
Abstract
Autism spectrum disorder (ASD) is a high cost neurodevelopmental condition; and there are currently no effective pharmacological treatments for its core symptoms. This has led some families and researchers to trial alternative remedies – including the non-intoxicating Cannabis sativa-derived compound cannabidivarin (CBDV). However, how CBDV affects the human brain is unknown. Previous (pre)clinical evidence suggests that CBDV may modulate brain excitatory-inhibitory systems, which are implicated in ASD. Hence, our main aim was to test, for the first time, if CBDV shifts glutamate and/or GABA metabolites – markers of the brain’s primary excitatory and inhibitory system - in both the ‘typical’ and autistic brain. Our subsidiary aim was to determine whether, within ASD, brain responsivity to CBDV challenge is related to baseline biological phenotype. We tested this using a repeated-measures, double-blind, randomized-order, cross-over design. We used magnetic resonance spectroscopy (MRS) to compare glutamate (Glx = glutamate + glutamine) and GABA + (GABA + macromolecules) levels following placebo (baseline) and 600 mg CBDV in 34 healthy men with (n = 17) and without (n = 17) ASD. Data acquisition from regions previously reliably linked to ASD (dorsomedial prefrontal cortex, DMPFC; left basal ganglia, BG) commenced 2 h (peak plasma levels) after placebo/CBDV administration. Where CBDV significantly shifted metabolite levels, we examined the relationship of this change with baseline metabolite levels. Test sessions were at least 13 days apart to ensure CBDV wash-out. CBDV significantly increased Glx in the BG of both groups. However, this impact was not uniform across individuals. In the ASD group, and not in the typically developing controls, the ‘shift’ in Glx correlated negatively with baseline Glx concentration. In contrast, CBDV had no significant impact on Glx in the DMPFC, or on GABA+ in either voxel in either group. Our findings suggest that, as measured by MRS, CBDV modulates the glutamate-GABA system in the BG but not in frontal regions. Moreover, there is individual variation in response depending on baseline biochemistry. Future studies should examine the effect of CBDV on behaviour and if the response to an acute dose of CBDV could predict a potential clinical treatment response in ASD.
Similar content being viewed by others

Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental condition estimated to affect up to 1 in 59 individuals1. ASD incurs a high cost. For instance, the average life expectancy in individuals with high functioning ASD is 12 years shorter than that in so-called ‘neurotypicals’2. Despite this, there are no effective pharmacological treatments for the core symptoms of ASD. Researchers are therefore increasingly exploring alternative remedies, such as the non-intoxicating Cannabis sativa-derived compounds cannabidiol (CBD) and cannabidivarin (CBDV)3,4. For instance, in a recently published study using magnetic resonance spectroscopy (MRS) we examined, for the first time, the effects of CBD on brain excitatory (E) glutamate and inhibitory (I) γ-aminobutyric acid (GABA) – given that E-I abnormalities may be one of the key mechanisms underpinning ASD3. We found that CBD modulated glutamate-GABA systems in both ASD and neurotypicals, but that prefrontal GABA systems responded differently in ASD. Specifically, CBD decreased prefrontal GABA levels in ASD but not in controls. These findings contributed to increasing evidence that an important effect of therapeutic compounds derived from Cannabis sativa may be the ‘shifting’ of brain E-I systems.
There is a growing number of clinical trials of these compounds, including the first trial in ASD, which investigates the impact of CBDV on irritability in autistic children (clinicaltrials.gov, identifier: NCT03202303). Unfortunately, however, most clinical trials which address ‘core’ social and/or repetitive behaviour symptoms of ASD fail. There are many reasons for this – but principal among them are that the candidate treatment may not ‘shift’ key mechanisms implicated in the disorder, and/or that they use an ‘all-comers’ approach (i.e., all patients meeting a broad set of eligibility criteria are included)5. This is relevant to ASD, which is a highly heterogeneous condition, where the response to candidate treatments may vary between individuals and where no single approach is likely to succeed for everyone5. A pressing goal should therefore be to determine objectively how candidate treatments, such as CBDV, impact upon the typical and atypical (autistic) brain; and who may be (biologically) responsive prior to clinical trial. Establishing an objective marker of biological response or ‘target engagement’ (e.g., using a marker of brain biochemistry) may be useful when a change in a biological target is linked to a change in behaviour. This may improve the success of drug trials by helping to predict if someone will respond – or not – to a pharmacological treatment with a change in behaviour, i.e., it may inform stratification approaches. This has not yet been achieved for CBDV.
We know from preclinical studies that CBDV may act on several neuroglial targets. Many of these effects are thought to converge to modulate glutamatergic and GABAergic pathways. For instance, CBDV binds to several members of the Transient Receptor Potential (TRP) family, including vanilloid type 1 (TRPV1), vanilloid type 2 (TRPV2), and ankyrin type 1 (TRPA1) receptors6 and, upon binding, CBDV is thought to activate and rapidly desensitize these receptors. TRP receptors are located on cortical and subcortical excitatory pyramidal cells7, inhibitory interneurons7, and microglia8, including in the basal ganglia (BG) and the dorsomedial prefrontal cortex (DMPFC)9,10,11,12,13. Thus, CBDV could potentially modulate the activity of neurons and glia directly involved in E-I regulation6 in key cortical and subcortical neural hubs. This is especially relevant to ASD, where subtle E-I alterations in several cortical-subcortical circuits14,15,16, including in the BG and DMPFC, have been linked to ASD symptoms17,18. Despite this, no study to date has examined the effect of CBDV on cortical and/or subcortical glutamate and/or GABA indices in-vivo in neurotypical individuals, let alone those with ASD.
Previous evidence suggests that autistic individuals may respond atypically to pharmacological challenge. For instance, in our recent study we discovered that, in the BG and DMPFC, CBD elicited a similar response in glutamate systems, but an opposite effect in GABA systems in the neurotypical compared to the autistic adult brain3. In addition to this between-group difference, our work has also revealed that there is response variability to pharmacological challenge within the autistic population3,18. These findings raise the question as to whether the biological response to CBDV also varies within the autistic population; and if so, which phenotypic measures may help predict treatment response. However, whether biology helps predict a (drug-induced) shift in E-I metabolite concentrations in ASD remains to be investigated.
Therefore, we examined for the first time: (1) the biological impact of CBDV on the typical and autistic brain, (2) if the response to CBDV is uniform across groups, and (3) the potential biological (baseline) correlates of response variability within ASD.
We tested this using a repeated-measures, randomized-order, placebo-controlled, double-blind, cross-over design. We compared magnetic resonance spectroscopy (MRS) measures of Glx (glutamate + glutamine) and GABA + (GABA+ macromolecules, see Methods) in adult men with and without a diagnosis of ASD following a single oral dose of 600 mg CBDV or a matched placebo. Data were collected from the BG and DMPFC because these regions have been reliably implicated in ASD, and alterations in E-I dynamics can be detected in these targets using MRS3,13,14,15,16,17,18. To explore variability of the drug response (where CBDV significantly shifted metabolite levels), we examined the relationship of any CBDV induced shift in metabolite with baseline metabolite levels.
Materials and Methods
Procedure
This research was conducted in accordance with the Declaration of Helsinki, at the Institute of Psychiatry, Psychology, and Neuroscience (IoPPN) at De Crespigny Park, SE5 8AF, London, UK (August 2016 to August 2018). All our participants provided written informed consent. Every participant took part in all aspects of this case-control, placebo-controlled, randomised, double-blind, repeated-measures, cross-over study. This research was conducted as part of a larger study into the role of phytocannabinoids in ASD (ethical approval provided by the King’s College London Research Ethics Committee, reference HR15/162744). Although the UK Medicines and Health Regulatory Authority (MHRA) confirmed that our experimental design was not a Clinical Trial, for transparency the study was registered on clinicaltrials.gov (identifier: NCT03537950, entry name: HR15-162744).
Drugs (placebo, PLC; or CBDV) were allocated in a pseudo-randomised order so that approximately half in each group attended a placebo visit before CBDV; and half attended a CBDV visit before placebo. This randomisation was conducted by Prof. McAlonan using a random number generator (https://www.random.org/). All participants and researchers directing the study were blinded to the order of drug/placebo exposure. Participants attended for two visits, which were separated by a minimum of 13 days to allow for drug wash-out. On each visit, we collected urine samples to screen for illicit substances (a full list is included below). Subsequently, participants underwent a brief health check, received a liquid oral dose of the pharmacological probe (600 mg of CBDV; in line with previous single dose studies of non-psychoactive cannabinoids in adults (e.g., ref. 19) or a matched placebo, both provided by GW Research Ltd, Cambridge, UK), and then underwent a second brief health check to test for potential acute adverse reactions/side effects. Participants commenced scanning timed to coincide with peak plasma (2 h) concentration. After the scan, participants underwent a final health check to ensure they had experienced no ill-effects and were fit to leave the department. Participants experienced no ill/unintended effects.
Participants
We excluded all potential participants that had a comorbid major psychiatric or medical disorder affecting brain development (e.g. schizophrenia or epilepsy), a known genetic condition associated with ASD (e.g. Fragile X syndrome), a history of brain/head injury, a full-scale intelligence quotient (FSIQ) below 70, or who were reliant on receiving regular medication known to directly influence Glx or GABA levels+, such as benzodiazepines. Participants were asked to refrain from using cannabis and/or other illicit substances in the month before scanning, and from drinking alcohol on the day before testing. Data from all individuals that screened positive for illicit substances in the urine drug screening were excluded. As a result, we retained data from 34 subjects (17 neurotypicals, 17 individuals with ASD) (see Table 1 for demographics). Power analyses suggested that this sample size was enough to detect a drug-induced 10% change in Glx (e.g. after ketamine administration20) at a power of 0.8 and a significance level of α = 0.05. All autistic participants had a clinical diagnosis of ASD according to ICD10 research criteria21,22,23, and their severity of symptoms was confirmed using standardised research diagnostic instruments where appropriate (ADOS Autism Diagnostic Observation Schedule23; and ADI-R Autism Diagnostic Interview-Revised22).
Imaging data acquisition
We acquired all our imaging data on a 3 T GE Excite II magnetic resonance imaging (MRI) scanner (GE Medical Systems, Milwaukee, WI, USA). Our scanning protocol included a structural MRI scan acquired using a 3D inversion recovery prepared fast spoiled gradient recalled (IR-FSPGR) sequence (slice thickness = 1.1 mm, spatial positions = 124, flip angle = 20°, field of view (FoV) = 280 mm, echo time (TE) = 2.844 ms, repetition time (TR) = 7.068 ms, inversion time = 450 ms, matrix = 256 × 256). This structural scan was conducted to obtain information used during the preprocessing of the spectroscopy scan. The scanning protocol further included a spectroscopy scan based on the MEshcher-GArwood Point RESolved Spectroscopy (MEGA-PRESS) sequence. We acquired data (352 datapoints) from two voxels: the first was positioned in the BG (echo time (TE) = 68 ms, repetition time (TR) = 1800 ms, voxel size = 35 × 30 × 25 mm3). The second voxel was positioned in the DMPFC (TE = 68 ms, TR = 2000 ms, voxel size = 25 × 40 × 30 mm3).
Urine test
We performed liquid chromatography–mass spectrometry (LC–MS) analysis on urine samples provided by each subject before the drug administration to evaluate presence or absence of illicit substances that could confound our results. Therefore, all participants that screened positive for any of the drugs tested, including Amphetamines (Amphetamine, Methamphetamine, MDMA/Ecstasy), Benzodiazepines, Cannabis, Cocaine (as benzoylecgonine), Methadone and its metabolite EDDP, and Opioids (6-monoacetylmorphine, morphine, codeine, dihydrocodeine) were excluded from further analysis. This led to the exclusion of four subjects (two typically developing controls, two ASD) from the original sample.
Data processing
Structural data processing
We inspected all T1-weighted structural MRI volumes manually to ensure adequate signal-to-noise ratio (SNR) and the absence of motion artefacts. Subsequently, we normalised our structural volumes to Montreal Neurological Institute (MNI) space and segmented them into grey matter (GM), white matter (WM), and cerebrospinal fluid (CSF), to obtain percentage measures of tissue composition in each individual MRS voxel, using positional coordinates embedded in the raw spectra data files.
Magnetic resonance spectroscopy data processing
We pre-processed our MRS data using in-house scripts adapted from FID-A24, which prepared the data for reading into the main processing software. This pre-processing comprised converting data to the correct file format, combining receiver channels, removing ‘bad’ averages (>4 standard deviations), correcting frequency drift (alignment of averages), separating and visualizing the edit on/off spectra, and subtracting them to produce the difference spectrum. We further inspected all spectra manually to ensure adequate SNR as well as the absence of artefacts25,27.
We processed our MRS data using LCModel v6.3-1L software (Stephen Provencher Incorporated, Oakville, Canada). This software uses a linear combination of model spectra derived from metabolite solutions in vitro to analyse the major resonances of in vivo spectra. Here, we used a basis set (mega-press-3T-1) to determine concentrations of GABA + (40% of the GABA signal represents macromolecules)25 and Glx (glutamate + glutamine). The spectra also included glutathione (GSH), N-acetyl-aspartate (NAA), N-acetyl-aspartylglutamate (NAAG), NAA + NAAG, and GSH + Glu + Gln in each voxel; however, for the purposes of this study, we focused only on GABA+ and Glx.
Partial volume effects (different proportions of GM, WM, and CSF in the MRS voxels) are a potential confound in MRS, especially in light of previously reported volumetric differences between autistic and neurotypical individuals26. Therefore, to account for partial volume effects, we corrected all metabolite values for GM, WM, and CSF percentages. Assuming that CSF only contains negligible quantities of the metabolites of interest, the calculations were as follows: LCModel assumes a voxel consists to 100% of WM with a water concentration (WCONC) of 35880 mM and corrects each metabolite value (where F stands for fraction) using the factor: (43300 ∗ FGM + 35880 ∗ FWM + 55556 ∗ FCSF)/(1 − FCSF). To correct for the value of water concentration being used in the processing through LCModel, we divided values by an individual correction factor (35880), arriving at (1.207 ∗ FGM + FWM + 1.548 ∗ FCSF)/(1 − FCSF). Thus, our corrected metabolite values were obtained by multiplying the raw metabolite values by this correction. Since we did not measure relaxation times for tissue water and metabolites, these were not corrected for - with the exception of assuming the tissue water relaxation time (T2 = 80 ms)27.
Moreover, to improve the robustness of our findings, we excluded all measurements of Glx (glutamate + glutamine) and GABA+ where the Cramér-Rao lower bound (CRLB) estimates exceeded 15% from further analysis (LCModel manual, Stephen Provencher Incorporated, Oakville, Canada). Based on this, we excluded eight data points from three subjects (1 neurotypical, 2 ASD): neurotypicals: 1: DMPFC GABA+CBDV; ASD: 2: BG GlxPLC; 3: DMPFC GABA+PLC & GlxPLC, DMPFC GABA+CBDV & GlxCBDV, and BG GABA+CBDV & GlxCBDV.
Statistical analysis
We compared demographic measures (age, FSIQ) and baseline levels of Glx and GABA+ in each region of interest using a one-way ANOVA (significance level p < 0.05).
To examine our primary hypothesis that CBDV shifts Glx and/or GABA+ in our two brain regions of interest (BG and DMPFC), we conducted 2 × 2 mixed-model ANOVAs with group (neurotypicals, ASD) as the between-subject factor, and drug (PLC, CBDV) as the within-subject factor for each metabolite in each region separately. We included the following subject numbers: for Glx measures in BG, ASD n = 12, neurotypicals n = 11; for Glx measures in DMPFC, ASD n = 12, neurotypicals n = 13; for GABA+ measures in BG, ASD n = 12, neurotypicals n = 13; and for GABA+ measures in the DMPFC, ASD n = 11, neurotypicals n = 11. With the caveat that Bonferroni correction can be overly conservative, we also report Bonferroni corrected p-values alongside any significant uncorrected results.
We examined the relationship between significant drug-induced shifts in metabolite levels (CBDV-PLC) and baseline metabolite measures across the ASD group using Pearson’s correlation analyses (p < 0.05).
Analyses were carried out using SPSS 24.00 software (SPSS, Chicago, IL, USA), and graphs were generated using GraphPad Prism version 7 for Mac, GraphPad Software, La Jolla, CA, USA, www.graphpad.com.
Results
Demographics
We retained data from 17 neurotypicals (three of those only had a scan after placebo and not after CBDV), and from 17 individuals with ASD (four of those only had a scan after placebo and not after CBDV). Groups were similar in age (F(1) = 0.956, p = 0.335), but, as is commonly reported, autistic individuals had a slightly lower FSIQ than typically developing controls and this difference was significant (F(1) = 5.781, p = 0.022). However, FSIQ did not correlate with drug-induced metabolite shifts across groups (all r ≤ 0.34, all p ≥ 0.122). FSIQ correlated with Glx shift in the BG within the ASD group (r = 0.70, p = 0.016, n = 11), but this correlation was not significant in the neurotypicals (r = −0.10, p = 0.765, n = 11) and did not differ significantly between groups (p > 0.05). This suggests that the between-group difference in FSIQ did not influence our results (Table 1).
Tissue composition and data quality
Tissue percentages (not excluding omitted spectra) differed significantly between groups for BG PLC GM (F(1) = 7.307, p = 0.011), BG PLC WM (F(1) = 9.345, p = 0.004), BG CBDV GM (F(1) = 6.70, p = 0.016), and for BG CBDV WM (F(1) = 7.89, p = 0.010); but not for other tissues, as summarised in Table 2. This is in line with previous studies that have demonstrated morphological differences in the BG in autistic compared to neurotypical individuals26,28. Moreover, as the composition properties of these tissues differed significantly at baseline and during the drug condition, this difference was not induced through CBDV. In the statistical analysis we corrected all metabolite values accordingly.
To ensure that the MRS data quality did not differ between groups, we compared the LCModel Cramér-Rao Lower Bound estimates for each metabolite (Glx, GABA+) in each voxel (excluding omitted spectra), using a one-way ANOVA29. As predicted, there were no significant differences (all F(1) ≤ 2.751, all p ≥ 0.107), as depicted in Table 3.
Extended MRS studies can be confounded by ‘drift’, where metabolite estimates on the same scanner change across long periods of time. Therefore, we compared the inter-scan duration (days between PLC and CBDV scan) between groups; and found no significant difference (F(1) = 0.487, p = 0.492), as shown in Table 1. Moreover, scan date for each drug condition (PLC, CBDV) did not correlate with metabolite values at that drug condition (all Pearson’s r ≤ 0.329, all p ≥ 0.087), confirming that data acquisition was stable over time.
Metabolite differences
Glx (glutamate + glutamine)
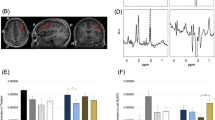
In the BG, we observed a significant effect of drug (F(1, 21) = 7.268, puncorr = 0.014, η2 = 0.257). Across groups, CBDV increased Glx compared to PLC, as displayed in Fig. 1a, b. This effect survived correction for multiple comparisons across voxels (pcorr = 0.03). In contrast, there was no significant effect of group (F(1) = 0.763, p = 0.392, η2 = 0.035), and no group × drug interaction effect (F(1, 21) = 0.235, p = 0.633, η2 = 0.011). In the DMPFC, there was no significant effect of drug or group, and no group × drug interaction effect (Fig. 1a).

a Glx (glutamate + glutamine) in the basal ganglia and dorsomedial prefrontal cortex for both groups in both drug conditions. Glx concentration (y-axis) represents the ratio of the Glx metabolite resonance area to the unsuppressed water resonance area. b Drug-induced shift in Glx in the basal ganglia. ΔGlx (y-axis) represents the cannabidivarin (CBDV)-induced shift in Glx concentration compared to placebo (PLC), i.e. CBDV-PLC. Group means are indicated by black horizonal bars. Error bars represent standard deviations. c gamma-aminobutyric acid + macromolecules (GABA+) in the basal ganglia and dorsomedial prefrontal cortex for both groups in both drug conditions. GABA+ concentration (y-axis) represents the ratio of the GABA+ metabolite resonance area to the unsuppressed water resonance area. d Correlation between Glx at baseline and CBDV-induced shift in Glx (ΔGlx) in the basal ganglia. ASD Autism spectrum disorder, BG Basal ganglia, CBDV Cannabidivarin, DMPFC Dorsomedial prefrontal cortex, Glx glutamate + glutamine, ns Not significant, PLC Placebo, TD Typically developing controls, * indicates a significance level at p ≤ 0.05.
GABA+
We observed no significant main or interaction effects within either voxel (Fig. 1c).
Relationship to baseline metabolite measures
There was a significant negative correlation between the CBDV-induced BG Glx shift and baseline Glx levels in ASD (r = −0.749, p = 0.005, n = 12), but not in the typically developing controls (r = 0.014, p = 0.967, n = 11). This correlation differed significantly between groups (p = 0.04). (Fig. 1d). In interpreting this result, we caution that recent studies suggest that correlations between baseline and change scores in randomized controlled trials are common and may represent a regression to the mean, rather than the treatment effect30. To account for this effect in our analyses, we re-ran our repeated-measures ANOVAs as ANCOVAs with baseline metabolite levels as a covariate and found that the directionality and significance of our results remained the same. We still observed an opposite direction of correlation between baseline levels and drug-induced changes in Glx in the two groups. We emphasize that our study is not designed to investigate the degree to which any correlation with baseline reflects underlying neurobiology or a purely statistical effect. However, it is possible that a biological group differences at least partly contributes to the different relationships found in each group.
Discussion
We have demonstrated that a single acute dose of CBDV ‘shifts’ subcortical levels of Glx, the brain’s primary excitatory neurotransmitter, in the living adult human brain. The direction of shift was the same across the neurotypical and autistic participants, i.e., CBDV induced a mean increase in BG Glx in both groups. However, the response to CBDV varied within the ASD group and correlated negatively with baseline BG Glx levels. In contrast, CBDV had no impact on Glx in DMPFC, nor on GABA+ levels in either voxel. Moreover, congruent with some18,30, but not all previous MRS studies of glutamate and GABA in ASD18,31,32 in the BG and DMPFC, there were no between-group differences in baseline metabolite levels.
Thus, our study suggests that CBDV targets subcortical excitatory glutamate systems both in autistic and neurotypical adults; but that individual responses in autistic brains vary depending on baseline Glx levels.
Neurobiological underpinnings of the effect of CBDV on Glx in the BG
Our findings suggest that CBDV may serve as a tool to shift Glx in specific subcortical regions (the left BG) both in the neurotypical and autistic brain. The neurobiological underpinnings of this effect, however, are not entirely clear. Previous evidence suggests several possible neurobiological mechanisms through which CBDV may influence Glx pathways in the BG. For instance, preclinical studies have shown that the BG contain a wealth of excitatory pyramidal (and inhibitory projection) neurons33, which are densely surrounded by TRP receptors (including TRPV1, TRPV2, and TRPA1)10,34,35. CBDV may have bound to and subsequently activated these pyramidal neuron-bound TRP receptors, thereby increasing Glx in this region. This is supported by previous evidence that CBDV may modulate excitatory neurotransmission through a TRP receptor-dependent mechanism6. The BG also contain a particularly high number of microglia36; and it is possible that CBDV increased Glx levels here by activating microglial TRP receptors. Specifically, activation of TRP receptors has been shown to upregulate microglial activity and migration8, which is known to enhance extracellular vesicular shedding and subsequent glutamate release8,37. The prefrontal cortex has a different configuration of receptors and cells compared to the BG, which might explain a different response to CBDV here. We have previously reported that CBD causes a differential effect on prefrontal GABA+ in the autistic compared to neurotypical brain. Unfortunately, the cellular mechanisms underpinning the effects of cannabinoids have not yet been fully uncovered. Consequently, we do not yet know which cellular response differences contribute to their divergent effects on excitatory and inhibitory metabolites at ‘bulk tissue’ level. In sum, across groups, CBDV may have shifted Glx through multiple neuroglial mechanisms.
Drug-response variability in ASD in relation to baseline measures
Although the mean effect of CBDV on Glx was uniform across groups, the drug response varied within groups. In ASD (but not in the neurotypicals), the drug-induced shift in BG Glx correlated significantly (negatively) with baseline Glx. Specifically, in the autistic individuals with the lowest baseline Glx measures, CBDV increased Glx levels; whereas the individuals with the highest baseline Glx measures experienced a decrease in Glx. Thus, these findings highlight the presence of intra-group response variability within ASD, where subjects may respond to drug challenge not just to different extents but also in opposite directions. Consequently, clinical trials of CBDV may find that not every participant responds similarly to CBDV treatment. Further research efforts may need to focus on identifying participant subgroups that will respond to treatment in a similar way.
Implications of a BG Glx shift for cognition and behaviour
The utility of this observed shift in BG Glx for predicting an individual’s long term (clinical) treatment response is unclear; and was not part of our study. Previous research suggests that the BG are connected with other cortical and subcortical regions in the form of subcortical-cortical-thalamic loops, which are regulated and maintained by excitatory and inhibitory neurotransmission38,39. These loops support a range of cognitive functions and behaviours, such as reward, learning, memory, and motor processing39,40,41,42,43,44,45,46,47,48,49,50, which can be impaired in ASD51,52. Future studies should therefore examine the impact of CBDV on these cognitive processes and behaviours to determine if CBDV may offer clinical benefits in specific autistic subgroups.
Limitations
Our findings must be considered alongside several limitations. We measured metabolite concentrations using MRS, which is unable to distinguish between the specific metabolite contributions to both the Glx and GABA+ signal. Moreover, the poor spatial resolution of MRS only allows ‘bulk’ assessments, which limited our ability to discern intra- and extra-cellular metabolite levels. Additional studies using more advanced technologies, such as (ultra-) high-field MRS are required to address this limitation.
MRS further has the disadvantage of relying on pre-selected regions-of-interest to be defined before the study. Thus, we cannot know how CBDV might modulate metabolites in other parts of the brain. This may change in the future with the development of sequences that allow whole-brain scanning53. At this point however, we are not aware of a scanning sequence that allows whole-brain spectroscopy of glutamate and GABA in a reasonable time frame.
Also, our sample size in this pilot study was modest. This was due to (i) our strict inclusion and exclusion criteria (e.g. exclusion of participants using glutamate and GABA-acting drugs); (ii) our time-intensive repeated-measures testing (involving drug administration), which was not always practical for participants; and (iii) our rigorous data quality control, e.g. exclusion of datasets based on head motion, which is known to be an issue in ASD. However, our repeated-measures design mitigated against this by reducing inter-subject variability (each subject acted as their own ‘control’) and thus increasing statistical power. Also, our sample size was comparable to (or bigger than) that in previous MRS studies in ASD18,30.
Moreover, CBDV remains an under-investigated compound. For instance, although plasma levels of CBDV are thought to peak on average two hours after administration, the exact times and concentrations may vary between individuals depending on several factors such as age, body-weight, size, genetics, absorption, distribution, and metabolism54. This made capturing the maximum drug effects for each participant challenging. Therefore, to minimize variability in drug levels, all efforts were made to match participants (e.g. by age) and to keep study procedures and timings consistent. Nonetheless, future studies should explore in more detail the pharmacodynamics of CBDV (e.g. effects of acute vs steady-state/long-term dosing), origins of inter-subject variability, and how this can be accounted for in pharmacological studies.
Finally, this was a first study of potential tissue level changes in metabolites following CBDV. Achieving even this modest sample size required a total of around 76 study visits. Therefore, we prioritized sample homogeneity and applied strict exclusion criteria. However, this approach has the disadvantage of potentially limiting the generalizability of our findings. We hope that having observed a biological response to CBDV in adult men with and without ASD (i.e. a Proof of Concept) will encourage subsequent studies to explore the effects of CBDV in women, children, those with intellectual difficulties, and other groups.
Conclusions
Here we report that CBDV can ‘shift’ subcortical levels of the brain’s primary excitatory metabolite glutamate (measured as Glx) both in the neurotypical and autistic brain; but that there may be significant response variability in ASD. These findings add to our understanding of the effects of CBDV in the adult human brain. Nonetheless, future studies will need to explore (i) the mechanisms of action of CBDV; (ii) the impact of CBDV on (ASD-related) cognition and behaviour; (iii) if single-dose responsivity could facilitate the identification of pharmacologically homogeneous sub-groups; and (iv) if acute CBDV effects are indicative of the impact of long-term treatment in ASD.
References
Baio J. et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014 (vol 67, pg 1, 2018). Mmwr-Morbid Mortal W. 2018;67(19):564.
Hirvikoski, T. et al. Premature mortality in autism spectrum disorder. Br. J. Psychiatry 208, 232–238 (2016).
Pretzsch C. M., et al. Effects of cannabidiol on brain excitation and inhibition systems; a randomised placebo-controlled single dose trial during magnetic resonance spectroscopy in adults with and without autism spectrum disorder. Neuropsychopharmacology 44, 1398–1405 (2019).
Pretzsch C. M. et al. The effect of cannabidiol (CBD) on low-frequency activity and functional connectivity in the brain of adults with and without autism spectrum disorder (ASD). J. Psychopharmacol. 269881119858306 33, (2019).
Loth, E. et al. Identification and validation of biomarkers for autism spectrum disorders. Nat. Rev. Drug Discov. 15, 70–73 (2016).
Iannotti, F. A. et al. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem. Neurosci. 5, 1131–1141 (2014).
Gibson, H. E., Edwards, J. G., Page, R. S., Van Hook, M. J. & Kauer, J. A. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 57, 746–759 (2008).
Miyake, T., Shirakawa, H., Nakagawa, T. & Kaneko, S. Activation of mitochondrial transient receptor potential vanilloid 1 channel contributes to microglial migration. Glia 63, 1870–1882 (2015).
Liapi, A. & Wood, J. N. Extensive co-localization and heteromultimer formation of the vanilloid receptor-like protein TRPV2 and the capsaicin receptor TRPV1 in the adult rat cerebral cortex. Eur. J. Neurosci. 22, 825–34. (2005).
Musella, A. et al. TRPV1 channels facilitate glutamate transmission in the striatum. Mol. Cell Neurosci. 40, 89–97 (2009).
Toth, A. et al. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res. Mol. Brain Res. 135, 162–168 (2005).
Fogaca, M. V., Aguiar, D. C., Moreira, F. A. & Guimaraes, F. S. The endocannabinoid and endovanilloid systems interact in the rat prelimbic medial prefrontal cortex to control anxiety-like behavior. Neuropharmacology 63, 202–210 (2012).
Lagatta, D. C., Kuntze, L. B., Ferreira-Junior, N. C. & Resstel, L. B. M. Medial prefrontal cortex TRPV1 and CB1 receptors modulate cardiac baroreflex activity by regulating the NMDA receptor/nitric oxide pathway. Pflug. Arch. 470, 1521–42. (2018).
Nelson, S. B. & Valakh, V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698 (2015).
Rubenstein, J. L. & Merzenich, M. M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267 (2003).
Yizhar, O. et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178 (2011).
Cochran, D. M. et al. Relationship among glutamine, gamma-aminobutyric acid, and social cognition in autism spectrum disorders. J. Child Adolesc. Psychopharmacol. 25, 314–322 (2015).
Horder, J. et al. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: a [(1)H]MRS study. Transl. Psychiatry 3, e279 (2013).
Bhattacharyya, S. et al. Cannabinoid modulation of functional connectivity within regions processing attentional salience. Neuropsychopharmacology 40, 1343–1352 (2015).
Stone, J. M. et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol. Psychiatry 17, 664–665 (2012).
World Health Organisation. International Statistical Classification of Diseases and Related Health Problems. Geneva, Switzerland (2016).
Lord, C., Rutter, M. & Le Couteur, A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 24, 659–685 (1994).
Lord, C. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J. Autism Dev. Disord. 19, 185–212 (1989).
Simpson, R., Devenyi, G. A., Jezzard, P., Hennessy, T. J. & Near, J. Advanced processing and simulation of MRS data using the FID appliance (FID-A)An open source, MATLAB-based toolkit. Magn. Reson Med. 77, 23–33 (2017).
Rothman, D. L., Petroff, O. A., Behar, K. L. & Mattson, R. H. Localized 1H NMR measurements of gamma-aminobutyric acid in human brain in vivo. Proc. Natl Acad. Sci. USA 90, 5662–5666 (1993).
Hollander, E. et al. Striatal volume on magnetic resonance imaging and repetitive behaviors in autism. Biol. Psychiatry 58, 226–232 (2005).
Tong, Z. Y., Yamaki, T., Harada, K. & Houkin, K. In vivo quantification of the metabolites in normal brain and brain tumors by proton MR spectroscopy using water as an internal standard. Magn. Reson. Imaging 22, 735–742 (2004).
Schuetze, M. et al. Morphological alterations in the thalamus, striatum, and pallidum in autism spectrum disorder. Neuropsychopharmacology 41, 2627–2637 (2016).
Kreis, R. The trouble with quality filtering based on relative cramer-rao lower bounds. Magn. Reson Med. 75, 15–18 (2016).
Ajram, L. A. et al. Shifting brain inhibitory balance and connectivity of the prefrontal cortex of adults with autism spectrum disorder. Transl. Psychiatry 7, e1137 (2017).
Bejjani, A. et al. Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PLoS ONE 7, e38786 (2012).
Gaetz, W. et al. GABA estimation in the brains of children on the autism spectrum: measurement precision and regional cortical variation. Neuroimage 86, 1–9 (2014).
Conn, P. J., Battaglia, G., Marino, M. J. & Nicoletti, F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat. Rev. Neurosci. 6, 787–798 (2005).
Cristino, L. et al. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 139, 1405–1415 (2006).
Kunert-Keil C., Bisping F., Kruger J., Brinkmeier H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics. 7, 159 (2006).
Lawson, L. J., Perry, V. H., Dri, P. & Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170 (1990).
Marrone, M. C. et al. TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat. Commun. 8, 15292 (2017).
DeLong, M. & Wichmann, T. Changing views of basal ganglia circuits and circuit disorders. Clin. EEG Neurosci. 41, 61–67 (2010).
DeLong, M. R. & Wichmann, T. Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 64, 20–24 (2007).
Jaramillo, T. C., Liu, S., Pettersen, A., Birnbaum, S. G. & Powell, C. M. Autism-related neuroligin-3 mutation alters social behavior and spatial learning. Autism Res. 7, 264–272 (2014).
D’Mello, A. M. & Stoodley, C. J. Cerebro-cerebellar circuits in autism spectrum disorder. Front Neurosci. 9, 408 (2015).
Moseley R. L. et al. Lost for emotion words: what motor and limbic brain activity reveals about autism and semantic theory. Neuroimage. 104, 413–422 (2015).
Sepeta, L. et al. Abnormal social reward processing in autism as indexed by pupillary responses to happy faces. J. Neurodev. Disord. 4, 17 (2012).
el Kaliouby, R., Picard, R. & Baron-Cohen, S. Affective computing and autism. Ann. N. Y Acad. Sci. 1093, 228–248 (2006).
Kennedy, D. P. & Courchesne, E. Functional abnormalities of the default network during self- and other-reflection in autism. Soc. Cogn. Affect Neurosci. 3, 177–190 (2008).
Boucher J. & Bowler D. M. Memory in autism. Cambridge, UK; New York: Cambridge University Press; 2008. xxiv, 358.
Baron-Cohen, S., Wheelwright, S., Hill, J., Raste, Y. & Plumb, I. The “Reading the Mind in the Eyes” Test revised version: a study with normal adults, and adults with Asperger syndrome or high-functioning autism. J. Child Psychol. Psychiatry 42, 241–51. (2001).
Calabresi, P., Picconi, B., Tozzi, A. & Ghiglieri, V. Interaction between basal ganglia and limbic circuits in learning and memory processes. Parkinsonism Relat. Disord. 22(Suppl 1), S65–S68 (2016).
Ikeda, H. et al. Investigating complex basal ganglia circuitry in the regulation of motor behaviour, with particular focus on orofacial movement. Behav. Pharmacol. 26, 18–32 (2015).
Haber, S. N. & Knutson, B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology 35, 4–26 (2010).
Scott-Van Zeeland, A. A., Dapretto, M., Ghahremani, D. G., Poldrack, R. A. & Bookheimer, S. Y. Reward processing in autism. Autism Res. 3, 53–67 (2010).
Delmonte, S. et al. Social and monetary reward processing in autism spectrum disorders. Mol. Autism 3, 7 (2012).
Zhang, Y. et al. Comparison of reproducibility of single voxel spectroscopy and whole-brain magnetic resonance spectroscopy imaging at 3T. NMR Biomed. 31, e3898 (2018).
Luscombe, D. K. Factors influencing plasma drug concentrations. J. Int Med Res. 5, 82–97 (1977).
Acknowledgements
This study was an Investigator Initiated Study (G.M.) which received funding and product from GW Research Ltd (Cambridge, UK). GW Research Ltd (Cambridge, UK) had no role in the data collection or analysis of results, nor in the decision to publish. The authors also acknowledge infrastructure and training support from the National Institute for Health Research (NIHR) Mental Health Biomedical Research Centre (BRC) at the South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health, UK. Additional sources of support included the Sackler Institute for Translational Neurodevelopment at King’s College London, Autistica, a Medical Research Council (MRC) Centre grant (MR/N026063/1), and EU-AIMS (European Autism Interventions)/EU AIMS-2-TRIALS, a European Innovative Medicines Initiative Joint Undertaking under Grant Agreements No. 115300 and 777394, the resources of which are composed of financial contributions from the European Union’s Seventh Framework Programme (Grant FP7/2007–2013). R.A.E.E. receives salary support from NIH R01 MH106564 and U54 HD079123.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pretzsch, C.M., Voinescu, B., Lythgoe, D. et al. Effects of cannabidivarin (CBDV) on brain excitation and inhibition systems in adults with and without Autism Spectrum Disorder (ASD): a single dose trial during magnetic resonance spectroscopy. Transl Psychiatry 9, 313 (2019). https://doi.org/10.1038/s41398-019-0654-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0654-8
This article is cited by
-
Cross-sectional and longitudinal neuroanatomical profiles of distinct clinical (adaptive) outcomes in autism
Molecular Psychiatry (2023)
-
Review: Cannabinoids as Medicinals
Current Addiction Reports (2022)
-
The Feasibility of Magnetic Resonance Imaging in a Non-Selective Comprehensive Clinical Trial in Pediatric Autism Spectrum Disorder
Journal of Autism and Developmental Disorders (2022)
-
Modulation of striatal functional connectivity differences in adults with and without autism spectrum disorder in a single-dose randomized trial of cannabidivarin
Molecular Autism (2021)
