
Abstract
Cannabis, the most widely used illicit drug, can induce hallucinations. Our understanding of the biology of cannabis-induced hallucinations (Ca-HL) is limited. We used the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA) to identify cannabis-induced hallucinations (Ca-HL) among long-term cannabis users (used cannabis ≥1 year and ≥100 times). A genome-wide association study (GWAS) was conducted by analyzing European Americans (EAs) and African Americans (AAs) in Yale-Penn 1 and 2 cohorts individually, then meta-analyzing the two cohorts within population. In the meta-analysis of Yale-Penn EAs (n = 1917), one genome-wide significant (GWS) signal emerged at the CHRM3 locus, represented by rs115455482 (P = 1.66 × 10−10), rs74722579 (P = 2.81 × 10−9), and rs1938228 (P = 1.57 × 10−8); signals were GWS in Yale-Penn 1 EAs (n = 1092) and nominally significant in Yale-Penn 2 EAs (n = 825). Two SNPs, rs115455482 and rs74722579, were available from the Collaborative Study on the Genetics of Alcoholism data (COGA; 3630 long-term cannabis users). The signals did not replicate, but when meta-analyzing Yale-Penn and COGA EAs, the two SNPs’ association signals were increased (meta-P-values 1.32 × 10−10 and 2.60 × 10−9, respectively; n = 4291). There were no significant findings in AAs, but in the AA meta-analysis (n = 3624), nominal significance was seen for rs74722579. The rs115455482*T risk allele was associated with lower CHRM3 expression in the thalamus. CHRM3 was co-expressed with three psychosis risk genes (GABAG2, CHRNA4, and HRH3) in the thalamus and other human brain tissues and mouse GABAergic neurons. This work provides strong evidence for the association of CHRM3 with Ca-HL and provides insight into the potential involvement of thalamus for this trait.
Similar content being viewed by others

Introduction
Cannabis is the most widely used illicit drug. It has acute and chronic effects on physical and mental health; adverse effects include rapid heartbeat, disorientation, lack of physical coordination, panic attacks or anxiety, depression or sleepiness, deterioration in cognitive function, and brain abnormalities after long-term use1,2,3. The pharmacological effects of cannabis are due primarily to tetrahydrocannabinol (THC), which mimics the activity of endocannabinoids such as anandamide. Both THC and endocannabinoids efficiently bind to the G-protein-coupled cannabinoid receptor, CB1, in the brain and transiently inhibit the release of either the inhibitory neurotransmitter γ-aminobutyric acid (GABA) or the excitatory transmitter glutamate4. Although there is another well-characterized cannabinoid G-protein-coupled receptor, CB2, only CB1 receptors are abundantly expressed in the brain, where they are localized specifically on axons and axon terminals. These effects are largely responsible for the psychoactive effects of cannabis, which include potentially therapeutic ones (e.g., analgesia) and reinforcing effects (e.g., relaxation, hallucination, or altered perception)3,5. Our understanding of the biology of cannabis-induced hallucinations (Ca-HL) remains limited.
Genome-wide association study (GWAS) is a useful strategy to study the genetics and ultimately the biology of hallucinations. Here we carried out the first GWAS of Ca-HL among European American (EA) (total N = 4291) and African American (AA) (total N = 3624) long-term cannabis users. We identified one genome-wide significant (GWS) signal close to the gene cholinergic receptor muscarinic 3 (CHRM3), predisposing to Ca-HL in EAs, with the finding nominally replicated in AAs.
Materials and methods
Subjects, genotyping, and imputation
Subjects were selected from among previously described samples, Yale-Penn 1 (n = 5540) and 2 (n = 3675)6, which were recruited from five eastern US sites to participate in studies of the genetics of drug (opioid or cocaine) or alcohol dependence6. All participants were given written informed consent that was approved by the institutional review board at each recruiting site. Certificates of confidentiality were provided by the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism. Yale-Penn 1 samples were genotyped on the Illumina (San Diego, CA, USA) HumanOmni1-Quad v1.0 microarray. A total of 1,140,419 single-nucleotide polymorphisms (SNPs) were genotyped in Yale-Penn 1. Samples for Yale-Penn 2 were genotyped with the Illumina HumanCore Exome array, which includes a total of 550,601 SNPs, including 268,631 exonic SNPs and 281,970 tagging SNPs. Quality control (QC) for microarrays in each cohort was carried out using PLINK1.97 based on the following criteria: (1) individual genotype missing rate < 2%, (2) SNP genotype missing rate < 2%, (3) Hardy–Weinberg P > 1 × 10−6, and (4) minor allele frequency (MAF) > 3%. After QC, samples from Yale-Penn 1 and 2 were subjected to ancestry analysis by comparison with the 1000 Genomes Project phase 1 reference panel8. Eigensoft9 was used for principal components (PCs) analysis with the first ten PC scores serving to differentiate EAs and AAs through K-means clustering10. For each Yale-Penn cohort, SNPs from EAs and AAs were imputed together using Minimac3 implemented in the Michigan Imputation Server11 with the 1000 Genomes phase 3 reference panel. We transformed dosage data into best-estimate genotypes using PLINK1.9, retaining high-quality genotyping data by filtering imputed data with genotype imputation probability (GP) ≥ 0.9; the resulting genotype data were transformed into plink binary format data, which can be used directly in association tests with the GWAS software, Genome-wide Efficient Mixed Model Association (GEMMA)12. After retaining genotypes with GP ≥ 0.9, individual genotyping missing rate < 5%, MAF > 3%, and missing call frequency < 5%, there were 8,200,853 and 5,916,265 remaining variants for Yale-Penn 1 and 2 EAs, respectively, and 14,134,502 and 10,346,266 variants for Yale-Penn 1 and 2 AAs, respectively.
Apart from the above Yale-Penn samples used in our discovery GWAS, an independent cohort, the Collaborative Study on the Genetics of Alcoholism data (COGA; see Supplementary Information for detailed description of COGA) was assigned as the replication cohort for the top hits that emerged from Yale-Penn samples.
Definition of Ca-HL
Ca-HL was defined using similar questions in the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA)13 (for Yale-Penn) and the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA)14 (for COGA). For the SSADDA, this question is, “Has your use of [marijuana] ever caused you emotional or psychological problems like: Hearing, seeing, or smelling things that weren’t really there?” In the SSAGA, the question is “Because of your marijuana use, did you ever experience any of the following: Hearing, seeing or smelling things that weren’t really there?”. As in Yale-Penn samples >90% of participants with Ca-HL used cannabis ≥1 year and ≥100 times, these two criteria were used to define comparison subjects (controls) who were long-term cannabis users without Ca-HL. The same criteria were used to select samples from COGA. The sample sizes of our GWAS were not predesigned for ensuring adequate power to detect a pre-specific effect size.
Ca-HL sample description
For Ca-HL samples in Yale-Penn cohorts, there were 1092 Yale-Penn 1 EAs (cases = 51 and controls = 1041), 825 Yale-Penn 2 EAs (cases = 40 and controls = 785), 1610 Yale-Penn 1 AAs (cases = 149 and controls = 1461), and 758 Yale-Penn 2 AAs (cases = 71 and controls = 687). Meanwhile, for replication samples from COGA, 2374 EAs (cases = 256 and controls = 2118) and 1256 (cases = 142 and controls = 1114) AAs had genotypic data and reported long-term use of cannabis.
GWAS analysis
GWAS software
GEMMA12 was used separately in each Yale-Penn subgroup of EAs and AAs, with adjustment for sex, age, body mass index (BMI), the first three PCs of ancestry, and the degree of relatedness among subjects. BMI was used as covariant because of the consideration that it could be a potential confounding factor affecting Ca-HL. GEMMA uses linear mixed model to determine the association between SNP and phenotype; it can account for relatedness among participants and can control for population stratification and other confounding factors12. We used GEMMA because it allowed us to account for ancestry. A correction for inflation was not necessary, because the inflation was low (all λ < 1.1) in our GWAS. For summary statistics from GEMMA, the inverse variance method implemented in PLINK1.9 was used to generate fixed-effects meta-analysis P-values (meta-P) for all variants by matching their chromosomal positions and two alleles among the GWAS datasets from EAs and AAs separately. We used a GWS threshold of P < 5.0 × 10−8. GWAS summary statistics data for Yale-Penn samples are freely available upon request.
For the two GWS variants that emerged in the meta-analysis of Yale-Penn cohorts, we used logistic regression via generalized estimating equation implemented in the R package GWAF15, to correlate the two GWS variants’ genotypes from COGA with Ca-HL (defined exactly as for Yale-Penn) and adjust for sex, age, the first three PCs of ancestry, and array types. As BMI was not available in COGA, it was not included as a covariate in the analysis. The association results for these two GWS variants were combined with the summary statistics of Yale-Penn 1 and 2, and COGA in the final meta-analysis using PLINK1.9.
Correlation between rs115455482 genotype and CHRM3 expression
To facilitate the biological interpretation of our top association signal, we explored the potential relationship between rs115455482 and CHRM3 expression across ten brain tissues. We associated rs115455482 genotype with CHRM3 expression in Braineac16, where genotype and gene expression data of ten brain tissues, including occipital cortex (OCTX), frontal cortex (FCTX), temporal cortex, hippocampus (HIPP), intralobular white matter, cerebellar cortex (CRBL), thalamus (THAL), putamen (PUTM), substantia nigra (SNIG), and medulla (inferior olivary nucleus, MEDU), were obtained from 134 healthy human brains from the UK Biobank17.
CHRM3 co-expression analysis and disease gene-enrichment analysis
Gene co-expression information can be used to evaluate a gene’s function and identify related pathways. To accomplish this for CHRM3 in Ca-HL, a novel bioinformatics pipeline was created to search for genes co-expressed with CHRM3 in an unbiased way genomewide. First, CHRM3 co-expressed genes were identified with COXPRESdb18, a database that provides gene co-expression relationships for animal species, including human, mouse, rat, and others. Next, the top 100 CHRM3 co-expressed genes, as well as CHRM3 itself (total 101 genes), were subjected to enrichment analysis of disease-associated genes using the 2013 version WEB-based GEne SeT AnaLysis Toolkit (WebGestalt)19 with default settings of a minimum number of four genes (out of 101) required for a gene category to be included in the enrichment analysis. Enrichment analysis method “Disease Association Analysis” of WebGestalt was used to test the enrichment of disease-associated genes among these 101 genes. Finally, CHRM3 co-expressed genes that emerged in the top ten enriched gene categories of disease-associated genes were validated for co-expression with CHRM3 using ten different types of human healthy brain tissues (downloaded from Braineac database)17 and the homologous gene in mouse, Chrm3, using six types of cortical GABAergic neurons from mouse FCTX (gene expression omnibus accession number GSE92522)20. The ten brain tissues (total samples = 1340) are the same as these listed above. In addition, we considered six types of mouse single neurons (total samples = 584): martinotti cells, interneuron selective cells, cholecystokinin expressing (CCK)-basket cells, parvalbumin expressing (PV) basket cells, chandelier cells upper layer (CHC1) and deep layer (CHC2) of mouse brain, and long projecting cells. All of the cells were isolated from mouse FCTX. The correlation between CHRM3 and its co-expressed genes emerged in the enrichment of disease-associated genes was analyzed separately among ten brain tissues for Pearson’s correlation by using MATLAB (Statistics Toolbox Release 2015b, The MathWorks, Inc., Natick, Massachusetts, USA). The same correlation analysis was performed for the mouse homologous Chrm3 among six types of single neurons. The adjusted P-value for significance was 0.05/(10*24) = 2 × 10−4 for the co-expression analysis with 10 brain tissues for 24 genes, and in the co-expression analysis of 6 mouse GABAergic neurons for 14 genes, the adjusted significance P-value was 0.05/(6*14) = 5 × 10−4.
Results
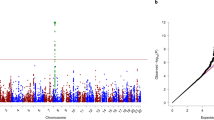
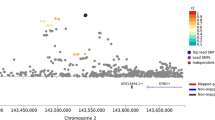
In the discovery GWAS, we meta-analyzed Yale-Penn 1 and 2 EAs and AAs separately by population (Fig. 1). In the meta-analysis of Yale-Penn EAs, three GWS SNPs representing one signal were identified (see GWAS results in Fig. 1 for EAs): rs115455482, rs74722579, and rs1938228, which are highly correlated (r2 ≥ 0.95, MAF ~ 0.06 in EAs when cases and controls were considered together; Table 1 and Supplementary Table S1). The association P-values were 1.66 × 10−10, 2.81 × 10−9, and 1.57 × 10−8 for rs115455482, rs74722579, and rs1938228, respectively; the leading SNP rs115455482 is ~230 kb upstream of transcript NM_000740 of CHRM3 (muscarinic acetylcholine receptor (AChR) M3). There were no GWS SNPs identified in AAs (Supplementary Figs. 1 and 2 for AAs). Low inflation was observed in the meta-analysis of Yale-Penn 1 and 2 for EAs (λ = 1.06; Supplementary Fig. 3) and AAs (λ = 1.02; Supplementary Fig. 2).

a Manhattan plot with three significant variants, rs115455482, rs74722579, and rs1938228, on chromosome 1, in a meta-analysis of two EA cohorts. The three variants are in high linkage disequilibrium (all r2 ≥ 0.95) in EAs. The line in the plot represents the genome-wide significance cutoff (5 × 10−8). b Regional Manhattan plot demonstrates rs115455482 is close to gene CHRM3 (and regulates CHRM3 expression; see Fig. 2 and text). According to a published schizophrenia genome-wide association study36, these top three SNPs associated with cannabis-induced hallucinations were not significantly associated with schizophrenia (all associations P ~ 0.25); one SNP rs12081830, associated with cannabis-induced hallucination (P = 8.5 × 10−5), was nominally associated with schizophrenia (P = 4.3 × 10−2). rs115455482 and rs12081830 are not highly linked with each other (r2 < 0.2 in EAs). The light blue line and right Y-axis show the observed recombination rate in the 1000 Genomes Project European samples (EUR, hg19). The SNPs are colored in accordance to r2 with rs115455482.
The independent COGA sample was used to attempt to replicate these top signals in the discovery GWAS. Two of these SNPs, rs115455482 and rs74722579, were available in the COGA data; neither of these were significantly associated with trait, although their β-effects were both in the same direction (0.05 and 0.02, respectively). Further meta-analysis of both Yale-Penn discovery EA samples with the COGA EA replication sample (Table 1) for these SNPs increased the statistical significance of the association signals (meta-P-values of 1.32 × 10−10 and 2.60 × 10−9 for rs115455482 and rs74722579, respectively). In addition, meta-analysis for rs74722579 (MAF ~ 0.10) including all available AA samples from Yale-Penn 1 and 2, and COGA (total cases = 362 and controls = 3404) yielded a nominally significant meta-P-value (meta-P = 4.58 × 10−2); the analysis for rs115455482 was not significant. For the latter variant, the minor allele is rare in AAs (MAF = 0.01, meta-P = 1.37 × 10−1) (Table 1). Taken together, the robust association signals for the two top SNPs in EAs and the nominal significance for rs74722579 in AAs (Fig. 1 and Table 1) support the validity of the GWAS results.
To evaluate a potential underlying mechanism for the association of rs115455482 genotype with Ca-HL, the regulatory relationship between rs115455482 and CHRM3 expression was evaluated in the Braineac database. In expression quantitative trait locus (eQTL) analysis, CHRM3 was variably expressed among ten brain tissues and differentially expressed among rs115455482*T risk allele carriers in the THAL (P = 4.5 × 10−4; adjusted P = 4.5 × 10−3 across ten brain tissues) and PUTM (P = 1.6 × 10−2; adjusted P = 1.6 × 10−1 across ten brain tissues) (Fig. 2). Rs115455482 risk genotype TC (TT homozygotes were too rare to be observed in this sample) was correlated with lower mRNA expression of CHRM3 in THAL, with the same trend found in other tissues, including PUTM, OCTX, MEDU, SNIG, FCTX, HIPP, and CRBL.

The correlation between rs115455482 genotype and CHRM3 expression across ten brain tissues in European samples. Line within each boxplot represents median and box region indicates the range from first quantile to third quantile. Up and down whiskers, as well as dots outside box region, represent maximum value, minimum value, and outliers, respectively. The boxplot was generated in Braineac (http://www.braineac.org/). The abbreviations for ten brain tissues are CRBL (cerebellar cortex), FCTX (frontal cortex), HIPP (hippocampus), MEDU (medulla specifically inferior olivary nucleus), OCTX (occipital cortex), PUTM (putamen), SNIG (substantia nigra), TCTX (temporal cortex), THAL (thalamus), and WHMT (intralobular white matter). Affymetrix ID t2387606 represents transcript IDs for CHRM3 at mRNA level.
To infer the potential biological function of CHRM3, we carried out co-expression analysis. We obtained the top 100 co-expressed genes (Supplementary Table S2) with respect to CHRM3 by application of COXPRESdb18 and then performed enrichment analysis in disease-associated gene categories in WebGestalt19, which showed that 24 genes were significantly enriched within the top 10 disease-associated gene categories (all raw P-values < 1 × 10−2 and adjusted P-values < 1 × 10−2), including psychotic disorders, epilepsy, and schizophrenia (Supplementary Information “CHRM3 Co-Expression Analysis” and Supplementary Table S3). According to GWAS database GRASP21, CHRM3 is an epilepsy risk gene and CHRM3 SNP rs10925980 was nominally associated with genetic generalized epilepsies (P = 1.90 × 10−6) and Ca-HL (P = 0.03) in EAs (see Supplementary Table S4). In addition, 24 CHRM3 co-expressed genes were significantly (all P-values < 1 × 10−2) correlated with CHRM3 in at least 1 of 10 brain tissues, with the same being true for 14 Chrm3 co-expressed genes expressed in GABAergic neurons (Supplementary Fig. 4).
The most frequently observed co-expression was between CHRM3 and GABRG2, which was seen in eight brain tissues (all P-values < 1 × 10−10 and r2 ≥ 0.6). CHRM3 was also significantly co-expressed with CHRNA4 and HRH3 in THAL (both P-values < 1 × 10−10 and r2 ≥ 0.7), and with HRH3 (P-value < 1 × 10−10 and r2 = 0.6) in PUTM. Furthermore, at the single-cell level, the homologous mouse genes Chrm3 and Gabrg2 were significantly co-expressed with one another in six different GABAergic neuronal cells (all P-values < 1 × 10−5 and r2 ≥ 0.6), whereas Hrh3 and Chrna4 were both significantly co-expressed with Chrm3 at least in three different GABAergic single neurons (all P-values < 1 × 10−5 and r2 ≥ 0.4). Taken together, these results show that CHRM3 is strongly co-expressed with GABRG2, HRH3, and CHRNA4 (risk genes for psychotic disorders, epilepsy, and schizophrenia) in THAL and other brain tissues.
Discussion
We report here biologically interesting GWS results from a case–control GWAS of Ca-HL. There was one GWS association signal at the CHRM3 locus in Yale-Penn 1 EAs, represented by rs115455482, rs74722579, and rs1938228 (all r2 ≥ 0.95 in EAs; rs115455482 is rare in AAs), with all three SNPs nominally significant in Yale-Penn 2 EAs. Only rs115455482 and rs74722579 were available in COGA, which when meta-analyzed with Yale-Penn 1 and 2 samples increased the significance of the association. We further evaluated the association of rs115455482 and rs74722579 with Ca-HL in the Yale-Penn 1 and 2, and COGA AA samples. The nominally significant association for rs74722579 (a common variant in both AAs and EAs) in the AA meta-analysis further supports the association. Cis-eQTL analysis of CHRM3 in brain showed the rs115455482*T risk allele to be associated with lower expression of CHRM3 in brain tissue THAL. Further co-expression analysis in large brain tissue expression datasets and mouse single neurons demonstrated a significant correlation between the expression of CHRM3 or its mouse homologous Chrm3, and that of three genes (GABRG2, CHRNA4, and HRH3) that have been shown to affect risk for psychotic disorders, including schizophrenia. Similar to CHRM3 itself, these are all biologically relevant.
The protein product of CHRM3 is the muscarinic AChR M3, which is localized to multiple tissues, including the brain, smooth muscle, endocrine and exocrine glands, and lungs. In human, mutation of CHRM3 causes disease of the urinary bladder and a prune-belly-like syndrome22. In mice, cannabinoids consistently increase acetylcholine (ACh) and decrease ACh turnover in the HIPP23. Pharmacological evidence has also implicated cholinergic dysfunction in the manifestation of psychotic symptoms. Muscarinic ACh receptors (AChRs) play important roles in animal models that are used to examine sensory gating, which is known to be disrupted in schizophrenic patients, and the activation of muscarinic AChRs was suggested as an alternative to classical antipsychotics for the treating of psychotic symptoms23. Chrm3 knockout mice treated with the antipsychotic drug oxotremorine that acts as a selective muscarinic ACh receptor agonist24,25,26 displayed increased dopamine release, which is consistent with Chrm3 playing an inhibitory role in dopamine release24. In animal models, oxotremorine can reverse methamphetamine-, ketamine-, and cocaine-induced hyperlocomotion26. Blockade of cholinergic receptors, particularly muscarinic receptors, causes a psychosis characterized by hallucinations and cognitive impairment in normal human subjects and exacerbates symptoms in schizophrenic patients26. This is consistent with the assumption that cannabis use is an environmental risk factor in the etiology of schizophrenia, as THC may directly affect T-type calcium channels in the THAL by increasing the excitability of THAL neurons27. In our study, the risk rs115455482*T allele is most significantly associated with lower expression of CHRM3 in THAL, suggesting potential excitatory-to-inhibitory imbalance in the THAL may predispose to cannabis-induced psychosis or schizophrenia.
In the analysis of several brain tissue samples, CHRM3 expression was associated with the expression of three other genes related to neuropsychiatric traits, GABRG2, CHRNA4, and HRH3. GABRG2 was co-expressed with CHRM3 across eight brain tissues and the same co-expression pattern was observed for its mouse homologous Chrm3 with Gabrg2 in six GABAergic single neurons. GABRG2 encodes a γ-aminobutyric acid receptor subunit; the receptor has chloride channel activity. Mutations in GABRG2 have been associated with epilepsy and febrile seizures28. Meanwhile, CHRNA4 and HRH3 are co-expressed with CHRM3 in specific brain tissues (particularly in THAL) and the same pattern is observed for the mouse homologs Chrna4 and Hrh3 with different GABAergic neurons. CHRNA4 encodes CHRNA4 (the α4 nicotinic ACh receptor), which belongs to a superfamily of ligand-gated ion channels that play a role in fast synaptic signal transmission. CHRNA4 interacts with CHRNB2 (the β2 nicotinic ACh receptor), which are critical for dopamine-dependent locomotor activation after repeated nicotine administration29. Mutations in CHRNA4 cause nocturnal frontal lobe epilepsy type 130 and polymorphisms of CHRNA4 have been reported in association with nicotine dependence31,32,33. HRH3 encodes the histamine receptor H3, which is ubiquitously released from neurons, mast cells, and enterochromaffin-like cells, and can regulate neurotransmitter release34. The significant co-expression between CHRM3 and GABRG2, CHRNA4, and HRH3 reflects the critical function of CHRM3 in the brain, which raises the question of whether, in response to long-term cannabis use, there is crosstalk between CHRM3 and the three noted genes in Ca-HL.
Multiple lines of evidence support the involvement of CHRM3 in schizophrenia, which has some symptoms phenotypically close to Ca-HL. In a neural connectivity GWAS in a Chinese schizophrenia case–control sample, CHRM3 variant rs6700381 was significantly associated with abnormal thalamo-orbital FCTX functional connectivity in first-episode schizophrenia patients35. In a multi-stage schizophrenia GWAS of up to 36,989 cases and 113,075 controls36, 1 SNP rs72769124 near CHRM3 was possibly associated with schizophrenia (P = 5.5 × 10−7; Supplementary Fig. S5). According to the Brainiac database, neither rs6700381 nor rs72769124 is a CHRM3 eQTL among ten healthy brain tissues. These risk variants may, however, be CHRM3 eQTLs under different conditions or environmental exposures or in different brain regions, or in different populations.
In our study, despite the lack of independent replication of rs115455482 in COGA, when we meta-analyzed Yale-Penn and COGA samples, the association of rs115455482 genotype with Ca-HL improved. The lack of significant replication in COGA may be due to the differential distributions of cannabis-dependence (CAD) criterion counts in Ca-HL cases and controls between Yale-Penn and COGA samples (Supplementary Fig. S6). We found that Ca-HL was significantly associated with CAD criterion counts both in Yale-Penn and COGA samples (Supplementary Fig. S7), which raises the question of potential involvement of the interaction between rs115455482 genotype and CAD severity in Ca-HL. As shown in Supplementary Fig. S7, we observed that the interaction between rs115455482*T and CAD criterion count was significantly associated with Ca-HL in Yale-Penn 1 EAs (P = 1.25 × 10−5) but not in Yale-Penn 2 EAs (P = 8.94 × 10−2) or COGA EAs (P = 0.72). This may be a potential reason for the lack of replication of rs115455482 in COGA, as the interaction between rs115455482*T and CAD criterion counts were different among Yale-Penn and COGA samples.
We note also a recent cannabis GWAS publication37 implicated CHRNA2 (and no other locus) as associated with cannabis-use disorder. In our study, CHRM3 was demonstrated to be significantly co-expressed with CHRNA4 in the brain tissue THAL. CHRNA2 and CHRNA4 encode the nicotinic AChR subunit α-2 and α-4, respectively. Our result considered in that context provides additional weight to considering cholinergic signaling as a mechanism of risk for cannabis-use disorder and cannabis-induced harmful effects.
This study has limitations. The sample size (number of Ca-HL cases) is small in the discovery GWAS, indicating that the power of our GWAS is likely to be low except for risk loci of large effect. Genes close to other prominent SNP association signals (P < 5 × 10−7) in the meta-analysis of Yale-Penn samples may warrant further study, including APBA2, EFCAB3, MTRR, PRPRN2, SPATA6, and FLJ12825 (SNPs mapped to these genes are included in Supplementary Table S5). A continuous measure (e.g., frequency of having experienced hearing, seeing or smelling things that were not really there after marijuana use) would provide more statistical power and should be considered for future surveys eliciting information on the trait.
In summary, we report one GWS association signal, represented by rs115455482 and rs74722579 (r2 ≥ 0.95 in EAs; rs74722579 is a common variant in both EAs and AAs) at the CHRM3 locus, with Ca-HL. The findings were GWS in our Yale-Penn 1 taken separately, with nominally significant associations of two variants in one of two other available samples of EAs. Additional support was obtained for rs74722579 in the meta-analysis of AA samples. Our findings are consistent with a model in which the regulatory variant rs115455482 affects the expression of CHRM3 in THAL, which could alter the stimulatory efficiency of cannabis on hallucinations, a potential underlying mechanism for its association with Ca-HL. Validation of these findings and the putative mechanism is warranted to increase our understanding of the biology of Ca-HL.
References
Yucel, M. et al. Regional brain abnormalities associated with long-term heavy cannabis use. Arch. Gen. Psychiatry 65, 694–701 (2008).
Zvolensky, M. J. et al. Marijuana use and panic psychopathology among a representative sample of adults. Exp. Clin. Psychopharmacol. 18, 129–134 (2010).
Mackie, K. Understanding cannabinoid psychoactivity with mouse genetic models. PLoS Biol. 5, e280 (2007).
Alger, B. E. Endocannabinoids: getting the message across. PNAS 101, 8512–8513 (2004).
Schoeler, T. et al. Association between continued cannabis use and risk of relapse in first-episode psychosis a quasi-experimental investigation within an observational study. JAMMA Psychiatry 73, 1173–1179 (2016).
Cheng, Z. et al. Genome-wide association study identifies a regulatory variant of RGMA associated with opioid dependence in European Americans. Biol. Psychiatry 84, 762–770 (2018).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Sherva, R. et al. Genome-wide association study of cannabis dependence severity, novel risk variants, and shared genetic risks. JAMA Psychiatry 73, 472–480 (2016).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824 (2012).
Pierucci-Lagha, A. et al. Reliability of DSM-IV diagnostic criteria using the semi-structured assessment for drug dependence and alcoholism (SSADDA). Drug Alcohol Depend. 91, 85–90 (2007).
Bucholz, K. K. et al. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J. Stud. Alcohol 55, 149–158 (1994).
Chen, M. H. & Yang, Q. GWAF: an R package for genome-wide association analyses with family data. Bioinformatics 26, 580–581 (2010).
Hernandez, D. G. et al. Integration of GWAS SNPs and tissue specific expression profiling reveal discrete eQTLs for human traits in blood and brain. Neurobiol. Dis. 47, 20–28 (2012).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Okamura, Y. et al. COXPRESdb in 2015: coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res. 43(D1), D82–D86 (2015).
Wang, J. et al. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 41(W1), W77–W83 (2013).
Paul, A. et al. Transcriptional architecture of synaptic communication delineates GABAergic neuron identity. Cell 171, 522–539 e20 (2017).
Eicher, J. D. et al. GRASPv2.0: an update on the Genome-Wide Repository of Associations between SNPs and phenotypes. Nucleic Acids Res. 43(Database issue), D799–D804 (2015).
Weber, S. et al. Muscarinic acetylcholine receptor M3 mutation causes urinary bladder disease and a prune-belly-like syndrome. Am. J. Hum. Genet. 89, 668–674 (2011).
Tripathi, H. L. et al. Effects of cannabinoids on levels of acetylcholine and choline and on turnover rate of acetylcholine in various regions of the mouse-brain. Alcohol Drug Res. 7, 525–532 (1987).
Zhang, W. L. et al. Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M-1-M-5 muscarinic receptor knock-out mice. J. Neurosci. 22, 6347–6352 (2002).
Tang, C., Castoldi, A. F. & Costa, L. G. Effects of the muscarinic agonist oxotremorine on membrane fluidity in rat lymphocytes. Biochem Mol. Biol. Int. 29, 1047–1054 (1993).
Maehara, S. et al. Antipsychotic property of a muscarinic receptor agonist in animal models for schizophrenia. Pharm. Biochem. Behav. 91, 140–149 (2008).
Vukadinovic, Z., Herman, M. S. & Rosenzweig, I. Cannabis, psychosis and the thalamus: a theoretical review. Neurosci. Biobehav. Rev. 37, 658–667 (2013).
Boillot, M. et al. Novel GABRG2 mutations cause familial febrile seizures. Neurol. Genet. 1, e35 (2015).
King, S. L., Caldarone, B. J. & Picciotto, M. R. Beta2-subunit-containing nicotinic acetylcholine receptors are critical for dopamine-dependent locomotor activation following repeated nicotine administration. Neuropharmacology 47(Suppl 1), 132–139 (2004).
Leniger, T. et al. A new CHRNA4 mutation with low penetrance in nocturnal frontal lobe epilepsy. Epilepsia 44, 981–985 (2003).
Han, S. et al. Meta-analysis of 15 genome-wide linkage scans of smoking behavior. Biol. Psychiatry 67, 12–19 (2010).
Han, S. et al. Association of CHRNA4 polymorphisms with smoking behavior in two populations. Am. J. Med Genet. B Neuropsychiatr. Genet. 156B, 421–429 (2011).
Xie, p. et al. Rare nonsynonymous variants in alpha-4 nicotinic acetylcholine receptor gene protect against nicotine dependence. Biol. Psychiatry 70, 528–536 (2011).
Ellenbroek, B. A. Histamine H(3) receptors, the complex interaction with dopamine and its implications for addiction. Br. J. Pharm. 170, 46–57 (2013).
Wang, Q. et al. The CHRM3 gene is implicated in abnormal thalamo-orbital frontal cortex functional connectivity in first-episode treatment-naive patients with schizophrenia. Psychol. Med. 46, 1523–1534 (2016).
Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Demontis, D. et al. Genome-wide association study implicates CHRNA2 in cannabis use disorder. Nat. Neurosci. 22, 1066–1074 (2019).
Acknowledgements
This study was supported by National Institutes of Health grants R01 DA12690, R01 AA11330, R01 AA017535, and the VA Connecticut Healthcare Center and Philadelphia VA MIRECCs. We appreciate the work in recruitment and assessment by James Poling, PhD, at Yale University School of Medicine and the APT Foundation; Roger Weiss, MD, at McLean Hospital; by Kathleen Brady, MD/PhD, and Raymond Anton, MD, at the Medical University of South Carolina, and David Oslin, MD, at the University of Pennsylvania. Genotyping services for a part of our GWAS were provided by the Center for Inherited Disease Research and Yale University (Center for Genome Analysis), which is fully funded by Federal contract N01-HG-65403 from the NIH to The Johns Hopkins University. We thank Ann Marie Lacobelle, MS, and Christa Robinson, BS, who provided technical assistance. We thank Yaira Nunez and Michelle Slivinsky, who led the QC effort for the Semi-Structured Assessment for Drug Dependence and Alcoholism interviews, and phenotyping for the study sample. We thank John Farrell, PhD, Section of Biomedical Genetics, Boston University School of Medicine, who provided database management assistance. We also thank Drs Hang Zhou, Renato Polimanti, and Daniel Levey for their constructive suggestions on the statistical analysis. COGA: The Collaborative Study on the Genetics of Alcoholism (COGA), Principal Investigators B. Porjesz, V. Hesselbrock, H. Edenberg, L. Bierut; includes 11 different centers: University of Connecticut (V. Hesselbrock), Indiana University (H.J. Edenberg, J. Nurnberger Jr., T. Foroud, Y. Liu), University of Iowa (S. Kuperman, J. Kramer), SUNY Downstate (B. Porjesz), Washington University in St. Louis (L. Bierut, J. Rice, K. Bucholz, A. Agrawal), University of California at San Diego (M. Schuckit), Rutgers University (J. Tischfield, A. Brooks), Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia; Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia PA (L. Almasy), Virginia Commonwealth University (D. Dick), Icahn School of Medicine at Mount Sinai (A. Goate), and Howard University (R. Taylor). Other COGA collaborators include the following: L. Bauer (University of Connecticut); J. McClintick, L. Wetherill, X. Xuei, D. Lai, S. O’Connor, M. Plawecki, S. Lourens (Indiana University); G. Chan (University of Iowa; University of Connecticut); J. Meyers, D. Chorlian, C. Kamarajan, A. Pandey, J. Zhang (SUNY Downstate); J.-C. Wang, M. Kapoor, S. Bertelsen (Icahn School of Medicine at Mount Sinai); A. Anokhin, V. McCutcheon, S. Saccone (Washington University); J. Salvatore, F. Aliev, B. Cho (Virginia Commonwealth University); and Mark Kos (University of Texas Rio Grande Valley). A. Parsian and H. Chen are the NIAAA Staff Collaborators. COGA investigators continue to be inspired by memories of Henri Begleiter and Theodore Reich, founding PI and Co-PI of COGA, and also owe a debt of gratitude to other past organizers of COGA, including Ting-Kai Li, P. Michael Conneally, Raymond Crowe, and Wendy Reich, for their critical contributions. This national collaborative study is supported by NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA). Arpana Agrawal also acknowledges support from K02DA032573.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Dr. Kranzler has been a consultant or advisory board member for Alkermes, Indivior, and Lundbeck. He is a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by AbbVie, Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer, Arbor, and Amygdala Neurosciences. Drs. Kranzler and Gelernter are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed 24 January 2018. The other authors have no biomedical financial interests or potential conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, Z., Phokaew, C., Chou, YL. et al. A regulatory variant of CHRM3 is associated with cannabis-induced hallucinations in European Americans. Transl Psychiatry 9, 309 (2019). https://doi.org/10.1038/s41398-019-0639-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0639-7
This article is cited by
-
G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders
Signal Transduction and Targeted Therapy (2023)
