression in rats submitted to a neurodevelopmental model of schizophrenia: reversal by clozapine. Schizophr Res. 2013;151:12–9.
Ding S, Hu Y, Luo B, Cai Y, Hao K, Yang Y, et al. Age-related changes in neuroinflammation and prepulse inhibition in offspring of rats treated with Poly I:C in early gestation. Behav Brain Funct. 2019;15:3.
Xia Y, Zhang Z, Lin W, Yan J, Zhu C, Yin D, et al. Modulating microglia activation prevents maternal immune activation induced schizophrenia-relevant behavior phenotypes via arginase 1 in the dentate gyrus. Neuropsychopharmacology. 2020;45:1896–1908.
Li X, Tian X, Lv L, Hei G, Huang X, Fan X, et al. Microglia activation in the offspring of prenatal Poly I: C exposed rats: a PET imaging and immunohistochemistry study. General Psychiatry. 2018;31:e000006.
Van den Eynde K, Missault S, Fransen E, Raeymaekers L, Willems R, Drinkenburg W, et al. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res. 2014;258:179–86.
Liaury K, Miyaoka T, Tsumori T, Furuya M, Wake R, Ieda M, et al. Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. J Neuroinflammation. 2012;9:56.
Liaury K, Miyaoka T, Tsumori T, Furuya M, Hashioka S, Wake R, et al. Minocycline improves recognition memory and attenuates microglial activation in Gunn rat: a possible hyperbilirubinemia-induced animal model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2014;50:184–90.
Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. 2017;7:e1120.
Shelton HW, Gabbita SP, Gill WD, Burgess KC, Whicker WS, Brown RW. The effects of a novel inhibitor of tumor necrosis factor (TNF) alpha on prepulse inhibition and microglial activation in two distinct rodent models of schizophrenia. Behav Brain Res. 2021;406:113229.
MacDowell KS, Munarriz-Cuezva E, Caso JR, Madrigal JL, Zabala A, Meana JJ, et al. Paliperidone reverts Toll-like receptor 3 signaling pathway activation and cognitive deficits in a maternal immune activation mouse model of schizophrenia. Neuropharmacology 2017;116:196–207.
Hui CW, St-Pierre A, El Hajj H, Remy Y, Hebert SS, Luheshi GN, et al. Prenatal immune challenge in mice leads to partly sex-dependent behavioral, microglial, and molecular abnormalities associated with schizophrenia. Front Mol Neurosci. 2018;11:13.
Hui CW, Vecchiarelli HA, Gervais E, Luo X, Michaud F, Scheefhals L, et al. Sex differences of microglia and synapses in the hippocampal dentate gyrus of adult mouse offspring exposed to maternal immune activation. Front Cell Neurosci. 2020;14:558181.
Chamera K, Trojan E, Kotarska K, Szuster-Gluszczak M, Bryniarska N, Tylek K, et al. Role of polyinosinic:polycytidylic acid-induced maternal immune activation and subsequent immune challenge in the behaviour and microglial cell trajectory in adult offspring: a study of the neurodevelopmental model of schizophrenia. Int J Mol Sci. 2021;22:1558.
Manitz MP, Plumper J, Demir S, Ahrens M, Esslinger M, Wachholz S, et al. Flow cytometric characterization of microglia in the offspring of PolyI:C treated mice. Brain Res. 2016;1636:172–82.
Meyer U, Feldon J. Prenatal exposure to infection: a primary mechanism for abnormal dopaminergic development in schizophrenia. Psychopharmacology. 2009;206:587–602.
Meyer U, Feldon J. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology 2012;62:1308–21.
Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–62.
Heider J, Vogel S, Volkmer H, Breitmeyer R. Human iPSC-derived glia as a tool for neuropsychiatric research and drug development. Int J Mol Sci. 2021;22:10254.
Hasselmann J, Blurton-Jones M. Human iPSC-derived microglia: a growing toolset to study the brain’s innate immune cells. Glia 2020;68:721–39.
Acknowledgements
FCT (UID/NEU/04539/2019, UIDB/04539/2020, and UIDP/04539/2020); COMPETE-FEDER (POCI-01-0145-FEDER-007440); Centro 2020 Regional Operational Programme (CENTRO-01-0145-FEDER-000008: BrainHealth 2020); and Faculty of Medicine, Coimbra University and Santander Totta Bank (FMUC-BST-2019).
Author information
Authors and Affiliations
Contributions
ACRN and CAG wrote the manuscript. ACRN elaborated all images and tables. Both CAG and AFA contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodrigues-Neves, A.C., Ambrósio, A.F. & Gomes, C.A. Microglia sequelae: brain signature of innate immunity in schizophrenia. Transl Psychiatry 12, 493 (2022). https://doi.org/10.1038/s41398-022-02197-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02197-1
This article is cited by
-
Regulation of Zbp1 by miR-99b-5p in microglia controls the development of schizophrenia-like symptoms in mice
The EMBO Journal (2024)
-
Minocycline and antipsychotics inhibit inflammatory responses in BV-2 microglia activated by LPS via regulating the MAPKs/ JAK-STAT signaling pathway
BMC Psychiatry (2023)
-
Molecular mapping of a core transcriptional signature of microglia-specific genes in schizophrenia
Translational Psychiatry (2023)
