
Abstract
Genome-wide association studies (GWAS) identify genetic variants associated with a trait, regardless of how those variants are associated with the outcome. Characterizing whether variants for psychiatric outcomes operate via specific versus general pathways provides more informative measures of genetic risk. In the current analysis, we used multivariate GWAS to tease apart variants associated with problematic alcohol use (ALCP-total) through either a shared risk for externalizing (EXT) or a problematic alcohol use-specific risk (ALCP-specific). SNPs associated with ALCP-specific were primarily related to alcohol metabolism. Genetic correlations showed ALCP-specific was predominantly associated with alcohol use and other forms of psychopathology, but not other forms of substance use. Polygenic scores for ALCP-total were associated with multiple forms of substance use, but polygenic scores for ALCP-specific were only associated with alcohol phenotypes. Polygenic scores for both ALCP-specific and EXT show different patterns of associations with alcohol misuse across development. Our results demonstrate that focusing on both shared and specific risk can better characterize pathways of risk for substance use disorders. Parsing risk pathways will become increasingly relevant as genetic information is incorporated into clinical practice.
Similar content being viewed by others

Introduction
Genome-wide association studies (GWAS) are rapidly advancing our ability to detect genetic loci associated with psychiatric disorders [1,2,3,4,5,6]. However, GWAS of any given outcome will detect genetic loci related to that outcome via correlated traits. This non-specificity is evident in the ubiquitous genetic correlations detected across psychiatric traits [1,2,3,4,5,6]. Using approaches that partition variance in traits can be useful in disentangling pleiotropic effects from those that are specific to a phenotype of interest. In the current analysis, we use the etiology of alcohol use disorders (AUD) as a primary example of how this approach can be useful.
AUDs are moderately heritable (~50%) [7], with most of the heritability of AUD shared with other externalizing phenotypes [8,9,10]. Externalizing generally refers to a broad liability towards behavioral disinhibition and dysregulation. The externalizing spectrum includes disorders such as attention-deficit/hyperactivity disorder (ADHD), conduct disorder, substance use disorders, and antisocial behavior personality disorder, as well as personality traits like impulsivity and sensation seeking [10,11,12]. Prior research estimates that the majority of the genetic variance for AUD is shared with other externalizing disorders with a much smaller proportion of genetic variation specific to alcohol use outcomes [8]. Additionally, genetic influences on alcohol use outcomes change over time, with externalizing liability being more important in adolescence and alcohol-specific risk becoming more important as individuals age [13, 14]. In total, research on the etiology of AUD suggests differing pathways through which problems can develop.
Herein, we apply new multivariate methods [15, 16] to tease apart specific versus shared pathways by which genetic loci are associated with problematic alcohol use and demonstrate how moving beyond GWAS that focus on a single outcome may help us better understand the manner in which risk for psychiatric problems unfolds. Specifically, we expand upon a recent multivariate GWAS of externalizing [17] to differentiate the genetic variants that impact problematic alcohol use through this broad externalizing liability (EXT), from variants that are specific to problematic alcohol use (ALCP-specific). We compare our multivariate results to those from a previously published meta-analysis of GWAS of problematic alcohol use [18, 19], which, by definition, combined all genetic pathways that impact risk for alcohol problems (ALCP-total). Across these three GWAS results, we compare: (1) the genetic correlations with other relevant phenotypes; (2) the biological annotations of each genetic signal, and (3) the associations of polygenic scores (PGS) for each component with a variety of substance use phenotypes. Our analyses aim to better characterize the genetic pathways associated with problematic alcohol use and illustrate the use of multivariate genomic analyses to differentiate patterns of risk.
Methods
GWAS of problematic alcohol use specific variance (ALCP-specific)
Our current analysis builds on results from a recently published multivariate GWAS of externalizing [17]. We used GenomicSEM [15] to fit a common factor model using summary statistics from seven externalizing-related phenotypes: attention-deficit/hyperactivity disorder (ADHD), problematic alcohol use (ALCP-total), lifetime cannabis use (CANN), age at first sexual intercourse (FSEX), number of sexual partners (NSEX), general risk tolerance (RISK), and lifetime smoking initiation (SMOK). This analysis suggested a single underlying latent genetic factor (EXT), with residual genetic variance on each phenotype. The original paper presented GWAS results for that latent factor EXT. Here, we extended the original model to partition SNP effects on ALCP-total into two pathways: those that are shared among externalizing phenotypes and those that are specific to problematic alcohol use (ALCP-specific) [17].
The details for how GWAS traits were selected are presented in detail in the original EXT analysis [17]. Briefly, we included GWAS of externalizing traits if studies had 50,000 or more participants, included relevant covariates (e.g., sex, age, ancestral principal components), were successfully genotyped genome-wide (individual genotyping rate: >95%), passed the standard quality controls, and were limited to unrelated individuals or used techniques to correct for relatedness. We standardized input summary statistics for each of the externalizing phenotypes and filtered to a set of common SNPs across each of the GWAS for a total of 6,132,068 common SNPs (MAF > 0.5%). Next, we performed association tests across all available SNPs, where EXT and ALCP-specific were sequentially regressed onto each SNP. All analyses were limited to samples of European ancestries. See Supplementary Note 2 for detailed information.
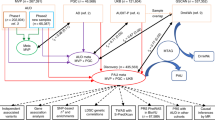
Figure 1 displays the three sets of GWAS results used to make comparisons. Figure 1A displays the univariate GWAS, focusing on a single phenotype (ALCP-total). Figure 1B displays the multivariate GWAS, with the seven indicators used to estimate the model. From these two models, we derive three sets of GWAS summary statistics: the univariate GWAS results for ALCP-total (path 1) [18, 19], the common factor GWAS results for EXT (path 2) [17], and the problematic alcohol use specific GWAS (path 3; ALCP-specific). These results allow us to dissect genetic variance in problematic alcohol use by comparing the results of ALCP-total to both EXT and ALCP-specific.

Univariate (A) and multivariate (B) GWAS models for problematic alcohol use and externalizing. The SNP associations with the yellow box labeled ALCP are referred to herein as “ALCP-total” (original problematic alcohol use), the SNP associations with the purple circle are referred to herein as “EXT” (shared risk towards externalizing), and the SNP associations with the teal circle are referred to herein as “ALCP-specific” (problematic alcohol user-specific).
Bioannotation
We compared biological annotations from each of the GWAS using a previously established pipeline [17]. First, we used FUMA [20] v1.2.8 to identify independent SNPs (LD threshold of r2 < 0.1) and conducted competitive gene-set, tissue and pathway analysis using MAGMA v1.08 [21]. Next, we used an extension of MAGMA, Hi-C coupled MAGMA (H-MAGMA) [22], to assign non-coding (intergenic and intronic) SNPs to genes based on their chromatin interactions. Lastly, we used S-PrediXcan v0.6.2 [23] to predict transcript abundance in 13 brain tissues, and to test whether the predicted transcripts showed divergent correlation patterns with each of the genetic factors. Supplementary Note 3 contains detailed information on the bioannotations.
Genetic correlations
We estimated genetic correlations between EXT, ALCP-specific, ALCP-total and 99 preregistered phenotypes, again using GenomicSEM. A full list of the genetic correlations is available in Supplementary Table 2. Genetic correlations allowed us to examine how patterns of associations differed across EXT, ALCP-total, and ALCP-specific. In determining which genetic correlations to compare, we limited results to those which were significantly associated with ALCP-total after correcting for multiple testing [24].
Polygenic scores
We created polygenic scores (PGS) from each set of GWAS summary statistics in particpants of primarily European ancestries from two independent cohorts: the National Longitudinal Study of Adolescent to Adult Health [25] (Add Health; N = 5107) and the Collaborative Study on the Genetics of Alcoholism [26] (COGA; N = 7594). Within each sample we created PGS for (1) ALCP-total; (2) EXT; and (3) ALCP-specific using PRS-CS, a Bayesian approach that uses a continuous shrinkage parameter to adjust GWAS summary statistics for linkage disequilibrium (LD) [27].
Within each of the holdout samples, we compared the effect size (ΔR2 above a model with covariates only) of the association of ALCP-total and ALCP-specific PGS with five substance categories, including alcohol, cannabis, nicotine, opioids (COGA only), and other illicit substances (e.g., cocaine, sedatives, stimulants, methamphetamine). Outcomes included “ever use” and SUD criterion counts. All models included age, sex, the first ten ancestral principal components, and study-specific covariates.
Finally, we compared the association between the EXT and ALCP-specific PGSs with an alcohol use index (AUI) [28] across time using a linear growth model [29] in Add Health. This composite index included five alcohol phenotypes ranging from normative to problematic use, scaled to a value of 0 to 10 [28] and is well suited to capture the developmentally contingent definition of substance “misuse” (e.g., early life initiation, drinking to intoxication in adolescence, developing problems in adulthood). Detailed descriptions of the holdout samples, phenotypes, and analyses are presented in Supplementary Note 4.
Results
Lead SNPs and bioannotations
Table 1 presents lead SNPs from the ALCP-total GWAS across the three GWAS. In the ALCP-total GWAS, we identified 542 genome-wide significant (p < 5 × 10–8) SNPs before pruning for LD. Table 1 includes the 11 independent (LD threshold of r2 < 0.1) lead SNPs for ALCP-total, and their corresponding estimates in the EXT and ALCP-specific GWAS results. Of these, only the locus on chromosome 3 (rs10511087), in the CADM2 region, was significant in the EXT GWAS, and the large number of SNPs before pruning were likely the result of a long-range LD region near CADM2. The lead SNP from another locus, located on chromosome 11, was in LD with one of the top SNPs from the EXT GWAS in NCAM1 (rs9919558, p < 6.50 × 10–59). Notably, none of the SNPs on chromosome 4 were significant (or in LD) in the EXT GWAS. Instead, 8 of the 9 lead SNPs on chromosome 4 were significant in the ALCP-specific GWAS. The top SNPs for ALCP-specific are in ADH1B and ADH1C, which are involved in alcohol metabolism, as well as other genes previously associated with alcohol phenotypes including KLB [30].
Gene-based analyses identified 13 genes associated with ALCP-total, but only 2 genes associated with ALCP-specific. Analysis of tissue expression in MAGMA and H-MAGMA did not allow for comparisons because of the limited power in the ALCP-specific results. In S-PrediXcan, only ADH1C was significantly associated with ALCP-specific. While these results do not point to any new biological pathways of risk, the biological sources of genetic variance across these different pathways reaffirm that EXT is capturing a broader risk domain, while ALCP-specific is identifying genes primarily associated with the pharmacokinetics of alcohol (see Supplementary Tables 8–11 for full results).
Genetic correlations across ALCP-total and ALCP-specific
ALCP-total was significantly correlated with 64 of the 99 preregistered phenotypes. We focus on the 35 traits related to substance use, personality, and other psychiatric outcomes (full results in Supplementary Table 2). Figure 2 provides two depictions of the results. Panel A presents the (rg) estimates (and 95% confidence intervals) between ALCP-total (yellow), ALCP-specific (teal), and the other traits. Here, the asterisks denote genetic correlations that differ significantly from ALCP-total to ALCP-specific. Three of the genetic correlations for substance related phenotypes (drug exposure, maternal smoking, and age of smoking initiation) and four genetic correlations for psychiatric traits (stress-related disorders, manic symptoms, psychotic symptoms, and iPSYCH cross-disorder) differed across the two sets of GWAS. None of the estimates for the personality related traits differed significantly across the sets of results.

Panel (A) presents the genetic correlations (rg) and 95% CI between problematic alcohol use (ALCP-total, yellow dots), problematic alcohol use-specific (ALCP-specific, teal dots), and selected traits significantly correlated with ALCP-total (after correcting for an FDR of 5%, Supplementary Table 2 reports all preregistered genetic correlations). Asterisks (*) in Panel (A) represent genetic correlations for ALCP-specific that differ significantly from those with ALCP-total. Panel (B) presents changes in point estimates for genetic correlations from ALCP-total to ALCP-specific. In Panel (B), asterisks represent genetic correlations for ALCP-specific that are still significant (after correcting for an FDR of 5%).
Panel B presents the difference in the same genetic correlations, and the asterisks indicate a significant association with ALCP-specific after correcting for multiple testing. Overall, genetic correlations with other traits were attenuated for ALCP-specific compared to ALCP-total. For substance use, ALCP-total was genetically correlated with all other forms of substance use (rg = −0.22–0.82). Once we remove the shared variance due to EXT, ALCP-specific was only associated with drinks per week (rg = 0.69) and age of smoking initiation (rg = 0.14). ALCP-total was genetically correlated with most of the impulsivity and personality phenotypes (rg = −0.48–0.56). Only neuroticism (rg = 0.33), lack of perseverance (rg = 0.37), and positive urgency (rg = 0.32) were correlated with ALCP-specific. Finally, ALCP-total was genetically correlated with various psychiatric traits (rg = −31–0.49). ALCP-specific remained associated with most of these traits, notably bipolar disorder (rg = 0.18), major depressive disorder (rg = 0.23), post-traumatic stress disorder (rg = 0.26), and schizophrenia (rg = 0.17). Overall, a substantial portion of the observed genetic correlations between problematic alcohol use and other phenotypes is due to genetic variants that operate via externalizing liability.
Polygenic scores and substance use disorders
Figure 3 illustrates associations between polygenic scores and various substance use phenotypes in the forms of ever use and SUD criterion counts. Neither the ALCP-total PGS nor the ALCP-specific PGS were associated with ever using alcohol in Add Health or COGA. For AUD criteria, the ALCP-total PGS explained 0.52% of the variance (Δ\(R_{{\rm{ALCP}} {\mbox{-}} {\rm{total}}}^2\)) in Add Health and 1.72% of the variance in COGA, compared to the ALCP-specific PGS, which explained 0.28% of the variance in Add Health and 0.85% of the variance in COGA. Supplementary Table 3 presents a full comparison of the EXT, ALCP-specific, and ALCP-total PGS for AUD criterion counts. The ALCP-total PGS was associated with cannabis use and other substance use in Add Health (ORALCP-total = 1.15–1.20, ΔR2 = 0.56–0.90%) and all forms of use in COGA (ORALCP-total = 1.20–1.27, ΔR2 = 0.65–1.26%). Additionally, the ALCP-total PGS was associated with cannabis use disorder criteria in both COGA and Add Health (βALCP-total = 0.11–0.16, ΔR2 = 0.26–0.31%), and nicotine dependence criteria in COGA (βALCP-total = 0.20, ΔR2 = 0.45%). However, the ALCP-specific PGS was only associated with ever using illicit substances (other than cannabis) and the associations are attenuated compared to that of ALCP-total (ORALCP-specific = 1.12–1.15, ΔR2 = 0.33–0.44%).

Bar charts illustrating the incremental proportion of variance (incremental R2, or ΔR2 above model with age, sex, PCs, and study-specific covariates) explained by the polygenic score in Add Health (N = 5107) and COGA (N = 7594). Association between polygenic scores and lifetime SUD criterion counts for alcohol, cannabis, nicotine, other illicit substances, and opioids (COGA only). Asterisks (*) represent polygenic scores that are significant after correcting for an FDR of 5%.
Polygenic scores and longitudinal models of alcohol misuse
Lastly, we fit a series of longitudinal growth models for a composite alcohol use index (AUI) in Add Health (see Supplementary Note 4.5 for complete results). The best fitting model represented a quadratic change in AUI over time (with sex differences in slope), a significant association between EXT PGS and base levels of AUI (βEXT = 0.12, SEEXT = 0.01, PEXT = 1.46 × 10–18), and a significant association between ALCP-specific PGS and change in the linear component of age for AUI (βALCP-specific*AGE = 0.07, SEALCP-specific*AGE = 0.02, PALCP-specific*AGE = 5.70 × 10–5). We found no evidence of sex-specific effects of either PGS in stratified models. Figure 4 provides a visual representation of the results from this best-fitting longitudinal model across levels of each PGS (±1.5 SD). Individuals higher on EXT PGS experience higher levels of AUI across time and sex, whereas those with higher levels of ALCP-specific experience increased growth in AUI. Supplemental Table 7 presents comparisons of estimates from longitudinal models using ALCP-total PGS to those in the main text.

Predicted values for alcohol use index (AUI) from ages 12 to 32 using linear mixed models in Add Health (N = 5107). Values for EXT and ALCP-specific polygenic scores set to ±1.5 SD. The shaded areas represent 95% confidence intervals. Confidence intervals estimated using percentile method bootstrapping over 1000 bootstrap samples. All other covariates set to mean values.
Discussion
Genetic influences on SUDs can operate via both shared genetic risk with other forms of externalizing as well as via substance specific pathways. Conventional analyses focused on a single phenotype, what we would term the “classical” GWAS approach, are unable to differentiate these pathways; newer multivariate approaches begin to make this possible. In the current analysis, we demonstrated the potential of disaggregating genetic variance in problematic alcohol use into risk shared with other externalizing behaviors/disorders versus risk that is specific to problematic alcohol use. We demonstrate that multivariate genomic analyses of correlated traits can increase the specificity for characterizing how genetic risk unfolds.
When we compared the results from the univariate ALCP-total GWAS to those from the multivariate model (EXT and ALCP-specific), we found robust evidence of distinct risk pathways. We compared the genetic correlations for ALCP-total and ALCP-specific across 99 preregistered phenotypes, focusing here on phenotypes related to personality, substance use, and psychopathology. ALCP-total was correlated with a broad range of personality phenotypes, especially those related to impulsivity. However, after removing the variance due to the shared risk for EXT, most of these associations were no longer significant. Similarly, ALCP-total was genetically correlated with multiple forms of other substance use phenotypes, while ALCP-specific remained correlated only with alcohol consumption and age of smoking initiation, and this latter correlation switched direction. The change in direction could reflect the fact that ALCP-specific captures (1) risk for problematic alcohol use once alcohol becomes available, or (2) risk for other psychiatric problems around their median age of onset, both of which occur during young adulthood [31], forcing the correlation with age of smoking initiation to be positive. It is also possible that this change in direction is merely a statistical artifact. Lastly, ALCP-total was correlated with a variety of psychiatric phenotypes. ALCP-specific continued to yield small associations with many psychiatric traits, suggesting that ALCP-specific contains signal related to other disorders. It is important to note that in addition to removing shared variance with EXT, some of these association may no longer be significant, in part, due to reduced statistical power in the ALCP-specific results.
At the SNP level, our approach was further able to separate alcohol-specific biology from a general risk towards externalizing, given that alcohol metabolizing genes (e.g., ADH1B and ADH1C) were significant in the alcohol-specific, but not EXT, results. We identified 11 lead SNPs after pruning for LD: one on chromosome 3, nine on chromosome 4, and one on chromosome 11. In the ALCP-specific GWAS, 8 of the 9 lead ALCP SNPs on chromosome 4 were genome-wide significant. These top SNPs were in alcohol metabolism genes (ADH1B and ADH1C), and other genes previously associated with alcohol phenotypes including KLB [30, 32]. Additionally, SLC39A8 has been consistently identified as a risk variant for schizophrenia [5, 33, 34]. This association with SLC39A8 could indicate that the ALCP-specific contains variance that is not unique to alcohol (e.g., risk for internalizing or psychotic disorders). Two of these ALCP-total SNPs were in strong LD (r2 ~ 0.98) with top SNPs from the EXT results. The SNP on chromosome 3 was in LD with two SNPs in the CADM2 region, while the SNP on chromosome 11 was in LD with a SNP on NCAM1. CADM2 has been implicated in previous GWAS of other substance use phenotypes [35, 36], risky behaviors [36,37,38], and impulsivity [38]. Overall, the SNP level results broadly point to two distinct pathways of risk: one related to risk taking/impulsivity, and one specific to the body’s processing of alcohol, both of which are entwined in the univariate GWAS results for problematic alcohol use.
Finally, we evaluated polygenic scores in Add Health and COGA [14]. The PGS for ALCP-specific were almost exclusively related to alcohol phenotypes, indicating that the model successfully differentiates shared and specific risk. While there were differences in the magnitudes in effect sizes across COGA and Add Health, which could reflect differences in how the samples were ascertained (nationally representative sample vs clinically ascertained), the overall patterns were similar. In longitudinal models, EXT was associated with higher mean-levels of AUI while ALCP-specific was associated with increased growth in AUI. Notably, these results illustrate that externalizing genetic risk is associated with differences in AUI early in development. In contrast, during emerging adulthood, when alcohol use becomes legal and more readily accessible, there is further differentiation by alcohol-specific genetic risk. Therefore, alcohol-specific risk does not lead to alcohol problems without exposure to drinking, while broader externalizing risk captures propensity to drinking exposure across the life course. This longitudinal model reiterates the developmentally contextual nature of risk [13, 14]. Overall, the PGS results support the notion of a shared externalizing risk pathway and an alcohol-specific risk pathway.
Our analyses included several important limitations. First, they were limited to GWAS of European ancestries. Unfortunately, Genomic SEM requires larger sample sizes to obtain stable estimates. As larger sample sizes become available in non-European ancestries, we will extend these models to those populations. Genetic research in diverse ancestries is important scientifically, but also morally, as failure to diversify genetic discovery will result in the exacerbation of health disparities [39]. Second, while we considered externalizing phenotypes, we did not consider internalizing or psychotic conditions, which also show genetic overlap with AUD and other substance use disorders [4, 6, 18, 40, 41]. Finally, our estimates of SNPs associated with ALCP-specific were limited by the relatively small discovery sample size for ALCP-total (N ~ 150 K). We do note that our GWAS of ALCP-total was highly correlated with the largest meta-analysis of problematic alcohol use to date (rG = 0.94, p = 4.94 × 10–324) [3]. Future iterations with more powerful GWAS of problematic alcohol use may reveal additional variants associated specifically with ALCP. Additionally, including traits related to alcohol-specific biological processes may further help distinguish alcohol-specific from other processes through which AUD develops.
GWAS of psychiatric disorders contain a mixture of different signals. Moving beyond univariate to multivariate GWAS designs offers the potential to tease apart these signals. Herein, we decomposed the genetic variation of problematic alcohol use into that which is shared with other externalizing phenotypes from that which is specific to problematic alcohol use. Comparison of results at multiple levels showed that variance specific to problematic alcohol use was related to alcohol phenotypes while that which was shared was more strongly related to other forms of substance use and impulsivity. Differentiating these pathways of risk will become more important as genetic data becomes incorporated into clinical practice.
Data availability
All data sources are described in the manuscript and supplemental information. No new data were collected. Only data from existing studies or study cohorts were analyzed, some of which have restricted access to protect the privacy of the study participants. The GWAS summary statistics for the EXT GWAS, can be obtained by following the procedures detailed at https://externalizing.org/request-data/. Summary statistics are derived from analyses based in part on 23andMe data, for which we are restricted to only publicly available report results for up to 10,000 SNPs. The full set of externalizing GWAS summary statistics can be made available to qualified investigators who enter into an agreement with 23andMe that protects participant confidentiality. Once the request has been approved by 23andMe, a representative of the Externalizing Consortium can share the full GWAS summary statistics. Access to genetic data for COGA (dbGaP Study Accession: phs000763.v1.p1) and Add Health (dbGaP Study Accession: phs001367.v1.p1) are available through dbGaP.
Code availability
No custom algorithms or software was developed in this study. All code is available by request from the corresponding author.
References
Zhou H, Rentsch CT, Cheng Z, Kember RL, Nunez YZ, Sherva RM, et al. Association of OPRM1 functional coding variant with opioid use disorder: a genome-wide association study. JAMA Psychiatry 2020; https://doi.org/10.1001/jamapsychiatry.2020.1206.
Johnson EC, Demontis D, Thorgeirsson TE, Walters RK, Polimanti R, Hatoum AS, et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry 2020; https://doi.org/10.1016/S2215-0366(20)30339-4.
Zhou H, Sealock JM, Sanchez-Roige S, Clarke TK, Levey DF, Cheng Z, et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci. 2020; https://doi.org/10.1038/s41593-020-0643-5.
Levey DF, Stein MB, Wendt FR, Pathak GA, Zhou H, Aslan M, et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat Neurosci. 2021; https://doi.org/10.1038/s41593-021-00860-2.
Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022;2022:1–13.
Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet. 2021; https://doi.org/10.1038/s41588-021-00857-4.
Verhulst B, Neale MC, Kendler KS. The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychol Med. 2015;45:1061–72.
Kendler KS, Myers J. The boundaries of the internalizing and externalizing genetic spectra in men and women. Psychol Med. 2014;44:647–55.
Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry 2003;60:929–37.
Krueger RF, Hicks BM, Patrick CJ, Carlson SR, Iacono WGWG, McGue M. Etiological connections among substance dependence, antisocial behavior and personality: modeling the externalizing spectrum. J Abnorm Psychol. 2002;111:411–24.
Lahey BB, Rathouz PJ, van Hulle C, Urbano RC, Krueger RF, Applegate B, et al. Testing structural models of DSM-IV symptoms of common forms of child and adolescent psychopathology. J Abnorm Child Psychol. 2008;36:187–206.
Kotov R, Cicero DC, Conway CC, DeYoung CG, Dombrovski A, Eaton NR, et al. The hierarchical taxonomy of psychopathology (HiTOP) in psychiatric practice and research. Psychol Med. 2022;52:1666–78.
Kendler KS, Gardner C, Dick DM. Predicting alcohol consumption in adolescence from alcohol- specific and general externalizing genetic risk factors, key environmental exposures and their interaction. Psychol Med. 2011;41:1507–16.
Meyers JL, Salvatore JE, Vuoksimaa E, Korhonen T, Pulkkinen L, Rose RJ, et al. Genetic influences on alcohol use behaviors have diverging developmental trajectories: a prospective study among male and female twins. Alcohol Clin Exp Res. 2014;38:2869–77.
Grotzinger AD, Rhemtulla M, de Vlaming R, Ritchie SJ, Mallard TT, Hill WD, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav. 2019;3:513–25.
Grotzinger AD, Mallard TT, Akingbuwa WA, Ip HF, Adams MJ, Lewis CM, et al. Genetic architecture of 11 major psychiatric disorders at biobehavioral, functional genomic, and molecular genetic levels of analysis. Nat. Genet. 2020;54:548–59.
Karlsson Linn‚r R, Mallard TT, Barr PB, Sanchez-Roige S, Madole JW, Driver MN, et al. Multivariate analysis of 1.5 million people identifies genetic associations with traits related to self-regulation and addiction. Nat Neurosci. 2021;24:1367–76.
Walters RK, Polimanti R, Johnson EOECEO, McClintick JN, Adams MJ, Adkins AE, et al. Trans-ancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat Neurosci. 2018;21:1656–69.
Sanchez-Roige S, Palmer AA, Fontanillas P, Elson SL, Adams MJ, Howard DM, et al. Genome-wide association study meta-analysis of the alcohol use disorders identification test (AUDIT) in two population-based cohorts. Am J Psychiatry 2019;176:107–18.
Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1–11.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
Sey NYA, Hu B, Mah W, Fauni H, McAfee JC, Rajarajan P, et al. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. 2020;23:583–593.
Barbeira A, Pividori M, Zheng J, Wheeler H, Nicolae D, Im HK. Integrating predicted transcriptome from multiple tissues improves association detection. 2018; https://doi.org/10.1101/292649.
Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B (Methodol). 1995;57:289–300.
Harris KM. The add health study: design and accomplishments. 2013. https://doi.org/10.17615/C6TW87.
Edenberg HJ. The collaborative study on the genetics of alcoholism: an update. Alcohol Res Health 2002;26:214–8.
Ge T, Chen C-Y, Ni Y, Feng Y-CA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10:1776.
Vachon DD, Krueger RF, Irons DE, Iacono WG, McGue M. Are alcohol trajectories a useful way of identifying at-risk youth? A multiwave longitudinal-epidemiologic study. J Am Acad Child Adolesc Psychiatry 2017;56:498–505.
Fitzmaurice GM, Laird NM, Ware JH. Applied longitudinal analysis. Wiley. 2012.
Clarke TK, Adams MJ, Davies G, Howard DM, Hall LS, Padmanabhan S, et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N = 112 117). Mol Psychiatry. 2017;22:1376.
Solmi M, Radua J, Olivola M, Croce E, Soardo L, Salazar de Pablo G, et al. Age at onset of mental disorders worldwide: large-scale meta-analysis of 192 epidemiological studies. Mol Psychiatry 2021 27:1. 2021;27:281–95.
Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–44.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511:421–7.
Mealer RG, Jenkins BG, Chen CY, Daly MJ, Ge T, Lehoux S, et al. The schizophrenia risk locus in SLC39A8 alters brain metal transport and plasma glycosylation. Sci Rep. 2020;10. https://doi.org/10.1038/s41598-020-70108-9.
Pasman JA, Verweij KJH, Gerring Z, Stringer S, Sanchez-Roige S, Treur JL, et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat Neurosci. 2018;21:1161–70.
Arends RM, Pasman JA, Verweij KJH, Derks EM, Gordon SD, Hickie I, et al. Associations between the CADM2 gene, substance use, risky sexual behavior, and self-control: a phenome-wide association study. Addiction Biol. 2021;n/a:e13015.
Karlsson Linnér R, Biroli P, Kong E, Meddens SFW, Wedow R, Fontana MA, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51:245–57.
Sanchez-Roige S, Fontanillas P, Elson SL, Gray JC, de Wit H, MacKillop J, et al. Genome-wide association studies of impulsive personality traits (BIS-11 and UPPS-P) and drug experimentation in up to 22,861 adult research participants identify loci in the CACNA1I and CADM2 genes. J Neurosci. 2019;39:2562.
Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–91.
Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Hatoum AS, Johnson EC, Colbert SMC, Polimanti R, Zhou H, Walters RK, et al. The addiction risk factor: a unitary genetic vulnerability characterizes substance use disorders and their associations with common correlates. Neuropsychopharmacology 2021;2021:1–7.
Acknowledgements
The Externalizing Consortium: Principal Investigators: Danielle M. Dick, Philipp Koellinger, K. Paige Harden, Abraham A. Palmer. Lead Analysts: Richard Karlsson Linnér, Travis T. Mallard, Peter B. Barr, Sandra Sanchez-Roige. Significant Contributors: Irwin D. Waldman. The Externalizing Consortium has been supported by the National Institute on Alcohol Abuse and Alcoholism (R01AA015416-administrative supplement), and the National Institute on Drug Abuse (R01DA050721). Additional funding for investigator effort has been provided by K02AA018755, U10AA008401, P50AA022537, DP1DA054394, and a European Research Council Consolidator Grant (647648 EdGe to Koellinger). The content is solely the responsibility of the authors and does not necessarily represent the official views of the above funding bodies. The Externalizing Consortium would like to thank the following groups for making the research possible: Add Health, Vanderbilt University Medical Center’s BioVU, Collaborative Study on the Genetics of Alcoholism (COGA), the Psychiatric Genomics Consortium’s Substance Use Disorders working group, UK10K Consortium, UK Biobank, and Philadelphia Neurodevelopmental Cohort. We would also like to thank the research participants and employees of 23andMe, Inc. for making this work possible. The full set of externalizing GWAS summary statistics can be made available to qualified investigators who enter into an agreement with 23andMe that protects participant confidentiality. Once the request has been approved by 23andMe, a representative of the Externalizing Consortium can share the full set of summary statistics. All code necessary to replicate this study is available upon request. Finally, we thank The Collaborative Study on the Genetics of Alcoholism (COGA), Principal Investigators B. Porjesz, V. Hesselbrock, T. Foroud; Scientific Director, A. Agrawal; Translational Director, D. Dick, includes eleven different centers: University of Connecticut (V. Hesselbrock); Indiana University (H.J. Edenberg, T. Foroud, J. Nurnberger Jr., Y. Liu); University of Iowa (S. Kuperman, J. Kramer); SUNY Downstate (B. Porjesz, J. Meyers, C. Kamarajan, A. Pandey); Washington University in St. Louis (L. Bierut, J. Rice, K. Bucholz, A. Agrawal); University of California at San Diego (M. Schuckit); Rutgers University (J. Tischfield, A. Brooks, R. Hart); The Children’s Hospital of Philadelphia, University of Pennsylvania (L. Almasy); Virginia Commonwealth University (D. Dick, J. Salvatore); Icahn School of Medicine at Mount Sinai (A. Goate, M. Kapoor, P. Slesinger); and Howard University (D. Scott). Other COGA collaborators include: L. Bauer (University of Connecticut); L. Wetherill, X. Xuei, D. Lai, S. O’Connor, M. Plawecki, S. Lourens (Indiana University); L. Acion (University of Iowa); G. Chan (University of Iowa; University of Connecticut); D.B. Chorlian, J. Zhang, S. Kinreich, G. Pandey (SUNY Downstate); M. Chao (Icahn School of Medicine at Mount Sinai); A. Anokhin, V. McCutcheon, S. Saccone (Washington University); F. Aliev, P. Barr (Virginia Commonwealth University); H. Chin and A. Parsian are the NIAAA Staff Collaborators. We continue to be inspired by our memories of Henri Begleiter and Theodore Reich, founding PI and Co-PI of COGA, and also owe a debt of gratitude to other past organizers of COGA, including Ting- Kai Li, P. Michael Conneally, Raymond Crowe, and Wendy Reich, for their critical contributions. This national collaborative study is supported by NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA).
Author information
Authors and Affiliations
Consortia
Contributions
D.M.D., P.B.B., T.T.M. and S.S.-R. conceived the study. D.M.D. oversaw the study. D.M.D., P.B.B., T.T.M., S.S.-R. and H.E.P. led the writing of the manuscript, with substantive contributions to the writing from D.M.D., K.P.H, R.K.L. and A.A.P. PBB was the lead analyst, responsible for polygenic score analyses. T.T.M. was responsible for genetic correlations and multivariate analyses with genomic SEM. S.S.-R. led the bioinformatics analyses, and H.E.P. contributed to those analyses. R.K.L. provided GWAS summary statistics. P.B.B., T.T.M., S.S.-R. and H.E.P. prepared the tables and figures. I.D.W. provided helpful advice and feedback on various aspects of the study design. All authors contributed to and critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barr, P.B., Mallard, T.T., Sanchez-Roige, S. et al. Parsing genetically influenced risk pathways: genetic loci impact problematic alcohol use via externalizing and specific risk. Transl Psychiatry 12, 420 (2022). https://doi.org/10.1038/s41398-022-02171-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02171-x
This article is cited by
-
A Developmentally-Informative Genome-wide Association Study of Alcohol Use Frequency
Behavior Genetics (2024)
-
Alcohol Use Disorder Polygenic Risk Scores and Trajectories of Early Adolescent Externalizing Behaviors: Examining the Role of Parenting and Family Conflict in the Racially/Ethnically Diverse ABCD Sample
Behavior Genetics (2024)
-
Genetic Risk, Neighborhood Characteristics, and Behavioral Difficulties Among African American Adolescents Living in Very Low-Income Neighborhoods
Research on Child and Adolescent Psychopathology (2023)
-
Genomic risk for post-traumatic stress disorder in families densely affected with alcohol use disorders
Molecular Psychiatry (2023)
