
Abstract
Conduct disorder (CD), a psychiatric disorder characterized by a repetitive pattern of antisocial behaviors, results from a complex interplay between genetic and environmental factors. The clinical presentation of CD varies both according to the individual’s sex and level of callous-unemotional (CU) traits, but it remains unclear how genetic and environmental factors interact at the molecular level to produce these differences. Emerging evidence in males implicates methylation of genes associated with socio-affective processes. Here, we combined an epigenome-wide association study with structural neuroimaging in 51 females with CD and 59 typically developing (TD) females to examine DNA methylation in relation to CD, CU traits, and gray matter volume (GMV). We demonstrate an inverse pattern of correlation between CU traits and methylation of a chromosome 1 region in CD females (positive) as compared to TD females (negative). The identified region spans exon 1 of the SLC25A24 gene, central to energy metabolism due to its role in mitochondrial function. Increased SLC25A24 methylation was also related to lower GMV in multiple brain regions in the overall cohort. These included the superior frontal gyrus, dorsolateral prefrontal cortex, supramarginal gyrus, secondary visual cortex and ventral posterior cingulate cortex, which are regions that have previously been implicated in CD and CU traits. While our findings are preliminary and need to be replicated in larger samples, they provide novel evidence that CU traits in females are associated with methylation levels in a fundamentally different way in CD and TD individuals, which in turn may relate to observable variations in GMV across the brain.
Similar content being viewed by others

Introduction
Conduct disorder (CD) is a psychiatric disorder of childhood and adolescence characterized by persistent antisocial behaviors (i.e., violence towards others or animals, destruction of property, theft, and serious rule violations), which significantly impact the individual’s social, academic, or occupational functioning [1]. There is considerable variation in the possible combinations of symptoms that could lead to a CD diagnosis [2]. Therefore, to identify more homogeneous subgroups of youth with CD, several subtyping approaches are included within the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) [1]. One approach focuses on the ‘Limited Prosocial Emotions’ specifier, which indexes callous-unemotional (CU) traits (i.e., reduced empathy, callousness, a lack of guilt, and shallow effect). This specifier designates a particularly impaired subgroup of youths with CD who are at increased risk of developing psychopathy in adulthood [3, 4]. Levels of CU traits show moderate stability from adolescence to adulthood [5] and are also a predictor of more severe antisocial and aggressive behaviors both in adolescence and adulthood [6]. In this context, understanding the etiology of these CU traits in adolescents with CD is an important step towards identifying risk factors for a subgroup of youths with CD who are particularly susceptible to poorer outcomes in adulthood [7].
Research shows that both genetic and environmental risk factors are implicated in the development of conduct problems or CD [8, 9], with around 50% of the variance in CD risk attributable to heritable genetic influences [8]. Crucially, twin studies indicate that youths with CD symptomatology and high versus low levels of CU traits are characterized by different environmental and genetic risk vulnerabilities [4]. Indeed, Viding et al. (2005) demonstrated that antisocial behavior in youths with CD symptomatology and high levels of CU traits is highly heritable (0.76), whereas in youths with CD symptomatology and low levels of CU traits it is moderately heritable (0.64) and more influenced by environmental factors [10]. Along with CU traits, sex is an important factor to consider in youths with CD in relation to genetic vulnerability for this disorder. Indeed, heritability estimates for antisocial behavior in youths with CD are higher in males than females [11]. Furthermore, in males with CD and high levels of CU traits, heritable factors explain a high proportion of the variance in antisocial behavior [10]. Conversely, antisocial behavior in females with conduct problems (CP) and high levels of CU traits was shown to be entirely explained by environmental factors in one study [12]. These data suggest sex differences in the biological mechanisms underlying antisocial behavior in youths with CD depending on their levels of CU traits.
Gene–environment interplay in CD development
A key question in CD research is how genetic and environmental risk factors interact at the molecular level in relation to CU trait phenotypes [13]. One candidate mechanism is via epigenetic changes in the form of DNA methylation, which involves addition of a methyl group at a specific genomic location [14]. Depending on the pattern, location, and level of methylation within or proximal to the gene’s coding sequence, gene expression may be suppressed or amplified [14]. The genetic variation of an individual is also an important factor to consider in understanding how environmental factors are translated into methylation signatures. Recent research has highlighted that individual differences in heritable factors may influence methylation signatures [15] and thus gene regulation. These genetic variants that can affect DNA methylation are known as methylation quantitative trait loci (mQTLs) and may be further useful markers for genetic influence on gene regulation [16].
Altered regulation of genes expressed in brain tissues and/or implicated in behavior, may explain how methylation levels mechanistically mediate environmental influences, e.g. adverse life experiences to subsequent risk for CD [17] and CU traits [18]. A recent study suggests that exposure to adverse prenatal environmental factors has a large effect on the brain epigenome, and that epigenetic effects associated with brain development are also sex-specific [19].
Epigenetic studies of youths with CD or sub-clinical CP have provided initial evidence that DNA methylation patterns may mediate environmental factors associated with antisocial behavior [20, 21]. In males with CD, methylation of the oxytocin receptor gene (OXTR) correlates positively with CU traits [22]. Similarly, in a mixed-sex study, higher methylation of OXTR at birth was associated with higher CU traits in adolescence for participants with low levels of anxiety [23]. Alterations in the expression of genes that govern the oxytocin system, as a result of epigenetic modifications, may thus play an important biological role in the development of CD and CU traits [22, 23]. A recent small-scale epigenetic neuroimaging study on males with CD showed that OXTR methylation and levels of CU traits interacted to predict frontoparietal hyperactivity and weaker amygdalo-frontoparietal connectivity in males during a face-processing task [24]. This is consistent with previous reports of abnormalities in this circuitry in CD (e.g., [25]) and the fact that OXTR is highly expressed in both limbic and cortical brain tissues [26]. Interestingly, a fundamentally opposite association between brain functional connectivity and level of CU traits was observed in CD as compared to TD youths [24].
Study aims
To expand current knowledge on epigenetics in CD and limited research on females with CD, we adopted an exploratory approach and conducted the first Epigenome-Wide Association Study (EWAS) with salivary DNA data on females with CD and varying levels of CU traits. As previous research in psychiatric disorders has demonstrated differential methylation according to diagnostic status [27] and level of CU traits [22, 28], we first examined the main effects of CD diagnostic status and level of CU traits. Secondly, we [29] and others [24] have demonstrated an inverse association between biomarkers and the level of CU traits in clinical groups as compared to TD populations. Thus, we investigated whether there was a CDxCU traits interaction effect on DNA methylation. The relationship between CU traits and methylation level has been demonstrated in individuals with CD [22, 23] but the nature and direction of this relationship in TD youth is unknown. Finally, to investigate whether these methylation changes co-incidence with altered brain development, we related our methylation data to gray matter volume as measured using voxel-based morphometry (VBM).
Methods and materials
Participants
Fifty-one females with CD (mean age = 14.9, SD = 1.7) and 59 TD females (mean age = 14.7, SD = 2.4), recruited across five sites, were included as a subsample of the FemNAT-CD study [30] (see Supplementary Tables S1 for details). This study was conducted according to the legal regulations outlined by the European Union, national legislation, and the Declaration of Helsinki. For each site, written informed consent was obtained from all participants and their parents, in accordance with the site-specific ethical requirements. In addition to standard FemNAT-CD inclusion and exclusion criteria (see Supplementary materials), participants were required to be non-smokers, be medication-free, and have good quality saliva-DNA and structural MRI data. Participants were included in the CD group if they either; (a) met the DSM-5 criteria for a diagnosis of CD; (b) were 9–12 years old, met the criteria for a diagnosis of oppositional defiant disorder (ODD) and also had at least one current symptom of CD; or (c) were aged >12 years, met the criteria for ODD and also had at least 2 current CD symptoms. All TD participants had no diagnosable psychiatric disorders and no history of externalizing disorders (ADHD, ODD). The participants were aged 9–18 years and groups were matched on pubertal development status, performance IQ, ethnicity, and data-collection site (Table 1 and Supplementary Table S1).
Clinical and psychometric measures
Detailed information about these measures is provided in our previous work [31]. Briefly, trained staff interviewed the participants and their parents (or caregivers) separately using the Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime version (K-SADS-PL [32]) to assess for CD and other DSM-IV-TR psychiatric disorders. Supplementary questions from the K-SADS-PL (e.g. for ODD/ADHD) were completed if key items were endorsed during the initial screening. CU traits were assessed using the parent-version Inventory of Callous-Unemotional Traits (ICU [33]). Total, verbal and performance IQ was assessed using the Wechsler Abbreviated Scale of Intelligence [34] in the UK and the Wechsler Intelligence Scale for Children, Fifth Edition [35] at other sites. Pubertal status was determined using the Pubertal Development Scale (PDS) [36] completed by the participants (if aged >12 years) or by the parents/caregivers (for participants ≤ 12 years).
Genome-wide methylation data pre-processing
DNA was extracted from saliva within 7 days of collection using the Oragene OG-500 Kit. DNA quality cutoff was a 260/280 ratio above 1.8. DNA was stored at −80 °C immediately. Genome-wide methylation was measured using the Illumina Infinium HumanMethylationEPIC BeadChip Array at Life & Brain GmbH, Bonn, Germany. Pre-processing was performed in R version 3.6.0 [37]. Raw.idat files were pre-processed with the minfi [38] package (version1.32.0) following standard parameter settings (see Supplementary Methods). We removed failed and noisy probes as suggested [39], and also probes spanning an SNP with an SNP147 data-base annotated MAF > 10%. Finally, cross-reactive probes were eliminated. Between-array normalization was completed using the preprocessFunnorm() function [40] included in the minfi package, following standard recommendations. This unsupervised method uses control probes to identify unwanted variation. It then extends the idea of quantile normalization to regresses out components of variation captured by these control probes [40]. This has been shown to be an effective method for removing positional effects [41].
We used ANOVA testing in the normalized methylation data to ensure there were no residual batch effects. As an additional check, we also extracted the first principal component of the methylation data and performed pairwise T-tests (with Tukey’s correction for multiple testing) across the batches to confirm there were no correlations between the batch IDs and M values.
Heat maps and hierarchical clustering plots based on the Euclidean distance of the top 2000 loci selected by variance in methylation were generated to visually check for outliers and batch effects (Supplementary Fig. 1). The methylation M-values were calculated based on the log-transformed ratio of methylated to unmethylated signal-intensities for each locus in line with previous research [42] and we ensured these M values were normally distributed across the differentially methylated region (Supplementary Fig. 1). Probes were mapped to their genomic region using the human reference genome hg19.
MRI acquisition
T1-weighted structural scans were collected at five research sites using MRI scanners all operating with 3 T fields (either Siemens or Philips manufactured) and harmonized acquisition sequences (see refs. [29, 31] and Supplementary materials).
Pre-processing of the neuroimaging data
Consistent with our previous work [29], SPM12 (www.fil.ion.ucl.ac.uk/spm), Computational Anatomy 12 (CAT-12: http://dbm.neuro.uni-jena.de/cat/) and template-o-matic (TOM8 [42]) toolboxes were used to pre-process MRI data (see Supplementary materials).
Genome-wide methylation statistical analysis
To examine the associations between CD diagnostic status, level of CU traits and genome-wide methylation, we employed linear regression modelling: M-values for each CpG site was modelled as a function of CD status, CU traits (total ICU score), and the CDxCU traits interaction effect. Corrections for the effects of age and hormonal contraceptive use were included in the model. Socio-economic status (SES) was not included as a covariate in the DNA methylation analysis on statistical and conceptual grounds (see Supplementary materials for further details).
To identify components of extraneous variation due to unmodelled or unknown latent variables, surrogate variable analysis in R (sva package, “leek” method selected) was performed and the two factors identified were included in the final model as covariates. The effect sizes and p-value of each predictor (CD-case status, CU-trait levels and CDxCU) were calculated using the suggested Bayesian approach as implemented in the minfi ebayes function. P-values were then submitted to the Bumphunter algorithm [43] to identify differentially methylated regions (DMRs). We specified different coefficients from the linear regression modelling in the arguments of the Bumphunter function to test separately for: (i) the main effect of CD diagnosis, (ii) the main effect of CU score, and (iii) a CD × CU interaction effect on methylation, while controlling for the main effects of the other two factors. QQ plots were generated to confirm appropriate model fits for each EWAS model (see Supplementary Fig. 2). Correction for multiple testing using the false discovery rate (FDR [44]) was done across the individual probes tested as recommended [45].
VBM analysis
Since we identified a significant DMR associated with the group-by-CU traits interaction effect on methylation level, we employed the GLM framework to explore the association between GMV and average M-value across probes within the respective DMR. No DMR associated with main effects for CD or CU-traits was identified.
Specifically, GMV was analyzed on a voxel-by-voxel basis, via multiple regressions. PDS, SES, total intracranial volume (TIV), scanning site (dummy coded), and total IQ were included as covariates of no interest. Unlike in the epigenetic analysis, we include SES as a covariate here to allow us to investigate the association between methylation and GMV across our full cohort without the potential confounding effects of SES on GMV that are independent of methylation. At a whole-brain level, inferences were made using a statistical threshold of p < 0.05 after family-wise error (FWE) correction for multiple comparisons. We also investigated associations between GMV and M-value in four regions of interest (ROIs, bilaterally) where the identified gene of interest SLC25A24 is highly expressed (Genotype-Tissue Expression [46] GTEx project database, see supplementary material Fig. 3), namely the amygdala, hippocampus, basal ganglia and cerebellum (Supplementary Fig. 5). Masks of these regions were defined based on the Talairach Daemon database using the WFU PickAtlas tool in SPM12 [47]. The MarsBAR toolbox was used to extract mean-cluster and peak-voxel GMV values from significant clusters for each participant. All brain imaging coordinates are reported in the standardized Montreal Neurological Institute (MNI) space.
Results
Participant characteristics
As per matching on PDS and performance IQ, CD and TD females did not differ in terms of age, puberty, ethnicity, site and performance IQ, but the CD group had lower full-scale IQs than the TD group (Table 1). The number of ADHD symptoms did not differ between groups, but individuals with CD had significantly more symptoms of a generalized anxiety disorder (GAD) and major depressive disorder (MDD) than the TD participants. Females with CD also had higher total ICU and ICU subscale scores (see Table 1 and Supplementary Fig. 4).
Power calculation
While we acknowledge that our sample size is rather small for a genome-wide approach, power analysis using the online calculation tool epigenetics.essex.ac.uk/shiny/EPICDNAmPowerCalcs confirmed that our analysis with a sample size of n = 110 participants conferred each CpG site tested with ~80% power to detect a difference in methylation at the recommended level for the EPIC array (p < 6.21e−05). Two other recent studies have similarly adopted a genome-wide approach to investigating DNA methylation in relation to aggressive behaviours in youth, both using a sample size <n = 100 [48, 49].
Identification of differentially methylated regions
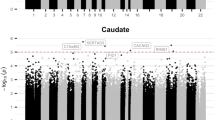
At the single probe level, DNA-methylation was not predicted by case-control status or level of CU traits (at a significance level of pFDR < 0.05). However, the CDxCU traits interaction significantly predicted differential methylation at one genomic region on chromosome 1 (hg19 chr1: 108,735,312–108,735,893, FDR = 0.004), spanning eight probes. The interaction was driven by a positive association between CU traits and methylation of the respective probes in females with CD (Pearson r(49) = 0.39, p = 0.006), but a negative association between CU traits and methylation in TD females (Pearson r(57) = −0.27, p = 0.042). The slopes of these correlations differed significantly (Z = 2.48, p = 0.007). The region identified includes exon 1 of the solute carrier SLC25A24 gene (see Fig. 1).

(Top) UCSC Genome Browser Illustration showing stacked annotation tracks beneath the genomic coordinates of the region which showed differential methylation according to the CD × CU traits interaction (from the hg19 human reference gene); (bottom) a scatter plot of this differentially methylated region highlighting the opposite relationship between methylation and level of CU traits in CD cases vs. control participants.
It is important to note that these methylation findings do not include the methylation values at common SNPs, as these were removed during the pre-processing stage of our analysis, thus our findings should be considered in light of this limitation.
Association between methylation and gray matter volume
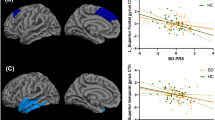
We then tested whether the SLC25A24 methylation levels observed for the interaction effect of CDxCU traits was also associated with GMV in any brain region. After correction for multiple comparisons, no significant (i.e. pFWE < 0.05) positive or negative associations between the average M-value of the SLC25A24-DMR and GMV were detected (in analysis across the whole cohort). However, given the exploratory nature of this study, we report findings at a more liberal significance level of p < 0.001 uncorrected with an extent threshold of k = 72 voxels empirically determined according to random field theory [50, 51]. At this level we observed a negative association with SLC25A24 methylation M-value for GMV in several clusters within the brain (please see Supplementary Table s2), indicating that higher SLC25A24 methylation is associated with lower GMV in these regions. We identified these clusters in multiple brain regions including the superior frontal gyrus (SFG), dorsolateral prefrontal cortex (dlPFC), supramarginal gyrus, the secondary visual cortex in the left hemisphere, and the ventral PCC and secondary visual cortex in the right hemisphere. All coordinates are reported in MNI space. Mean cluster GMV values were extracted for each participant and then plotted against the average methylation M-value across the DMR on chromosome 1 (i.e. exon 1 of gene SLC25A24 (see Fig. 2)). Across all regions, in both CD and TD groups, there was a negative association between GMV and the mean exon 1 SLC25A24 M-value.

Mean gray matter volume (GMV) values in the cluster significantly associated with methylation for p < 0.001, size > 72 voxels were extracted for each participant and then plotted against the average methylation M-value across the DMR on chromosome 1 corresponding to exon 1 of gene SLC25A24. CD participants (red) and TD (blue) participants are differentiated by color. In all clusters there is a negative association between GMV and M value in both CD and TD groups; the difference between groups in the strength of the correlation is not statistically significant at p < 0.05.
ROI analysis
No significant positive or negative association between SLC25A24 methylation and GMV could be detected in the amygdala, hippocampus, basal ganglia or cerebellum ROIs. (Please see Supplementary Fig. 5 for 3D visualization of the four brain regions tested as ROIs.)
Post-hoc testing of OXTR methylation
We did not observe a significant association between CU traits and methylation at any of the 12 CpG sites on the OXTR gene for which we had DNA methylation data. Even when the significance threshold was reduced to a nominal level of p < 0.001, uncorrected, the main effect of CU traits was not significant for any of the individual sites, or for this region as a whole.
Discussion
To our knowledge, this is the first EWAS and epigenetic neuroimaging study in females with CD. First, we examined the main effects of CD group status, level of CU traits and their interaction on saliva-based DNA methylation. Our analyses revealed that in CD and TD females there is a fundamentally opposite pattern of association between CU traits and methylation at a chromosome 1 genomic region, spanning exon 1 of the SLC25A24 gene. Second, we related the identified DMR to GMV, both in multiple brain regions implicated in CD and CU traits and in a whole-brain exploratory analysis. GMV in regions including the SFG, dlPFC and supramarginal gyrus was negatively correlated with methylation levels, however, these neuroimaging findings did not reach the minimum threshold for significance.
Genome-wide methylation
We found a significant CD × CU traits interaction effect on methylation level in exon 1 of the SLC25A24 gene, whereby methylation level was positively correlated with CU traits in CD participants, but negatively correlated with CU traits in TD controls. Elevated methylation at the first exon and promoter regions of genes has been demonstrated to decrease the expression of the respective gene [52, 53]. Thus, our results indicate that in adolescent females with CD, higher levels of CU traits are associated with reduced SLC25A24 gene expression, whereas in TD females, CU traits are positively associated with gene expression.
SLC25A24, a member of a solute-carrier gene family [54], is involved in adenosine triphosphate (ATP)-mediated Calcium buffering at the mitochondrial matrix and is potentially involved in protecting cells against oxidative stress-induced cell death. In mitochondria, ATP production is associated with the production of free oxidative radicals. These cellular redox scavengers, as well as nutrition-derived antioxidants, are crucial to neutralize these free radicals [55]. As the brain accounts for 25% of the body’s total energy expenditure [56], impaired mitochondrial function, as suggested by a reduced expression of SLC25A24, may lead to higher rates of cell death due to oxidative stress [57] and thus leave neuronal cells especially vulnerable to oxidative damage [58]. Increased cell death, due to an impaired redox-scavenger system in the brain’s mitochondria, may also, at least partially, explain the association we observed with GMV. Furthermore, unbalanced energy provision and reduced Calcium homeostasis in neurons may result in impaired functioning and ultimately lead to neurodegeneration [57]. Accordingly, mitochondrial dysfunction has been suggested to be associated with several neurodevelopmental disorders, including autism spectrum disorder (ASD) [59, 60] and ADHD [61]. Reduced expression of the SLC25A24 gene has been reported in the thalamus and motor cortex of patients with ASD and hypothesized to be associated with the impairments in sensory processing and response inhibition observed in this population [62].
As discussed, deficient mitochondrial functioning is a possible consequence of increased methylation and the resulting decreased expression of the SLC25A24 gene. Given that mitochondria work alongside the mitochondrial-bound monoamine oxidase A (MAO-A) enzyme to break down catecholaminergic neurotransmitters [63], altered functioning of either component in the degradation process may contribute to abnormally high or low levels of neurotransmitters in the brain [64]. Importantly, atypical levels of neurotransmitters have previously been associated with both CD [13] and CU traits [28]. Both elevated SLC25A24 methylation and variants of the MAO-A enzyme may contribute to disrupted catecholamine catabolism. This is reported to be the biological means by which variation of the MAOA gene contributes to the affective (e.g., emotion dysregulation) and behavioral (e.g., reactive aggression) features of females with CD [65]. Thus, SLC25A24 gene hypermethylation may also result in behavioral patterns associated with atypical levels of neurotransmitters in the brain in a similar way to that reported for variants of the MAO-A enzyme, which have previously been linked to aggressive/violent behaviors in both animals [66] and humans [67].
Environmental risk factors and SLC25A24 methylation
Childhood maltreatment, a key factor known to influence DNA methylation [68], has been shown to interact with MAOA variants to predict aggression in both sexes [69]. In females, the high activity allele has been shown to confer a risk for aggressive behavior following childhood maltreatment [69], but see ref. [70]. Future studies should further investigate the relationship between childhood maltreatment and methylation to determine whether experiences of child maltreatment alter DNA methylation levels and thereby increase the risk for aggressive behaviors.
More generally, mitochondrial dysfunction has been linked to exposure to environmental stressors [71]. Mitochondria are key components of the human body’s stress response system, providing intra-cellular energy and synthesizing stress hormones and neurotransmitters central to stress responding [72]. Experimental manipulation of mitochondrial function has been shown to influence physiological and behavioral responses to psychological stress [72]. Crucially, there is evidence that epigenetic markers of stress exposure are mitochondrially regulated [72]. Thus, reduced expression in genes governing mitochondrial function, such as SLC25A24, may arbitrate how environmental factors result in epigenetic modifications [73].
Individuals with CD are more likely to have experienced ‘stressful’ early life environments and thus to have elevated stress biomarkers associated with psychiatric symptoms [74]. CU traits may be another factor that moderates the association between environmental risk factors and the individual’s biological stress response [75]. Consequently, the combination of CD diagnostic status and level of CU traits may influence epigenetic markers associated with stress exposure. Altered methylation across genes in the energy metabolism system may represent an adaptive response to these variations. Thus, rather than being a unique marker of one stressor, we postulate that SLC25A24 gene methylation may reflect the cumulative effect of exposure to multiple early-life environmental factors triggering the biological stress response system.
Epigenetic neuroimaging data
Our neuroimaging analysis revealed trend-level negative associations between SLC25A24 methylation values and GMV in several brain regions, namely, the SFG, dlPFC, supramarginal gyrus and secondary visual cortex in the left hemisphere, and the ventral posterior cingulate cortex (PCC) and secondary visual cortex in the right hemisphere.
These results may suggest that higher levels of SLC25A24 gene methylation is linked to a reduction in GMV in these regions. This finding would be consistent with the theory that increased methylation has a silencing effect on the gene, leading to impaired mitochondrial function (and thus a reduced capacity for energy production and growth) during brain development. Many of the regions where reduced GMV was observed, such as the SFG, dlPFC, the supramarginal gyrus and the ventral PCC, are involved in higher cognitive functions, such as working memory [76], as well as socio-cognitive processes such as affective empathy, which have been shown to be impaired in CD [13, 77]. For example, in a recent meta-analysis of 13 VBM studies, we found that youths with CP had significantly reduced GMV in the left medial SFG [78]. Atypical cortical thickness and functional connectivity have also been reported in adults with psychopathy in several brain regions across the frontal cortices [79] and deficits in cortical folding in these regions are also reported in youths with CD [80].
In youths with CD, greater levels of methylation were observed in association with higher CU traits and greater levels of methylation were also related to reductions in GMV at trend-level. In TD youths, we see the inverse pattern (with individuals with higher CU traits having higher GMV in the observed brain regions). We speculate that in individuals with CD and high CU traits this increased methylation and the associated higher levels of oxidative stress during energy production contributes to a higher rate of neuronal death during neuronal pruning, and subsequently leads to a reduction in GMV in the observed brain regions in this group. However, currently, the underlying factors contributing to this mechanism are unknown, and further research with more highly powered studies is needed to determine whether the suggestive negative relationship between GMV and methylation we observed here holds true in larger samples.
Post-hoc testing of OXTR methylation
The fact that other studies have found an association between CU traits and methylation of the OXTR gene (e.g. refs. [22, 23]), but we did not can be explained by a number of factors. For example, this may be related to methodological differences between our study and previous studies, such as the use of different measures of CU traits (i.e., ICU here, but others [24] have used the Youth Psychopathic Traits Inventory (YPI [81]) or other different investigative approaches, i.e. candidate gene vs. epigenome-wide studies. Additionally, we focused on females only, which contrasts with previous studies that have relied on male-only or mixed-sex samples.
Strengths and limitations
As the first study integrating epigenetic and neuroimaging data from females with CD, this work is an important contribution to our understanding of the biological factors implicated in CD and CU traits in females. Using multi-site data allowed for a larger sample size than would have been possible at a single site, as CD females are difficult to recruit. Furthermore, as data were collected as part of the FemNAT-CD project, the sample is well-characterized, with all participants undergoing thorough assessment for psychiatric disorders and symptoms using a reliable measure based on DSM-IV-TR criteria. Finally, the two groups did not differ on PDS, performance IQ, ADHD symptoms, site and ethnicity, minimizing the potential confounding effects of these factors.
Nevertheless, this study has limitations. First, the sample size is relatively small. As mentioned above, power analysis confirmed our analysis with a sample size of n = 110 participants conferred each CpG site tested with ~80% power to detect a difference in methylation at the recommended level for the EPIC array (p < 6.21e−05). This power allows us to detect moderate-to-large effects, however smaller effects (f < 0.35) on genome-wide methylation levels or GMV were not detectable with this study design. Also, we only had data on childhood maltreatment for a small subset of participants (n = 31), so we were unable to include this information in our analysis. Second, while several previous studies report concordance of DNA methylation across saliva and brain tissues (e.g. ref. [82]), tissue-specific epigenetic modifications have also been reported [83]. Thus, it is possible that the differential methylation in salivary DNA demonstrated in this study does not accurately reflect brain-level methylation and might thus be specific to buccal cells only. We also did not correct for cell composition in our salivary DNA samples. Third, as the methylation findings we report do not include the methylation at common SNPs, we do not yet know whether the methylation differences we observe are themselves genetically influenced. Finally, due to funding limitations, we chose to focus solely on investigating genome-wide methylation in females. We felt this would maximise the novelty of our work and add to the knowledge base in this particularly under-researched group. However, as we only included female participants our findings may not apply to males with CD, as research indicates sex-specific influences of environmental and genetic factors on CD and CU traits [10, 12]. Thus similar studies in males and mixed-sex samples will be an important area of future research to investigate whether these mechanisms are sex-specific.
Conclusions
Methylation of the SLC25A24 gene was significantly associated with CU traits in both females with CD and TD females but in a fundamentally opposing pattern. Given its essential role in energy metabolism, SLC25A24 is a key component of the biological stress response system. We postulate that the combination of the individual’s level of CU traits and the number of stressful early life experiences may epigenetically modify the SLC25A24 gene thus influencing its functionality. Furthermore, we detected negative trends between SLC25A24 methylation values and GMV in several brain regions, many of which have also been implicated in CD and CU traits. While our findings are preliminary and need to be replicated in larger samples, they provide novel evidence that CU traits in females are associated with methylation levels in a fundamentally different way in CD and TD groups, which in turn relates to observable variations in GMV in the brain.
Change history
29 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41398-021-01643-w
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM-5®). American Psychiatric Association Publishing. Washington DC, United States 2013.
Nock MK, Kazdin AE, Hiripi E, Kessler RC. Prevalence, subtypes, and correlates of DSM-IV conduct disorder in the National Comorbidity Survey Replication. Psychol Med. 2006;36:699–710.
Frick PJ, Ray JV, Thornton LC, Kahn RE. Annual research review: a developmental psychopathology approach to understanding callous‐unemotional traits in children and adolescents with serious conduct problems. J Child Psychol Psychiatry. 2014;55:532–48.
Viding E, McCrory EJ. Understanding the development of psychopathy: progress and challenges. Psychol Med. 2018;48:566–77.
Loney BR, Huntenburg A, Counts‐Allan C, Schmeelk KM. A preliminary examination of the intergenerational continuity of maternal psychopathic features. Aggress Behav. 2007;33:14–25.
Marcus RF. The development of aggression and violence in adolescence. Springer; 2017. New York, United States p. 141–70.
Walters GD. Assessing the proactive and reactive dimensions of criminal thought process: divergent patterns of correlation with variable-and person-level measures of criminal risk and future outcome. J Personal Assess. 2020;102:223–30.
Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–9.
Salvatore JE, Dick DM. Genetic influences on conduct disorder. Neurosci Biobehav Rev. 2018;91:91–101.
Viding E, Blair RJR, Moffitt TE, Plomin R. Evidence for substantial genetic risk for psychopathy in 7‐year‐olds. J Child Psychol Psychiatry. 2005;46:592–7.
Caspi A, Taylor A, Moffitt TE, Plomin R. Neighborhood deprivation affects children’s mental health: environmental risks identified in a genetic design. Psychol Sci. 2000;11:338–42. https://doi.org/10.1111/1467-9280.00267
Fontaine NM, Rijsdijk FV, McCrory EJ, Viding E. Etiology of different developmental trajectories of callous-unemotional traits. J Am Acad Child Adolesc Psychiatry. 2010;49:656–64.
Fairchild G, Hawes DJ, Frick PJ, Copeland WE, Odgers CL, Franke B, et al. Conduct disorder. Nat Rev Dis Prim. 2019;5:43. https://doi.org/10.1038/s41572-019-0095-y
Anastasiadi D, Esteve-Codina A, Piferrer F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenet Chromatin. 2018;11:37. https://doi.org/10.1186/s13072-018-0205-1
van Dongen J, Ehli EA, Jansen R, van Beijsterveldt C, Willemsen G, Hottenga JJ, et al. Genome-wide analysis of DNA methylation in buccal cells: a study of monozygotic twins and mQTLs. Epigenet Chromatin. 2018;11:54. https://doi.org/10.1186/s13072-018-0225-x
Barker ED, Walton E, Cecil C, Rowe R, Jaffee SR, Maughan B, et al. A Methylome-Wide Association Study of trajectories of oppositional defiant behaviors and biological overlap with attention deficit hyperactivity disorder. Child Dev. 2018;89:1839–55. https://doi.org/10.1111/cdev.12957
Veroude K, Zhang-James Y, Fernàndez-Castillo N, Bakker MJ, Cormand B, Faraone SV. Genetics of aggressive behavior: an overview. Am J Med Genet B. 2016;171B:3–43. https://doi.org/10.1002/ajmg.b.32364
Henry J, Pingault JB, Boivin M, Rijsdijk F, Viding E. Genetic and environmental aetiology of the dimensions of callous-unemotional traits. Psychol Med. 2016;46:405–14. https://doi.org/10.1017/S0033291715001919
Mattern F, Post A, Solger F, O'Leary A, Slattery DA, Reif A, et al. Prenatal and postnatal experiences associated with epigenetic changes in the adult mouse brain. Behav Brain Res. 2019;359:143–8. https://doi.org/10.1016/j.bbr.2018.10.037
Gescher DM, Kahl KG, Hillemacher T, Frieling H, Kuhn J, Frodl T. Epigenetics in personality disorders: today’s insights. Front Psychiatry. 2018;9:579. https://doi.org/10.3389/fpsyt.2018.00579
Chiocchetti AG. (2020 (under review)).
Dadds MR, Moul C, Cauchi A, Dobson-Stone C, Hawes DJ, Brennan J, et al. Methylation of the oxytocin receptor gene and oxytocin blood levels in the development of psychopathy. Dev Psychopathol. 2014;26:33–40. https://doi.org/10.1017/S0954579413000497
Cecil CA, Lysenko LJ, Jaffee SR, Pingault JB, Smith RG, Relton CL, et al. Environmental risk, Oxytocin Receptor Gene (OXTR) methylation and youth callous-unemotional traits: a 13-year longitudinal study. Mol Psychiatry. 2014;19:1071–7. https://doi.org/10.1038/mp.2014.95
Aghajani M, Klapwijk ET, Colins OF, Ziegler C, Domschke K, Vermeiren R, et al. Interactions between oxytocin receptor gene methylation and callous-unemotional traits impact socioaffective brain systems in conduct-disordered offenders. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:379–91. https://doi.org/10.1016/j.bpsc.2017.12.010
Gillespie NA, Neale MC, Hagler DJ Jr, Eyler LT, Fennema-Notestine C, Franz CE, et al. Genetic and environmental influences on mean diffusivity and volume in subcortical brain regions. Hum Brain Mapp. 2017;38:2589–98. https://doi.org/10.1002/hbm.23544
Quintana DS, Rokicki J, van der Meer D, Alnæs D, Kaufmann T, Córdova-Palomera A, et al. Oxytocin pathway gene networks in the human brain. Nat Commun. 2019;10:668. https://doi.org/10.1038/s41467-019-08503-8
Lin D, Chen J, Perrone-Bizzozero N, Bustillo JR, Du Y, Calhoun VD, et al. Characterization of cross-tissue genetic–epigenetic effects and their patterns in schizophrenia. Genome Med. 2018;10:13. https://doi.org/10.1186/s13073-018-0519-4
Moul C, Dobson-Stone C, Brennan J, Hawes D, Dadds M. An exploration of the serotonin system in antisocial boys with high levels of callous-unemotional traits. PLoS ONE. 2013;8:e56619. https://doi.org/10.1371/journal.pone.0056619
Raschle NM, Menks WM, Fehlbaum LV, Steppan M, Smaragdi A, Gonzalez-Madruga K, et al. Callous-unemotional traits and brain structure: sex-specific effects in anterior insula of typically-developing youths. Neuroimage Clin. 2018;17:856–64. https://doi.org/10.1016/j.nicl.2017.12.015
Freitag CM, Konrad K, Stadler C, De Brito SA, Popma A, Herpertz SC, et al. Conduct disorder in adolescent females: current state of research and study design of the FemNAT-CD consortium. Eur Child Adolesc Psychiatry. 2018;27:1077–93. https://doi.org/10.1007/s00787-018-1172-6
Rogers JC, Gonzalez-Madruga K, Kohls G, Baker RH, Clanton RL, Pauli R, et al. White matter microstructure in youths with conduct disorder: effects of sex and variation in callous traits. J Am Acad Child Adolesc Psychiatry. 2019;58:1184–96. https://doi.org/10.1016/j.jaac.2019.02.019
Kaufman J, Birmaher B, Brent D, Rao U, Flynn C, Moreci P, et al. Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL): initial reliability and validity data. J Am Acad Child Adolesc Psychiatry. 1997;36:980–8. https://doi.org/10.1097/00004583-199707000-00021
Frick PJ, Cornell AH, Barry CT, Bodin SD, Dane HE. Callous-unemotional traits and conduct problems in the prediction of conduct problem severity, aggression, and self-report of delinquency. J Abnorm Child Psychol. 2003;31:457–70. https://doi.org/10.1023/a:1023899703866
Wechsler D. Wechsler Abbreviated Scale of Intelligence–2nd Edition. San Antonio, TX: NCS Pearson; 2011.
Wechsler D. Wechsler intelligence scale for children. 5th ed (WISC-V). San Antonio: Psychological Corporation; 2014.
Petersen AC, Crockett L, Richards M, Boxer A. A self-report measure of pubertal status: reliability, validity, and initial norms. J Youth Adolesc. 1988;17:117–33.
Team, RCR. A language and environment for statistical computing—version 3.6.0. Team, RCR; Vienna, Austria 2019.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9. https://doi.org/10.1093/bioinformatics/btu049
McCall MN, Almudevar A. Affymetrix GeneChip microarray preprocessing for multivariate analyses. Brief Bioinform. 2012;13:536–46. https://doi.org/10.1093/bib/bbr072
Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. https://doi.org/10.1186/s13059-014-0503-2
Jiao C, Zhang C, Dai R, Xia Y, Wang K, Giase G, et al. Positional effects revealed in Illumina methylation array and the impact on analysis. Epigenomics. 2018;10:643–59. https://doi.org/10.2217/epi-2017-0105
Wilke M, Holland SK, Altaye M, Gaser C. Template-O-Matic: a toolbox for creating customized pediatric templates. Neuroimage. 2008;41:903–13. https://doi.org/10.1016/j.neuroimage.2008.02.056
Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41:200–9. https://doi.org/10.1093/ije/dyr238
Genovese CR, Wasserman L. Exceedance control of the false discovery proportion. J Am Stat Assoc. 2006;101:1408–17.
Li D, Xie Z, Pape ML, Dye T. An evaluation of statistical methods for DNA methylation microarray data analysis. BMC Bioinforma. 2015;16:217. https://doi.org/10.1186/s12859-015-0641-x
Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. https://doi.org/10.1038/ng.2653
Lancaster JL, Woldorff MG, Parsons LM, Liotti M, Freitas CS, Rainey L, et al. Automated Talairach atlas labels for functional brain mapping. Hum Brain Mapp. 2000;10:120–31. https://doi.org/10.1002/1097-0193(200007)10:3<120::aid-hbm30>3.0.co;2-8
Guillemin C, Provençal N, Suderman M, Côté SM, Vitaro F, Hallett M, et al. DNA methylation signature of childhood chronic physical aggression in T cells of both men and women. PLoS ONE. 2014;9:e86822. https://doi.org/10.1371/journal.pone.0086822
Provençal N, Suderman MJ, Guillemin C, Vitaro F, Côté SM, Hallett M, et al. Association of childhood chronic physical aggression with a DNA methylation signature in adult human T cells. PLoS ONE. 2014;9:e89839. https://doi.org/10.1371/journal.pone.0089839
Hayasaka S, Nichols TE. Combining voxel intensity and cluster extent with permutation test framework. Neuroimage. 2004;23:54–63. https://doi.org/10.1016/j.neuroimage.2004.04.035
Worsley KJ, Marrett S, Neelin P, Vandal AC, Friston KJ, Evans AC. A unified statistical approach for determining significant signals in images of cerebral activation. Hum Brain Mapp. 1996;4:58–73. https://doi.org/10.1002/(SICI)1097-0193(1996)4:1<58::AID-HBM4>3.0.CO;2-O
Li S, Li Y, Chen B, Zhao J, Yu S, Tang Y, et al. exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018;46:D106–D112.
Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE. 2011;6:e14524. https://doi.org/10.1371/journal.pone.0014524
He L, Dai B, Zeng B, Zhang X, Chen B, Yue B, et al. The complete mitochondrial genome of the Sichuan Hill Partridge (Arborophila rufipectus) and a phylo. Genet Anal Relat Species Gene. 2009;435:23–28. https://doi.org/10.1016/j.gene.2009.01.001
Fraunberger EA, Scola G, Laliberte VL, Duong A, Andreazza AC. Redox modulations, antioxidants, and neuropsychiatric disorders. Oxid Med Cell Longev. 2016;2016:4729192. https://doi.org/10.1155/2016/4729192
Herculano-Houzel S. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proc Natl Acad Sci USA. 2012;109:10661–8. https://doi.org/10.1073/pnas.1201895109
Kramer P, Bressan P. Our (mother’s) mitochondria and our mind. Perspect Psychol Sci. 2018;13:88–100. https://doi.org/10.1177/1745691617718356
de Oliveira DM, Ferreira Lima RM, El-Bacha RS. Brain rust: recent discoveries on the role of oxidative stress in neurodegenerative diseases. Nutr Neurosci. 2012;15:94–102. https://doi.org/10.1179/1476830511Y.0000000029
Chiocchetti AG, Haslinger D, Boesch M, Karl T, Wiemann S, Freitag CM, et al. Protein signatures of oxidative stress response in a patient specific cell line model for autism. Mol Autism. 2014;5:10. https://doi.org/10.1186/2040-392-5-10
Varga NÁ, Pentelényi K, Balicza P, Gézsi A, Reményi V, Hársfalvi V, et al. Mitochondrial dysfunction and autism: comprehensive genetic analyses of children with autism and mtDNA deletion. Behav Brain Funct. 2018;14:4. https://doi.org/10.1186/s12993-018-0135-x
Hwang IW, Hong JH, Kwon BN, Kim HJ, Lee NR, Lim MH, et al. Association of mitochondrial DNA 10398 A/G polymorphism with attention deficit and hyperactivity disorder in Korean children. Gene. 2017;630:8–12. https://doi.org/10.1016/j.gene.2017.08.004
Anitha A, Nakamura K, Thanseem I, Yamada K, Iwayama Y, Toyota T, et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism. 2012;3:12. https://doi.org/10.1186/2040-392-3-12
Floris G, Cadeddu R, Bortolato M. Handbook of behavioral neuroscience. vol. 31. Elsevier; 2020. p. 267–78.
Godar SC, Fite PJ, McFarlin KM, Bortolato M. The role of monoamine oxidase A in aggression: current translational developments and future challenges. Prog Neuropsychopharmacol Biol Psychiatry. 2016;69:90–100. https://doi.org/10.1016/j.pnpbp.2016.01.001
Holz N, Boecker R, Buchmann AF, Blomeyer D, Baumeister S, Hohmann S, et al. Evidence for a sex-dependent MAOAx childhood stress interaction in the neural circuitry of aggression. Cereb Cortex. 2016;26:904–14. https://doi.org/10.1093/cercor/bhu249
Bortolato M, Floris G, Shih JC. From aggression to autism: new perspectives on the behavioral sequelae of monoamine oxidase deficiency. J Neural Transm. 2018;125:1589–99. https://doi.org/10.1007/s00702-018-1888-y
Kolla NJ, Houle S. Single-photon emission computed tomography and positron emission tomography studies of antisocial personality disorder and aggression: a targeted review. Curr Psychiatry Rep. 2019;21:24. https://doi.org/10.1007/s11920-019-1011-6
Cecil CAM, Zhang Y, Nolte T. Childhood maltreatment and DNA methylation: a systematic review. Neurosci Biobehav Rev. 2020;112:392–409. https://doi.org/10.1016/j.neubiorev.2020.02.019
Byrd AL, Manuck SB. MAOA, childhood maltreatment, and antisocial behavior: meta-analysis of a gene-environment interaction. Biol Psychiatry. 2014;75:9–17.
Ducci F, Enoch MA, Hodgkinson C, Xu K, Catena M, Robin RW, et al. Interaction between a functional MAOA locus and childhood sexual abuse predicts alcoholism and antisocial personality disorder in adult women. Mol Psychiatry. 2008;13:334–47. https://doi.org/10.1038/sj.mp.4002034
Bennuri SC, Rose S, Frye RE. Mitochondrial dysfunction is inducible in lymphoblastoid cell lines from children with autism and may involve the TORC1 pathway. Front Psychiatry. 2019;10:269. https://doi.org/10.3389/fpsyt.2019.00269
Picard M, McEwen BS, Epel ES, Sandi C. An energetic view of stress: focus on mitochondria. Front Neuroendocrinol. 2018;49:72–85. https://doi.org/10.1016/j.yfrne.2018.01.001
Aon MA, Cortassa S, Juhaszova M, Sollott SJ. Mitochondrial health, the epigenome and healthspan. Clin Sci. 2016;130:1285–305. https://doi.org/10.1042/CS20160002
Horn SR, Leve LD, Levitt P, Fisher PA. Childhood adversity, mental health, and oxidative stress: a pilot study. PLoS ONE. 2019;14:e0215085. https://doi.org/10.1371/journal.pone.0215085
Hawes DJ, Brennan J, Dadds MR. Cortisol, callous-unemotional traits, and pathways to antisocial behavior. Curr Opin Psychiatry. 2009;22:357–62. https://doi.org/10.1097/YCO.0b013e32832bfa6d
du Boisgueheneuc F, Levy R, Volle E, Seassau M, Duffau H, Kinkingnehun S, et al. Functions of the left superior frontal gyrus in humans: a lesion study. Brain. 2006;129:3315–28. https://doi.org/10.1093/brain/awl244
Martin-Key N, Brown T, Fairchild G. Empathic accuracy in male adolescents with conduct disorder and higher versus lower levels of callous-unemotional traits. J Abnorm Child Psychol. 2017;45:1385–97. https://doi.org/10.1007/s10802-016-0243-8
Rogers JC, De Brito SA. Cortical and subcortical gray matter volume in youths with conduct problems: a meta-analysis. JAMA Psychiatry. 2016;73:64–72. https://doi.org/10.1001/jamapsychiatry.2015.2423
Yang Y, Raine A, Joshi AA, Joshi S, Chang YT, Schug RA, et al. Frontal information flow and connectivity in psychopathy. Br J Psychiatry. 2012;201:408–9. https://doi.org/10.1192/bjp.bp.111.107128
Hyatt CJ, Haney-Caron E, Stevens MC. Cortical thickness and folding deficits in conduct-disordered adolescents. Biol Psychiatry. 2012;72:207–14.
Pechorro P, Ribeiro da Silva D, Andershed H, Rijo D, Abrunhosa Goncalves R. The Youth Psychopathic Traits Inventory: measurement invariance and psychometric properties among Portuguese youths. Int J Environ Res Public Health 2016;13. https://doi.org/10.3390/ijerph13090852
Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry. 2019;9:47. https://doi.org/10.1038/s41398-019-0376-y
Gutierrez-Arcelus M, Ongen H, Lappalainen T, Montgomery SB, Buil A, Yurovsky A, et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015;11:e1004958. https://doi.org/10.1371/journal.pgen.1004958
Acknowledgements
This study was conducted as part of the FemNAT-CD consortium (Neurobiology and treatment of adolescent female conduct disorder: The Central Role of Emotion Processing, coordinator Christine M. Freitag). This collaborative project is funded by the European Commission under the 7th Framework Health Program with Grant Agreement no. 602407. We would like to thank all members of the FemNAT-CD consortium for their contributions to the project. We would also like to thank our participants, their families, and the numerous teachers, clinicians, social workers, foster carers and other professionals who gave their time generously to assist with recruitment and participate in the study. Elizabeth Farrow is a Ph.D. student funded by the Biotechnology and Biological Sciences Research Council (BBSRC)’s Midlands Integrative Biosciences Training Partnership (MIBTP). During the writing of the manuscript, Stephane A. De Brito was supported by a short-term Invitational Fellowship from the Japanese Society for the Promotion of Science (JSPS - S19103) and an International Academic Fellowship from the Leverhulme Trust (IAF-2019-032).
Author information
Authors and Affiliations
Contributions
The authors confirm that all individuals listed as authors meet authorship criteria. Authors who were also members of the FemNAT-CD Consortium steering committee were involved in the design of the FEMNAT-CD project methods and organisation of data collection. All listed authors reviewed the manuscript and provided critical inputs prior to submission for publication.
Corresponding authors
Ethics declarations
Competing interests
As stated above, all the authors of this paper were involved in the FemNAT-CD project, which was funded by the European Commission under the 7th Framework Health Program with Grant Agreement no. 602407. To the authors’ knowledge, there are no other biomedical financial interests or potential competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Farrow, E., Chiocchetti, A.G., Rogers, J.C. et al. SLC25A24 gene methylation and gray matter volume in females with and without conduct disorder: an exploratory epigenetic neuroimaging study. Transl Psychiatry 11, 492 (2021). https://doi.org/10.1038/s41398-021-01609-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01609-y
