
Abstract
Depression is a severe and debilitating mental disorder diagnosed by evaluation of affective, cognitive and physical depression symptoms. Severity of these symptoms strongly impacts individual’s quality of life and is influenced by a combination of genetic and environmental factors. One of the molecular mechanisms allowing for an interplay between these factors is DNA methylation, an epigenetic modification playing a pivotal role in regulation of brain functioning across lifespan. The aim of this study was to investigate if there are DNA methylation signatures associated with depression symptomatology in order to identify molecular mechanisms contributing to pathophysiology of depression. We performed an epigenome-wide association study (EWAS) of continuous depression symptomatology score measured in a cohort of 724 monozygotic Danish twins (346 males, 378 females). Through EWAS analyses adjusted for sex, age, flow-cytometry based blood cell composition, and twin relatedness structure in the data we identified depression symptomatology score to be associated with blood DNA methylation levels in promoter regions of neuropsin (KLK8, p-value = 4.7 × 10−7) and DAZ associated protein 2 (DAZAP2, p-value = 3.13 × 10−8) genes. Other top associated probes were located in gene bodies of MAD1L1 (p-value = 5.16 × 10−6), SLC29A2 (p-value = 6.15 × 10−6) and AKT1 (p-value = 4.47 × 10−6), all genes associated before with development of depression. Additionally, the following three measures (a) DNAmAge (calculated with Horvath and Hannum epigenetic clock estimators) adjusted for chronological age, (b) difference between DNAmAge and chronological age, and (c) DNAmAge acceleration were not associated with depression symptomatology score in our cohort. In conclusion, our data suggests that depression symptomatology score is associated with DNA methylation levels of genes implicated in response to stress, depressive-like behaviors, and recurrent depression in patients, but not with global DNA methylation changes across the genome.
Similar content being viewed by others

Introduction
Depression is a multifactorial common psychiatric disorder diagnosed by evaluation of various depressive symptoms, such as low mood, loss of interest and pleasure, fatigue and loss of energy, decline in cognitive functioning, poor concentration, increase in anxiety, inappropriate guilt, change in appetite, sleep disturbance, as well as changes in psychomotor activity1. Early diagnosis and treatment of depression is beneficial for the patient and individual’s later mental health outcome2. However, due to the complexity of depression symptomatology and its varying severity across the general population, depression is reported to be under-diagnosed and therefore under-treated in society3,4,5. Depression is estimated to be the leading global cause of years lost due to disability worldwide, with lifetime prevalence of the disorder estimated to be ~14%, and even reaching 21% in high-income countries6,7. Risk of suffering from depression is influenced by common genetic variants8,9,10,11, with twin studies attributing ~40% of the variation in depression liability to the additive genetic effects12,13, and change in depression symptomatology reported to be a heritable trait, with heritability estimates reaching 30%14. Apart from genetic factors, which contribute to but do not fully explain individual’s disorder risk, environmental exposures, such as social and socioeconomic factors (social isolation15, life events16, low income or financial problems17, level of education18,19), as well as lifestyle factors (such as diet20 or level of physical activity21) impact severity of depression symptomatology and risk of developing depression across the lifespan22.
One of the molecular mechanisms through which environmental factors can modulate phenotype outcome is epigenetics, with DNA methylation being one of the most studied modifications of the genome. DNA methylation is dynamic and changes across an individual’s lifespan, influenced by prenatal environmental factors23,24,25,26, life events27,28,29,30, lifestyle choices23,31,32,33,34,35,36,37, as well as puberty38,39 and aging40,41. DNA methylation plays a pivotal role in regulation of human brain development, its functioning, and aberrant changes in this modification are increasingly reported to be associated with mental disorders (schizophrenia, bipolar disorder, mental retardation, ADHD, autism)42,43,44,45,46,47,48,49,50,51,52, mental disorder trajectories53, and cognitive54,55,56, as well as social functioning57.
In this study we hypothesized that there are DNA methylation signatures in the genome that associate with depression symptomatology in the general population. As liability to depression in part constitutes the extreme of a quantitatively measurable depression symptomatology identification of such signatures could inform on genes and molecular pathways involved in progression of depression symptoms, and could allow in the future for earlier identification of individuals at risk of developing depression in the general population. Since both depression symptomatology and DNA methylation are not independent of individual’s genetic background we performed our epigenome-wide association study (EWAS) in a large cohort of Danish monozygotic twins, which allowed us to adjust the analyses for shared genetic and environmental factors.
Materials and methods
Study population
The study was performed on a sample of 724 Danish monozygotic twins (378 females and 346 males, representing 362 complete twin pairs) recruited as part of the Danish Twin Registry (DTR)58,59 for whom DNA methylation data and depression symptomatology score was available. Participants were enrolled in the survey as a part of the Middle Aged Danish Twin Study (MADT)60 and the Longitudinal Study of Aging in Danish Twins (LSADT)61, both designed, organized and performed by DTR. Depression symptomatology was assessed for all participating twins by using a nine-symptom ‘affect scale’, corresponding to the affective depression assessment, adapted from the Depression Section of the Cambridge Mental Disorders of the Elderly Examination (CAMDEX), as previously described62. The nine questions evaluate the current emotional state of study participants and a final affective depression symptomatology score was calculated as the sum of the nine items. Higher affective depression symptomatology score corresponds to more severe symptoms14,62. During the visit whole blood samples were collected for all study participants. All study participants gave informed consent. Permissions to collect blood samples and the usage of register-based information were granted by Regional Committees on Health Research Ethics for Southern Denmark (S-VF-19980072 and S-VF-20040241). Genomic DNA was extracted from buffy-coat fraction with the use of the semi-automated salt precipitation protocol with Autopure System (Qiagen, Hilden, Germany).
DNA methylation profiling
In total 500 ng of genomic DNA extracted from buffy coat from each individual was bisulfite converted with the use of EZ Methylation Gold Kit (Zymo Research, Irvine, California, United States). Bisulfite converted DNA was further analyzed using the Infinium Human Methylation 450 K array (Illumina, San Diego, California, United States) according to manufacturer’s protocol. Quality control of DNA methylation data was performed with a combination of MethylAid63 and minfi64 tools. In short probes with high detection p-value (>0.01), low bead count (<3 beads), zero signal, missing in >5% of samples and cross-reactive probes, as reported before65, were removed from the dataset. In order to reduce the technical variation methylome data was normalized with the use of functional normalization (FunNorm)66, which regresses out the technical variability estimated from control probes included in Infinium HumanMethylation450BeadChip. Obtained normalized beta-values were further logit transformed to obtain M-values, as recommended before by Du P. and co-authors67. According to the current Danish legislation transfer and sharing of individual-level data requires prior approval from the Danish Data Protection Agency and requires that data sharing requests are delt with on a case-by-case basis. To comply with the study’s ethical approval the data cannot be deposited in a public database, however, we welcome any enquiries regarding collaboration and individual requests for data sharing.
Blood cell composition
Blood cell counts were available for 477 individuals, where five blood leukocyte subtypes (monocytes, lymphocytes, basophils, neutrophils, eosinophiles) were measured using a Coulter LH 750 Hematology Analyzer (Beckman Coulter, Brea, California, United States). For the remaining individuals, where blood cell composition was not available, data was imputed by partial least squares regression with the use of wbccPredictor tool (https://github.com/mvaniterson/wbccPredictor), as described before55. In short, a regression model was first fitted based on the 477 samples for which measurements were available, and afterwards it was applied for prediction of missing cell counts. The model used log(cell count + 1) as response and included all beta values, that were available for all samples, as covariates. Sex, age, and sentrix position were also included as covariates in the model. The sentrix position was modeled by two categorical variables indicating the position in each of the two directions on the chip: column-wise and row-wise. All calculations were performed in R68.
Association of depression symptomatology score with covariates
We performed several exploratory analyses to investigate if measured affective depression symptomatology score is associated with age, sex, and blood cell counts in this cohort of MADT and LSADT monozygotic twins. The analyses were performed with the use of linear mixed models with depression symptomatology score as the outcome variable, age, sex, and blood cell composition as fixed effects, and twin pairing information as a random effect. The analyses were performed with the use of lmerTest69 R package.
Epigenome-wide association studies
In this study we examined if DNA methylation in whole blood is associated with depression symptomatology in middle-aged as well as elderly individuals, while adjusting for their genetic background with a monozygotic twin study design. Possible associations between DNA methylation levels and depression symptomatology score were investigated in two statistical models, first where we studied monozygotic twin intra-pair differences (paired analysis), and second model where all individuals were treated as singletons while adjusting for the relatedness structure in the dataset (unpaired analysis). For the paired analysis differences in the depression symptomatology score, DNA methylation and blood cell composition between twins were calculated for all twin pairs. DNA methylation differences were regressed on depression symptomatology score differences and were adjusted for age, sex and intra-pair differences in blood cell composition. The reason for including sex and age variables in the paired model was to adjust for sex and age effects on the intra-pair differences. This aspect is important as the intra-pair differences can increase with increasing age and may also differ between male and female twin pairs. For the unpaired regression analysis we used linear mixed models and adjusted for age, sex, individuals’ blood cell composition, and specified twin pairing as a random effect. All analyses were performed in R68. All probes with suggestive association p-value < 1×10−5 were annotated to gene symbols according to human genome assembly (hg19) to provide better overview of the most associated sites in the study. Identified genes were further used for Gene Ontology, KEGG and DisGeNET (human disease) pathway overrepresentation enrichment analysis (ORA) with the use of WebGestalt tool70 versus genes included at Infinium HumanMethylation450BeadChip as background.
We also performed a replication of the most associated sites (p-value < 1 × 10−5) identified in a recent EWAS meta-analysis study of depression symptomatology71 in our cohort of Danish monozygotic twins in results obtained from both paired and unpaired models. We performed replication for all results, from both discovery and meta-analysis, from Story et al. (2018) report71.
Differentially methylated regions
In order to expand the search of epigenetic signatures associated with depression symptomatology we extended our analyses to differentially methylated regions (DMRs) for both paired and unpaired statistical approaches. DMRs were identified with a comb-p tool72 reported to have consistently the best sensitivity and high control of false-positive rate when compared to other DMR tools (DMRcate, bumphunter, and probe lasso)73. Comb-p analyses were ran using Python 2.7 with parameters reported to achieve the best performance, as tested for DNA methylation array studies, seed <0.05 and dist = 75073. Identified DMRs, consisting of at least 3 probes and reaching unadjusted DMR p-value <0.05, were annotated to gene symbols according to human genome assembly (hg19). Additionally, p-value for each DMR was adjusted for multiple testing with Šidák correction method74 as implemented by default in the comb-p tool72.
DNA methylation age
Another approach to analyze epigenetic signatures of a studied trait is to investigate associations between DNA methylation age (DNAmAge) and the phenotype of interest. DNAmAge, known also as the epigenetic clock, represents age-related changes in DNA methylation at multiple sites in the genome and provides an alternative to performing single-site analysis as in the case of EWAS approach. DNAmAge was estimated for each individual with Horvath75 and Hannum76 biological clock estimators, both appropriate for methylome data obtained from blood tissue collected from adult individuals. Horvath and Hannum DNAmAge estimates were further correlated with affective depression symptomatology score with and without adjusting for chronological age of each individual. Additionally, we investigated if deltaDNAmAge (difference between DNAmAge and chronological age of each individual) and accelDNAmAge (residuals from DNAmAge regressed on chronological age of each individual) are associated with depression symptomatology score. The difference between deltaDNAmAge and accelDNAmAge is that the first measure represents age difference at individual level, while the latter one measures an alteration of aging of an individual when compared to the rest of the cohort56. All DNAmAge-related depression symptomatology score analyses were performed for both Horvath and Hannum DNAmAge estimates. Analyses were performed with linear mixed models, and were adjusted for sex as fixed effect and specified twin pairing as random effect.
Results
DNA methylation data for all 724 monozygotic twins included in this study passed all quality control steps performed with minfi and MethylAid pipelines. Overview of demographics of MADT and LSADT twins participating in this study is provided in Table 1. We observed significant associations between depression symptomatology score with sex and with chronological age (see Table 2). Depression symptomatology score was increased in females in comparison to males, and additionally increased with age (see Table 2). We also investigated if blood cell composition is associated with the depression symptomatology score. Out of five studied blood cell types we observed two (lymphocyte and neutrophil proportions) to be significantly associated with the score (see Table 2). Interestingly, levels of associations for these two cell types with depression symptomatology score were very comparable (in terms of absolute effect size and significance level), and the reason for this similarity was a high negative correlation between lymphocyte and neutrophil proportions (r = −0.93).
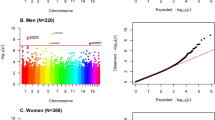
Next, we performed EWAS analyses of depression symptomatology score with both paired and unpaired twin models. In the paired analysis, where we investigated if within-twin-pair depression symptomatology score differences can be associated with within-twin-pair DNA methylation level differences, we identified cg05777061 probe as the most associated finding (p-value = 4.7 × 10−7, FDR p-value = 0.21, see Table 3). The probe is located on chromosome 19 and targets promoter region of kallikrein-8 (KLK8) gene, also known as neuropsin gene. The finding in KLK8 gene was the only one in the paired model analysis with p-value < 10−6, as depicted by a Manhattan plot (Fig. 1a). In the EWAS of depression symptomatology score performed with the unpaired approach, where all individuals were treated as singletons and we adjusted for the relatedness structure in the dataset, we identified cg00554948 probe as the most significant finding (p-value = 3.13 × 10−8, FDR p-value = 0.014). The probe is located on chromosome 12 in a promoter region of DAZ Associated Protein 2 (DAZAP2) gene, also known as Proline-Rich Transcript In Brain. This was the only finding with association p-value < 10−6, as depicted by a Manhattan plot (Fig. 1b). We identified suggestive associations (p-value < 10−5) for additional 12 loci from the paired model targeting MAD1L1, SLC29A2, AKT1, ATF6B, RGS12, LIG1, HCG11 genes, as well as intergenic regions. For the unpaired model we identified additional 5 loci with suggestive association p-value < 10−5, most of them located in intergenic regions and one located in ATG16L1. All differentially methylated sites with suggestive association p-value < 10−5 are presented in Table 3. Inflation factor λ was calculated for of each of the performed EWAS analysis with the use of ramwas R package77. λ was estimated to be 1.014 for the paired EWAS, and 0.92 for the unpaired EWAS indicating little deflation or inflation of our EWAS results from the expected distribution of p-values.

Manhattan plots of depression symptomatology EWAS results from paired A and unpaired B models adjusted for sex, age and blood cell composition. All loci with p-value < 10−5 are annotated to genes according to human genome assembly (hg19)
Top enrichment results for ORA pathway analysis from paired EWAS of depression symptomatology were: ‘Regulation of Myelination’ (p-value = 8.78 × 10−5) for Gene Ontology, ‘Longevity Regulating pathway’ (p-value = 9.43 × 10−4) for KEGG, ‘Schizophrenia’ (p-value = 1.34 × 10−3) for DisGeNET analysis (Supplementary Table 1). None of the pathways remained significant after FDR p-value correction. ORA performed on genes reported in the unpaired EWAS of depression symptomatology did not identify any significantly enriched pathways.
Replication of the most associated sites from a recent EWAS meta-analysis study of depression symptomatology by Story et al.71 in our cohort replicated cg07012687 in SLC16A3 (p-valuetwin cohort = 8.75 × 10−4, p-valueStory-discovery = 3.47 × 10−7, p-valueStory-replication = 1.58 × 10−1, p-value Story-meta-analysis = 4.45 × 10−6) as the most associated finding. The second most associated finding within the 51 probes investigated for replication was cg12764201 in CORT (p-valuetwin cohort = 3.01 × 10−3, p-valueStory-discovery = 7.15 × 10−6, p-valueStory-replication = 7.20 × 10−1, p-valueStory-meta-analysis = 7.29 × 10−5). Probe cg04987734 in CDC42BPB, identified as one of three genome-wide significant findings in the meta-analysis71, replicated as third most significant finding across the investigated 51 probes (p-valuetwin cohort = 8.64 × 10−3, p-valueStory-discovery = 4.93 × 10−8, p-valueStory-replication = 4.82 × 10−2, p-valueStory-meta-analysis = 1.57 × 10−8). In total 12 of the investigated 51 probes reached nominal p-value < 0.05 in either paired or unpaired twin analysis. Our most significant findings in KLK8 and DAZAP2 were not reported to have p-value < 10−5 in the Story et al. report71. Overview of replication results in our Danish monozygotic twin cohort for all 51 investigated probes from Story et al.71 is presented in Supplementary Table 2.
Results from paired and unpaired EWAS of depression symptomatology obtained from MADT and LSADT monozygotic twin cohorts were further used to identify possible DMRs across the genome associated with this trait. We identified 30 DMRs from paired analysis and 40 DMRs from the unpaired analysis to be associated with depression symptomatology score with unadjusted DMR p-value < 0.05. Overview of all DMRs and their annotated genes is presented in Table 4. Five genes overlapped between DMR paired and unpaired analysis (PCDHGA4, GLIPR1L2, STAM, VARS2, MAST3), 2 genes overlapped between paired EWAS and paired DMR analysis (MAD1L1 and RGS12), and 1 gene overlapped between paired EWAS and unpaired DMR analysis (ATF6B). Overview of all probes located within each of the identified DMRs is presented in Supplementary Table 3 (paired) and Supplementary Table 4 (unpaired).
Apart from studying epigenetic signatures of depression symptomatology score in a single-CpG-site resolution manner we also investigated if blood-derived DNAmAge is associated with the depression symptomatology score. We observed a high Pearson’s correlation of both Horvath (r = 0.80) and Hannum (r = 0.79) DNAmAge estimates with chronological age of study participants. Horvath DNAmAge measure underestimated (mean = 65.07 years ± 9.49, min = 43.35 years, max = 107.96 years), while Hannum DNAmAge measure overestimated individuals’ age (mean = 74.30 years ± 8.31, min = 54.85 years, max = 108.75 years) in comparison to chronological age of the twin cohort (mean = 69.89 years ± 7.86, min = 55 years, max = 89 years). Both Horvath and Hannum DNAmAge estimates were found to be significantly associated with depression symptomatology score (p-values < 0.05), however, these findings did not remain significant after adjusting the models for chronological age (p-values >0.05). This result was also reflected in further regression analyses where no association was observed between depression symptomatology score and deltaDNAmAge and accelDNAmAge (see Table 5), as both represent measures from which chronological age was either subtracted or regressed. Overview of all results from association analyses performed between depression symptomatology score with Horvath and Hannum DNAmAge estimates adjusted for sex and relatedness structure in the data is presented in Table 5.
Discussion
Depression is a complex mood disorder influenced by a combination of genetic and environmental factors. One of the molecular mechanisms that allows for an interplay between genes and environment is epigenetics, which through its dynamic nature has the potential of continuously contributing to the pathophysiology of depressive symptoms. In this study we performed an EWAS of depression symptomatology in a large cohort of monozygotic twins with two different statistical approaches and we identified DNA methylation levels in KLK8 and DAZAP2 genes to be most associated with the depression symptomatology score. The two EWAS data analysis approaches (paired and unpaired) differ in how they estimate and handle confounding factors, as described before55. In short the first model analyzes intra-twin-pair differences which are adjusted for the effects from pair-specific confounding factors (age, sex, genetics, shared environmental factors), while the second model, even though also corrects for age and sex, does that for the effects on individual DNA methylation levels and also allows for inclusion of incomplete twin pairs if such are present55.
KLK8, also known as neuropsin, encodes a serine protease and maps to chromosome 19q13, a region implicated in schizophrenia and bipolar disorder by genetic linkage studies78,79. Genetic variation in human neuropsin itself was associated in a candidate-gene study with bipolar disorder and cognitive functioning, however not with major depression80. Neuropsin exists in two forms: a regular neuropsin (type 1) and hominoid-specific neuropsin (type 2)81, the latter containing a 135-bp insertion in 5′ upstream region of exon 3. Both types of neuropsin are abundantly expressed in human brain, with type 2 reported to be preferentially expressed in the adult brain, including hippocampus, frontal lobe and cerebral cortex80,81,82. Neuropsin is involved in synaptogenesis, maturation of orphan and small synaptic boutons83, and is responsible for degradation of cell adhesion molecule L1 (CAM-L1)84. Interestingly, CAM-L1 levels were found to be increased in prefrontal cortex and decreased in parieto-occipital cortex in post mortem brains of depressed individuals85. Exposure of rats to prolonged stress resulted in decreased CAM-L1 brain levels, while antidepressant treatment increased its expression86. In the same line acute stress was shown to increase neuropsin mRNA levels in mouse hippocampus87, while inactivation of neuropsin was shown to have protective effects against depressive-like behaviors and memory impairment induced by chronic stress in mice88. Significant increase in expression levels of human neuropsin, measured in peripheral blood, was observed between patients suffering from recurrent depression episodes compared to patients with first episode of depression89. These findings were further supported by a follow-up study where increased mRNA levels of neuropsin were found in patients with recurrent depression in comparison to healthy controls90. These studies support our observation of association between differential levels of DNA methylation in the promoter region of neuropsin and depression symptomatology, and further suggest that these changes may be modulated by external factors (such as chronic stress, acute stress or use of antidepressants).
The second probe identified in this study was located in the promoter region of DAZAP2, a well-conserved gene known for inducing stress granule formation91,92. According to Enrichr database DAZAP2 directly interacts with HGS, NEDD4, UBQLN4, UBB, UBC, MAP3K7, SMURF2, CTNNB1, ATXN1, and RPS27A93, most of these genes have been reported before to be associated with major depression, depression symptomatology, as well as exposure to stress94,95,96,97,98,99,100,101. Recent methylome analysis of monozygotic twins discordant for childhood psychotic symptoms identified a differentially methylated site located closest to DAZAP2 among the top ten most associated findings with the phenotype, however the probe was located -19713 bp from DAZAP2 TSS, while our probe was located closer to the gene, within the -1500 bp region from its TSS102. Differential methylation of DAZAP2 promoter region was shown to regulate its expression in multiple myeloma cell lines103, and its decreased expression levels were found to contribute to pathogenesis of this cancer104. Therefore, possible link between DAZAP2 and depression symptomatology requires further investigation. Differences between the top results observed from paired and unpaired EWAS analyses may be attributed to different statistical approaches that they use to evaluate the associations between DNA methylation levels and the studied trait, therefore interpretation of results from these two models differs. However, it is worth to note the overlap between genes identified from paired and unpaired DMR analyses, both based on the initial EWAS findings. This overlap indicates that even though these two models are not statistically equal they are both capable of identifying the same epigenetic signatures of depression symptomatology score.
Further investigation of other most differentially methylated sites in the EWAS analyses identified epigenetic changes in additional genes of high interest to the depression phenotype, such as mitotic arrest deficient 1 like 1 (MAD1L1), solute carrier family 29 member 2 (SLC29A2), AKT serine/threonine kinase 1 (AKT1). Recent large genome-wide association studies reported genetic variation in MAD1L1 to be genome-wide significantly associated with ICD-coded major depressive disorder (MDD)105, schizophrenia106, and bipolar disorder107. Additionally, a study comparing monozygotic twins discordant for MDD reported affected twins to have greater variance in methylation in MAD1L1 than their unaffected co-twins108, supporting our observation of association between depression phenotype and DNA methylation levels of MAD1L1. Genetic polymorphism in SLC29A2 showed suggestive association in candidate-gene studies (p-value < 0.05) with depression and depression with fatigue phenotypes in men109, as well with childhood trauma score within MDD cases110. Genetic polymorphism in AKT1 was associated with severity of depression, anxiety symptoms and suicide attempts in patients with MDD from a Chinese Han population111, with late-onset depression in a Brazilian population112, and antidepressant treatment response in patients with depressive disorder in a Caucasian population113. Differential methylation of AKT1 in blood was associated with clinical post-traumatic stress disorder in combat veterans114, and in post-mortem brain of schizophrenia patients compared to unaffected controls115, while changes in its expression level in blood were positively correlated with improvement of depression symptoms among bipolar disorder patients treated with lithium116. Altogether these studies support involvement of genes identified as differentially methylated in this study in the development of depression symptoms.
Other studies that investigated DNA methylation signatures of depression in discordant monozygotic twins study design implicated epigenetic changes in VDR26, HOXB7, CACNA1C, STK32C, NR1C3, and MYC genes among others, but not in KLK8 or DAZAP247,108,117,118. These studies were performed mainly in blood samples, but also in buccal cells of monozygotic twins discordant for depression, and even though they were performed on smaller twin cohorts, they studied individuals from a more severe part of depression scale in comparison to the MADT and LSADT twins analyzed in this study. That is why it should be noted that our findings represent epigenetic results related to severity of depression symptomatology across the general population, rather than markers related to MDD diagnosis. We have also observed that apart from the signal from epigenetic associations the depression symptomatology score was increased in females in comparison to males, and increased additionally with age, as reported before for elderly individuals22. Depression symptoms were reported to be comparable between younger and elderly individuals suffering from depression119,120. However, it should be noted that depression symptomatology evaluated in elderly individuals, in comparison to the young ones in the general population, may be driven by different factors, such as cognitive decline or represent a prodromal feature of dementia121,122,123. Based on the available data in this study we cannot delineate if the observed epigenetic differences associated with depression symptomatology in the elderly are generalizable to younger individuals. Other studies that identified changes in KLK8 expression levels to be linked with recurrent depression were performed in a younger population aged 18–67 (mean = 47.64 years, sd = 11.16)89 than MADT and LSADT cohorts, but more research is required across even younger age groups to assess if findings from this study apply to the general population.
In this study we have also tested the hypothesis that increased individual’s biological age (measured with Horvath- and Hannum-based DNAmAge estimates) in comparison to individual’s chronological age, which may serve as a marker of accelerated aging and health deterioration, associates with more severe depression symptomatology score in elderly individuals. Similarly to previous studies on cohorts with comparable age-spans we observed that Horvath-based DNAmAge underestimated, while Hannum-based DNAmAge overestimated the age of our study participants40,56. This difference could be explained by different set of probes used in these two DNAmAge predictors, with only 6 probes out of 353 in Horvath and 71 in Hannum DNAmAge estimators overlapping between them75,76. We initially observed a positive association between both Horvath and Hannum DNAmAge estimates and depression symptomatology score, however it was no longer significant after adjusting for chronological age of these individuals, a variable associated with the score itself. This observation suggests that the initial effect was observed only due to high correlation between DNAmAge and chronological age, and further between chronological age and depression symptomatology score. A recent study reported accelerated epigenetic aging in individuals suffering from MDD in comparison to controls124. The reason why we did not detect any association between accelDNAmAge and depression symptomatology score in our study may be because the phenotype in our study differed from the one studied by Han and co-authors, as we studied only depression symptomatology measured in a general population of monozygotic twins, and not in patients diagnosed with MDD, who, as pointed out before, represent the most severe part of the depression symptomatology spectrum. Thus, further studies, preferably performed in a cohort of monozygotic twins discordant for MDD, are needed to elucidate if DNAmAge acceleration is associated with the disorder regardless of the genetic background of studied individuals.
Apart from many strengths of this study, such as use of a large cohort of monozygotic twins and adjustment of EWAS models for blood cell proportions measured with flow cytometry, there are also potential limitations of this study, such as use of blood samples to study a brain-related phenotype. We are aware that DNA methylation profiles are tissue-specific, that they substantially differ between brain and blood samples, and that if surrogate tissues should be informative on the molecular changes of tissue of interest their methylation levels at locus of interest should ideally co-vary125,126. For the two most associated sites with p-value < 10−6 (in KLK8 and DAZAP2 genes) we did not find any strong evidence for correlation of methylation levels between blood and prefrontal cortex (PFC), entorhinal cortex (EC), superior temporal gyrus and cerebellum regions (https://epigenetics.essex.ac.uk/bloodbrain/)126. The most significant correlation for methylation levels for cg05777061 was observed between blood and EC (p-value = 0.026, r = 0.26) and for cg00554948 between blood and PFC (p-value = 0.035, r = 0.25). Remaining comparisons for these four tissues for these two probes had p-value > 0.05. Interestingly, previous studies did report factors that impact brain methylome to also leave an epigenetic signature in blood, and that inter-individual differences detected in blood correlate with differences observed in brain125,127. The latter observation may be explained to some extent by cross-tissues mQTL signals126,128, however a recent study reported DNA methylation differences observed in buccal cells between monozygotic twins discordant for depression to be successfully replicated in independent brain samples, which supports the use of secondary tissues for research of mental disorders117. That is why further replication of our findings in brain samples collected from an independent monozygotic twin cohort with data on depression symptomatology score is of high interest.
Use of the monozygotic twin study design has many advantages, as it allows adjusting the analyses for various factors shared between analyzed co-twins. However, the results could still be influenced by the unmeasured non-shared environmental factors, such as the well-known confounder of epigenomic studies tobacco smoking. None of our top associated sites was located in the well-established tobacco smoking loci, such as AHRR or GPR15, and none of the recent tobacco smoking EWAS reported DNA methylation changes in KLK8 or DAZAP2 to be associated with this trait31,129,130,131,132,133,134,135. Therefore we believe that tobacco smoking was not a confounder in our EWAS of depression symptomatology, or at least it was not a strong one. Another possible confounder that could impact our results was use of drugs, especially use of antidepressants is of interest in the context of this phenotype. According to the Drug-Gene Interaction Database neither KLK8 nor DAZAP2 have any well-established drug interactions136, however, an antidepressant fluoxetine was reported before to alter expression of KLK8 in the mice hippocampus137. Therefore we cannot exclude that the reported results are not influenced by additional non-shared environmental factors between investigated monozygotic twins.
It should be also noted that both top findings reported and discussed in this study (KLK8 and DAZAP2) were identified by using a p-value cut-off < 1 × 10−6, however, there is a discrepancy in the scientific literature on the threshold for epigenome-wide significant findings. Initially EWAS findings with p-value < 1 × 10−6 were considered to be genome-wide significant138, but a recent study proposed a more stringent threshold with p-value < 2.4 × 10−7 for the 450 K Illumina methylation array significant findings threshold, and p-value < 3.6 × 10−8 for genome-wide significant findings to be used139. Inclusion of more individuals with higher depression symptomatology score could allow for identification of more significant and stronger signals. These signals could also be informative on the epigenetic changes most related to the diagnosis of MDD. Also it should be remembered that the MADT and LSADT cohorts used in this study were composed only of elderly individuals and that their depression symptomatology score was evaluated with an instrument adequate for this age-span (CAMDEX)62. However, whether there are different biological processes that impact depression symptomatology at younger ages is yet to be elucidated.
In conclusion, we have performed EWAS of depression symptomatology score in a unique cohort of elderly monozygotic twins and identified blood methylation levels at KLK8, DAZAP2, MAD1L1, SLC29A2, AKT1, and other genes, as well as several DMRs across the genome to be associated with this trait. Function of these genes suggests a possible link between exposure to stress, epigenetic regulation of their expression, and further change in depression symptomatology, however, this hypothesis needs to be tested by future studies.
References
ICD-10 Version: 2016. http://apps.who.int/classifications/icd10/browse/2016/en (Accessed 26 Apr 2018).
Halfin, A. Depression: the benefits of early and appropriate treatment. Am. J. Manag Care 13, S92–S97 (2007).
Davidson, J. R. & Meltzer-Brody, S. E. The underrecognition and undertreatment of depression: what is the breadth and depth of the problem? J. Clin. Psychiatry 60(Suppl 7), 4–9 (1999). discussion10–1.
Barry, L. C., Abou, J. J., Simen, A. A. & Gill, T. M. Under-treatment of depression in older persons. J. Affect Disord. 136, 789–796 (2012).
Thornicroft, G. et al. Undertreatment of people with major depressive disorder in 21 countries. Br. J. Psychiatry 210, 119–124 (2017).
World Health Organization. WHO, The Global Burden of Disease, 2004 Update. 2008.
Kessler, R. C. & Bromet, E. J. The Epidemiology of Depression Across Cultures. Annu Rev. Public Health 34, 119–138 (2013).
Sullivan, P. F. et al. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol. Psychiatry 14, 359–375 (2009).
PGC - Major Depressive Disorder Working Group of the, Wray N. R., Sullivan P. F. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. bioRxiv 2017; 167577.
Power, R. A. et al. Genome-wide association for major depression through age at onset stratification: major depressive disorder Working Group of the Psychiatric Genomics Consortium. Biol. Psychiatry 81, 325–335 (2017).
Flint, J. & Kendler, K. S. The genetics of major depression. Neuron 81, 484–503 (2014).
Farmer, A. et al. Cardiff depression study. Br. J. Psychiatry 176, 150–155 (2000).
Sullivan, P. F., Neale, M. C. & Kendler, K. S. Genetic epidemiology of major depression: review and meta-analysis. Am. J. Psychiatry 157, 1552–1562 (2000).
Petersen, I., McGue, M., Tan, Q., Christensen, K. & Christiansen, L. Change in depression symptomatology and cognitive function in twins: a 10-year follow-up study. Twin Res. Hum. Genet 19, 104–111 (2016).
Prince, M. J., Harwood, R. H., Blizard, R. A., Thomas, A. & Mann, A. H. Social support deficits, loneliness and life events as risk factors for depression in old age. The Gospel Oak Project VI. Psychol. Med 27, 323–332 (1997).
Negele, A., Kaufhold, J., Kallenbach, L. & Leuzinger-Bohleber, M. Childhood trauma and its relation to chronic depression in adulthood. Depress Res Treat. 2015, 650804 (2015).
Roberts, R. E., Kaplan, G. A., Shema, S. J. & Strawbridge, W. J. Prevalence and correlates of depression in an aging cohort: the Alameda County Study. J. Gerontol. B Psychol. Sci. Soc. Sci. 52, S252–S258 (1997).
Mezuk, B., Eaton, W. W., Golden, S. H. & Ding, Y. The influence of educational attainment on depression and risk of type 2 diabetes. Am. J. Public Health 98, 1480–1485 (2008).
Coryell, W., Endicott, J. & Keller, M. Major depression in a nonclinical sample. Demographic and clinical risk factors for first onset. Arch. Gen. Psychiatry 49, 117–125 (1992).
Rao, T. S. S., Asha, M. R., Ramesh, B. N. & Rao, K. S. J. Understanding nutrition, depression and mental illnesses. Indian J. Psychiatry 50, 77–82 (2008).
van Uffelen, J. G. Z. et al. Sitting-Time, Physical Activity, and Depressive Symptoms in Mid-Aged Women. Am. J. Prev. Med 45, 276–281 (2013).
Heun, R. & Hein, S. Risk factors of major depression in the elderly. Eur. Psychiatry 20, 199–204 (2005).
Heijmans, B. T. et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl Acad. Sci. USA 105, 17046–17049 (2008).
Tobi, E. W. et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 5, 5592 (2014).
Richmond, R. C. et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet 24, 2201–2217 (2015).
Braithwaite, E. C., Kundakovic, M., Ramchandani, P. G., Murphy, S. E. & Champagne, F. A. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 10, 408–417 (2015).
Peter, C. J. et al. DNA Methylation Signatures of Early Childhood Malnutrition Associated With Impairments in Attention and Cognition. Biol. Psychiatry 80, 765–774 (2016).
Saunderson, E. A. et al. Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc. Natl. Acad. Sci. 113, 4830–4835 (2016).
Bustamante, A. C. et al. Glucocorticoid receptor DNA methylation, childhood maltreatment and major depression. J. Affect Disord. 206, 181–188 (2016).
Essex, M. J. et al. Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. 84, 58–75 (2013).
Zeilinger, S. et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 8, e63812 (2013).
Meng, Q. et al. Systems nutrigenomics reveals brain gene networks linking metabolic and brain disorders. EBioMedicine 7, 157–166 (2016).
Denham, J., Marques, F. Z., Bruns, E. L., O’Brien, B. J. & Charchar, F. J. Epigenetic changes in leukocytes after 8 weeks of resistance exercise training. Eur. J. Appl Physiol. 116, 1245–1253 (2016).
Fragou, D. et al. Effect of chronic heroin and cocaine administration on global DNA methylation in brain and liver. Toxicol. Lett. 218, 260–265 (2013).
Palma-Gudiel, H., Córdova-Palomera, A., Eixarch, E., Deuschle, M. & Fañanás, L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: a meta-analysis. Epigenetics 10, 893–902 (2015).
Palacios-García, I. et al. Prenatal stress down-regulates reelin expression by methylation of its promoter and induces adult behavioral impairments in rats. PLoS ONE 10, e0117680 (2015).
Rzehak, P. et al. Maternal smoking during pregnancy and DNA-methylation in children at age 5.5 years: epigenome-wide-analysis in the european childhood obesity project (CHOP)-study. PLoS ONE 11, e0155554 (2016).
Chen, S. et al. Consistency and variability of DNA methylation in women during puberty, young adulthood, and pregnancy. Genet Epigenet 9, 1179237X17721540 (2017).
Lomniczi, A. et al. Epigenetic control of female puberty. Nat. Neurosci. 16, 281–289 (2013).
Marioni, R. E. et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 16, 25 (2015).
Jung, M. & Pfeifer, G. P. Aging and DNA methylation. BMC Biol. 13, 7 (2015).
Aberg, K. A. et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 71, 255–264 (2014).
Hannon, E. et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 17, 176 (2016).
Abdolmaleky, H. M. et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum. Mol. Genet 15, 3132–3145 (2006).
Montano, C. et al. Association of DNA Methylation Differences With Schizophrenia in an Epigenome-Wide Association Study. JAMA psychiatry 73, 506–514 (2016).
Dempster, E. L. et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum. Mol. Genet 20, 4786–4796 (2011).
Córdova-Palomera, A. et al. Genome-wide methylation study on depression: differential methylation and variable methylation in monozygotic twins. Transl. Psychiatry 5, e557 (2015).
Kato, T. & Iwamoto, K. Comprehensive DNA methylation and hydroxymethylation analysis in the human brain and its implication in mental disorders. Neuropharmacology 80, 133–139 (2014).
Mill, J. et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet 82, 696–711 (2008).
Ladd-Acosta, C. et al. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 19, 862–871 (2014).
Nardone, S. et al. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 4, e433 (2014).
Ciernia, A. V. & LaSalle, J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat. Rev. Neurosci. 17, 411–423 (2016).
Walton, E. et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol. Psychiatry 22, 250–256 (2017).
Ianov, L., Riva, A., Kumar, A. & Foster, T. C. DNA methylation of synaptic genes in the prefrontal cortex is associated with aging and age-related cognitive impairment. Front Aging Neurosci. 9, 249 (2017).
Starnawska, A. et al. Epigenome-wide association study of cognitive functioning in middle-aged monozygotic twins. Front Aging Neurosci. 9, 413 (2017).
Starnawska A., et al. Blood DNA methylation age is not associated with cognitive functioning in middle-aged monozygotic twins. Neurobiol Aging 2017. https://doi.org/10.1016/j.neurobiolaging.2016.10.025.
Jack, A., Connelly, J. J. & Morris, J. P. DNA methylation of the oxytocin receptor gene predicts neural response to ambiguous social stimuli. Front Hum. Neurosci. 6, 280 (2012).
Skytthe, A. et al. The Danish twin registry in the new millennium. Twin Res Hum. Genet 9, 763–771 (2006).
Skytthe, A., Kyvik, K. O., Holm, N. V. & Christensen, K. The Danish twin registry. Scand. J. Public Health 39, 75–78 (2011).
Skytthe, A. et al. The Danish Twin Registry: linking surveys, national registers, and biological information. Twin Res Hum. Genet 16, 104–111 (2013).
Christensen, K., Holm, N. V., Mcgue, M., Corder, L. & Vaupel, J. W. A Danish population-based twin study on general health in the elderly. J. Aging Health 11, 49–64 (1999).
McGue, M. & Christensen, K. Genetic and environmental contributions to depression symptomatology: evidence from Danish twins 75 years of age and older. J. Abnorm Psychol. 106, 439–448 (1997).
van Iterson, M. et al. MethylAid: visual and interactive quality control of large Illumina 450k datasets. Bioinformatics 30, 3435–3437 (2014).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Chen, Y. et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209 (2013).
Fortin, J.-P. et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503 (2014).
Du, P. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinforma. 11, 587 (2010).
R Core Team. R: a language and environment for statistical computing. 2015. http://www.gbif.org/resource/81287 (Accessed 1 May 2016).
Kuznetsova, A., Brockhoff, P. B. & Christensen, R. H. B. lmerTest Package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26 (2017).
Wang, J., Duncan, D., Shi, Z. & Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 41, W77–W83 (2013).
Story Jovanova, O. et al. DNA Methylation Signatures of Depressive Symptoms in Middle-aged and Elderly Persons. JAMA Psychiatry 75, 949 (2018).
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 28, 2986–2988 (2012).
Mallik S., et al An evaluation of supervised methods for identifying differentially methylated regions in Illumina methylation arrays. Brief Bioinform 2018. https://doi.org/10.1093/bib/bby085.
Šidák, Z. Rectangular confidence regions for the means of multivariate normal distributions. J. Am. Stat. Assoc. 62, 626–633 (1967).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
Shabalin A. A., Clark S. L., Hattab M. W., Aberg K. A. van den O. E. Package ‘ramwas’ Title Fast Methylome-Wide Association Study Pipeline for Enrichment Platforms. 2017.
Badenhop, R. F. et al. A genome screen of 13 bipolar affective disorder pedigrees provides evidence for susceptibility loci on chromosome 3 as well as chromosomes 9, 13 and 19. Mol. Psychiatry 7, 851–859 (2002).
Francks, C. et al. Population-based linkage analysis of schizophrenia and bipolar case–control cohorts identifies a potential susceptibility locus on 19q13. Mol. Psychiatry 15, 319–325 (2010).
Izumi, A. et al. Genetic variations of human neuropsin gene and psychiatric disorders: polymorphism screening and possible association with bipolar disorder and cognitive functions. Neuropsychopharmacology 33, 3237–3245 (2008).
Li, Y. et al. Recent origin of a hominoid-specific splice form of neuropsin, a gene involved in learning and memory. Mol. Biol. Evol. 21, 2111–2115 (2004).
Mitsui, S., Tsuruoka, N., Yamashiro, K., Nakazato, H. & Yamaguchi, N. A novel form of human neuropsin, a brain-related serine protease, is generated by alternative splicing and is expressed preferentially in human adult brain. Eur. J. Biochem. 260, 627–634 (1999).
Nakamura, Y., Tamura, H., Horinouchi, K. & Shiosaka, S. Role of neuropsin in formation and maturation of Schaffer-collateral L1cam-immunoreactive synaptic boutons. J. Cell Sci. 119, 1341–1349 (2006).
Matsumoto-Miyai, K. et al. NMDA-dependent proteolysis of presynaptic adhesion molecule L1 in the hippocampus by neuropsin. J. Neurosci. 23, 7727–7736 (2003).
Laifenfeld, D., Karry, R., Klein, E. & Ben-Shachar, D. Alterations in cell adhesion molecule L1 and functionally related genes in major depression: A postmortem study. Biol. Psychiatry 57, 716–725 (2005).
Laifenfeld, D., Karry, R., Grauer, E., Klein, E. & Ben-Shachar, D. Antidepressants and prolonged stress in rats modulate CAM-L1, laminin, and pCREB, implicated in neuronal plasticity. Neurobiol. Dis. 20, 432–441 (2005).
Harada, A., Shiosaka, S., Ishikawa, Y. & Komai, S. Acute stress increases neuropsin mRNA expression in the mouse hippocampus through the glucocorticoid pathway. Neurosci. Lett. 436, 273–277 (2008).
Chang, S., Bok, P., Sun, C.-P., Edwards, A. & Huang, G.-J. Neuropsin inactivation has protective effects against depressive-like behaviours and memory impairment induced by chronic stress. PLOS Genet. 12, e1006356 (2016).
Talarowska, M., Bobinska, K., Galecka, E., Szemraj, J. & Galecki, P. Human neuropsin gene and social cognition in depression. Neuropsychiatry 6, 396–402 (2016).
Bobińska, K. et al. Human neuropsin gene in depression. Psychiatr. Danub 29, 195–200 (2017).
Yang, W. & Mansour, S. L. Expression and genetic analysis ofprtb, a gene that encodes a highly conserved proline-rich protein expressed in the brain. Dev. Dyn. 215, 108–116 (1999).
Kim, J.-E. et al. Proline-rich transcript in brain protein induces stress granule formation. Mol. Cell Biol. 28, 803–813 (2008).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Wei, J. et al. Histone modification of Nedd4 ubiquitin ligase controls the loss of AMPA receptors and cognitive impairment induced by repeated stress. J. Neurosci. 36, 2119–2130 (2016).
Hori, H. et al. Blood-based gene expression signatures of medication-free outpatients with major depressive disorder: integrative genome-wide and candidate gene analyses. Sci. Rep. 6, 18776 (2016).
Guilloux, J.-P. et al. Testing the predictive value of peripheral gene expression for nonremission following citalopram treatment for major depression. Neuropsychopharmacology 40, 701–710 (2015).
Smagin, D. A. et al. Dysfunction in ribosomal gene expression in the hypothalamus and hippocampus following chronic social defeat stress in male mice as revealed by RNA-Seq. Neural Plast. 2016, 3289187 (2016).
Karssen, A. M. et al. Stress-induced changes in primate prefrontal profiles of gene expression. Mol. Psychiatry 12, 1089–1102 (2007).
Le-Niculescu, H. et al. Identifying blood biomarkers for mood disorders using convergent functional genomics. Mol. Psychiatry 14, 156–174 (2009).
Gould, T. D. et al. Generation and behavioral characterization of beta-catenin forebrain-specific conditional knock-out mice. Behav. Brain Res 189, 117–125 (2008).
Kéri, S., Szabó, C. & Kelemen, O. Blood biomarkers of depression track clinical changes during cognitive-behavioral therapy. J. Affect Disord. 164, 118–122 (2014).
Fisher, H. L. et al. Methylomic analysis of monozygotic twins discordant for childhood psychotic symptoms. Epigenetics 10, 1014–1023 (2015).
Luo, S.-Q. et al. The effects of promoter methylation on downregulation of DAZAP2 in multiple myeloma cell lines. PLoS ONE 7, e40475 (2012).
Shi, Y. et al. The structure, expression, and function prediction of DAZAP2, a down-regulated gene in multiple myeloma. Genom. Proteom. Bioinforma. 2, 47–54 (2004).
Howard, D. M. et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 9, 1470 (2018).
Ripke, S. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Ikeda, M. et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol. Psychiatry 23, 639–647 (2018).
Byrne, E. M. et al. Monozygotic twins affected with major depressive disorder have greater variance in methylation than their unaffected co-twin. Transl. Psychiatry 3, e269 (2013).
Gass, N. et al. Contribution of adenosine related genes to the risk of depression with disturbed sleep. J. Affect Disord. 126, 134–139 (2010).
Van der Auwera, S. et al. Genome-wide gene-environment interaction in depression: A systematic evaluation of candidate genes: The childhood trauma working-group of PGC-MDD. Am. J. Med Genet B Neuropsychiatr. Genet 177, 40–49 (2018).
Yang, C. et al. Association between AKT1 gene polymorphisms and depressive symptoms in the Chinese Han population with major depressive disorder. Neural Regen. Res 7, 235–239 (2012).
Pereira, P. A. et al. Genetic variant of AKT1 and AKTIP associated with late-onset depression in a Brazilian population. Int J. Geriatr. Psychiatry 29, 399–405 (2014).
Losenkov, I. S., Vyalova, N. M., Simutkin, G. G., Bokhan, N. A. & Ivanova, S. A. An association of AKT1 gene polymorphism with antidepressant treatment response. World J. Biol. Psychiatry 17, 239–242 (2016).
Hammamieh, R. et al. Whole-genome DNA methylation status associated with clinical PTSD measures of OIF/OEF veterans. Transl. Psychiatry 7, e1169 (2017).
Wockner, L. F. et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 4, e339–e339 (2014).
Machado-Vieira, R. et al. Decreased AKT1/mTOR pathway mRNA expression in short-term bipolar disorder. Eur. Neuropsychopharmacol. 25, 468–473 (2015).
Dempster, E. L. et al. Genome-wide methylomic analysis of monozygotic twins discordant for adolescent depression. Biol. Psychiatry 76, 977–983 (2014).
Malki, K. et al. Epigenetic differences in monozygotic twins discordant for major depressive disorder. Transl. Psychiatry 6, e839 (2016).
Mezuk, B. & Kendler, K. S. Examining variation in depressive symptoms over the life course: a latent class analysis. Psychol. Med 42, 2037–2046 (2012).
Stage, K. B., Bech, P., Kragh-Sørensen, P., Nair, N. P. & Katona, C. Differences in symptomatology and diagnostic profile in younger and elderly depressed inpatients. J. Affect Disord. 64, 239–248 (2001).
McGue, M. & Christensen, K. Growing old but not growing apart: twin similarity in the latter half of the lifespan. Behav. Genet 43, 1–12 (2013).
Vinkers, D. J., Gussekloo, J., Stek, M. L., Westendorp, R. G. J. & van der Mast, R. C. Temporal relation between depression and cognitive impairment in old age: prospective population based study. BMJ 329, 881 (2004).
Brommelhoff, J. A. et al. Depression as a risk factor or prodromal feature for dementia? Findings in a population-based sample of Swedish twins. Psychol. Aging 24, 373–384 (2009).
Han, L. K. M. et al. Epigenetic aging in major depressive disorder. Am. J. Psychiatry 175, 774–782 (2018). appi.ajp.2018.1.
Davies, M. N. et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 13, R43 (2012).
Hannon, E., Lunnon, K., Schalkwyk, L. & Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 10, 1024–1032 (2015).
Aberg, K. A. et al. Testing two models describing how methylome-wide studies in blood are informative for psychiatric conditions. Epigenomics 5, 367–377 (2013).
Smith, A. K. et al. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genom. 15, 145 (2014).
Besingi, W. & Johansson, Å. Smoke-related DNA methylation changes in the etiology of human disease. Hum. Mol. Genet 23, 2290–2297 (2014).
Tsaprouni, L. G. et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 9, 1382–1396 (2014).
Sayols-Baixeras, S. et al. Identification of a new locus and validation of previously reported loci showing differential methylation associated with smoking. The REGICOR study. Epigenetics 10, 1156–1165 (2015).
Ambatipudi, S. et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics 8, 599–618 (2016).
Wan, E. S. et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. Genet 21, 3073–3082 (2012).
Park, S. L. et al. Association of internal smoking dose with blood DNA methylation in three racial/ethnic populations. Clin. Epigenetics 10, 110 (2018).
Li, S. et al. Causal effect of smoking on DNA methylation in peripheral blood: a twin and family study. Clin. Epigenetics 10, 18 (2018).
Cotto, K. C. et al. DGIdb 3.0: a redesign and expansion of the drug–gene interaction database. Nucleic Acids Res 46, D1068–D1073 (2018).
Huang, G.-J. et al. Neurogenomic evidence for a shared mechanism of the antidepressant effects of exercise and chronic fluoxetine in mice. PLoS ONE 7, e35901 (2012).
Tsai, P. C. & Bell, J. T. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int J. Epidemiol. 44, 1429–1441 (2015).
Saffari, A. et al. Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol. 42, 20–33 (2018).
Acknowledgements
This study was funded by The Danish Council for Independent Research (DFF-6110-00016), The European Union’s Seventh Framework Programme (FP7/2007-2011) under grant agreement no. 259679, and the Lundbeck Foundation, Denmark (R155-2014-1724). The twin surveys are supported by grants from The National Program for Research Infrastructure 2007 (09-063256) from the Danish Agency for Science Technology and Innovation, the Velux Foundation and the US National Institute of Health (P01 AG08761).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Starnawska, A., Tan, Q., Soerensen, M. et al. Epigenome-wide association study of depression symptomatology in elderly monozygotic twins. Transl Psychiatry 9, 214 (2019). https://doi.org/10.1038/s41398-019-0548-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0548-9
This article is cited by
-
Upregulation of KLK8 contributes to CUMS-induced hippocampal neuronal apoptosis by cleaving NCAM1
Cell Death & Disease (2023)
-
Associations of stress and stress-related psychiatric disorders with GrimAge acceleration: review and suggestions for future work
Translational Psychiatry (2023)
-
Molecular pathways of major depressive disorder converge on the synapse
Molecular Psychiatry (2023)
-
Therapeutic Potential of the Purinergic System in Major Depressive Disorder Associated with COVID-19
Cellular and Molecular Neurobiology (2023)
-
DNA methylome-wide association study of genetic risk for depression implicates antigen processing and immune responses
Genome Medicine (2022)
