
Abstract
Family and twin studies have shown a genetic component to seasonal affective disorder (SAD). A number of candidate gene studies have examined the role of variations within biologically relevant genes in SAD susceptibility, but few genome-wide association studies (GWAS) have been performed to date. The authors aimed to identify genetic risk variants for SAD through GWAS. The authors performed a GWAS for SAD in 1380 cases and 2937 controls of European-American (EA) origin, selected from samples for GWAS of major depressive disorder and of bipolar disorder. Further bioinformatic analyses were conducted to examine additional genomic and biological evidence associated with the top GWAS signals. No susceptibility loci for SAD were identified at a genome-wide significant level. The strongest association was at an intronic variant (rs139459337) within ZBTB20 (odds ratio (OR) = 1.63, p = 8.4 × 10−7), which encodes a transcriptional repressor that has roles in neurogenesis and in adult brain. Expression quantitative trait loci (eQTL) analysis showed that the risk allele “T” of rs139459337 is associated with reduced mRNA expression of ZBTB20 in human temporal cortex (p = 0.028). Zbtb20 is required for normal murine circadian rhythm and for entrainment to a shortened day. Of the 330 human orthologs of murine genes directly repressed by Zbtb20, there were 32 associated with SAD in our sample (at p < 0.05), representing a significant enrichment of ZBTB20 targets among our SAD genetic association signals (fold = 1.93, p = 0.001). ZBTB20 is a candidate susceptibility gene for SAD, based on a convergence of genetic, genomic, and biological evidence. Further studies are necessary to confirm its role in SAD.
Similar content being viewed by others

Introduction
Seasonal affective disorder (SAD) is a form of mood disorder that typically occurs in late fall and winter when periods of daylight are shortest. The hypothesis that light-sensitivity is a core feature of this disorder is supported by the efficacy of bright light and dawn simulation therapies1. Family and twin studies of SAD have suggested a genetic component to its etiology. Studies of the relatives of SAD probands have shown higher than expected rates of the disorder in these family members, with rates in the range of 14–26%2,3,4. A twin study demonstrated that genetic effects account for at least 29% of the variance in seasonality, including mood variation5.
Association studies using the candidate gene approach have been applied to investigate the role of genetic variation within biologically relevant genes in SAD susceptibility. For example, variants in genes related to serotonergic transmission, such as the serotonin transporter gene SLC6A46 and the 5-HT2A receptor gene HTR2A, have been associated with SAD at a suggestive level of significance7,8. Variants in the circadian genes ARNTL, PER2, and NPAS2 have been reported to be associated with SAD9,10. A variant in the melanopsin gene, OPN4, which encodes a retinal protein essential for registering light intensity, was nominally associated with SAD in one report11.
Genome-wide association studies (GWAS) have been widely used for identifying common variants associated with complex diseases. In contrast to the candidate gene approach, GWAS allows for testing genetic variants across the genome without reliance on prior knowledge about disease biology. Genetic findings from GWAS are shedding new light on psychiatric disorders, such as depression, schizophrenia, and bipolar disorder12,13,14. However, GWAS of SAD has to date been limited to one study, which did not report any promising candidate genes15.
In the current study, we performed a meta-analysis of GWAS for SAD in 1380 cases and 2937 controls of European-American (EA) origin, selected from samples for GWAS of major depressive disorder (MDD) and of bipolar disorder (BD). We report that ZBTB20 is a candidate susceptibility gene for SAD, based on a convergence of genetic, genomic, and biological evidence.
Methods
Study samples
Case-control samples for SAD GWAS were selected from those originally ascertained and assessed for MDD or BD. Informed consent for participation in genetic studies was obtained from all subjects. Basic information for the study samples from each GWAS is summarized in Table 1. The following provides details on the samples included.
BD subjects were selected from the National Institute of Mental Health (NIMH) Bipolar Genetics Study (BiGS). The genotyping focused on European ancestry subjects with Bipolar I (BPI) disorder from among those collected by the NIMH Genetics Initiative for Bipolar Disorder in five waves at 11 sites across the United States, as described elsewhere in detail16. Only BPI cases were included in this analysis. All cases were interviewed with the Diagnostic Interview for Genetic Studies (DIGS), and these together with interviewer observations, medical records and family history data collected through the Family Interview for Genetic Study (FIGS), were used to assign diagnoses based on DSM-III or DSM-IV criteria following a best-estimate procedure. Details of the DIGS and FIGS are available at http://www.nimhgenetics.org.
MDD subjects were selected from the Genetics of Recurrent Early-Onset Depression (GenRED) study. GenRED I collected affected sibling pair families for linkage studies, while GenRED II recruited cases for association studies. Probands had MDD diagnosed with either >1 episode or a single episode lasting >3 years, onset before age 31, and a parent or a sibling with MDD and age of onset <41. Proband information was gathered through the DIGS, which together with an interviewer narrative summary, available medical records and family history data gathered through FIGS, was used to establish a diagnosis based on DSM-IV criteria. Ascertainment and assessment details for GenRED have been previously described17.
Healthy control samples were obtained from the NIMH Genetics Initiative repository. Some of these were selected from among those ascertained through an NIMH-supported contract mechanism between Dr. Pablo Gejman and Knowledge Networks, Inc.18. They were ascertained to be free of psychiatric illness based on self-report using the Composite International Diagnostic Interview—Short Form, which assesses major depression, various anxiety disorders, and alcohol and drug dependence. This was augmented with three items inquiring about lifetime diagnosis of, or treatment for, schizophrenia, schizoaffective disorder, and bipolar disorder. A total of 1515 of these healthy control European ancestry (EA) subjects were included for the BD GWAS. Another independent set of 1422 healthy EA controls were included for the MDD GWAS, which included 492 from the above data set, and 930 more from the Genomic Psychiatry Cohort or the Mayo Clinic Biobank.
Phenotype assignment
The assignment of the seasonal or nonseasonal case status was based on identifying all subjects with seasonal depressive episodes among BD or MDD patients. We used a broad definition of seasonal depression. The assignment of seasonal depression status was based on the subject’s answer to “Do your depressions tend to begin in any particular season?” question in DIGS. All subjects with winter or fall answers were assigned seasonal case status in our study, and all of the others with answers became nonseasonal case subjects (non-SAD).
GWAS and quality control
All BD GWAS samples were genotyped with the Affymetrix 6.0 genome-wide SNP array at the Broad Institute Center for Genotyping and Analysis. Different genotyping efforts led to a partition of BiGS data into two sets: those genotyped as a part of Genetic Association Information Network Bipolar Sample (GAIN) and those genotyped as a part of the Translational Genomic Research Institute Sample (TGEN). The GAIN set includes subjects from the first four waves of the NIMH study, and a small subset of wave five subjects. The TGEN set includes most of the wave five subjects. GenRED samples were genotyped in two waves. First wave samples were genotyped using the Affymetrix 6.0 genome-wide SNP array, and the second wave used the Illumina Omni1-Quad microarray (GenRED2).
Initial quality control (QC) for each GWAS data set removed subjects that failed set thresholds for call rates or heterozygosity. We carried out additional QC filtering for each data set with the following criteria: minor allele frequency (MAF) > 1%, missing rate per SNP < 5%, and Hardy−Weinberg equilibrium (HWE) p value > 10−6. After QC, there were 692,411 (GAIN), 701,183 (TGEN), 701,402 (TGEN2), 646,301 (GENRED), and 719,050 (GENRED2) autosomal SNPs retained for imputation. To investigate population stratification, we computed principal components (PC) for GWAS samples using EIGENSOFT19 based on a subset of SNPs that were in low linkage disequilibrium with one another (r2 < 0.2). We did not detect any outlier subjects from the PC analysis, which were defined as subjects whose ancestry was at least three standard deviations from the mean on one of the two largest PCs.
Imputation
Following the best practice guidelines of IMPUTE220, we imputed 1000 Genomes variants into each GWAS sample. Prephasing was first performed with SHAPEIT21 to infer haplotypes for samples based on autosomal SNPs with MAF > 0.01. Imputation was carried out on prephased haplotypes using IMPUTE2 against haplotype reference data from the 1000 Genomes Phase 3. After postimputation QC (SNP missing rate < 0.05, MAF > 0.01, imputation quality score > 0.5, and HWE > 10−6), there were 8,462,922 (GAIN), 8,458,360 (TGEN), 8,272,185 (TGEN2), 8,452,433 (GENRED), and 8,498,837 (GENRED2) autosomal variants, among which the majority of variants were imputed, with an average proportion of 92% across study samples. A common set of 8,153,767 SNPs were retained for meta-analysis.
Association analysis
We first carried out association analysis for each autosomal SNP with SAD in each sample, and then performed meta-analysis to combine association evidence across samples. In association analysis, to account for the uncertainty of imputed genotypes, expected allelic dosages were used in a logistic regression framework as implemented by SNPTEST22. Sex and the first three PCs of population structure were included as covariates. We carried out meta-analysis based on effect size and standard error using METAL under a fixed-effect model23. We evaluated the possibility of population stratification or other systemic biases by using the quantile−quantile (QQ) plots based on p values of SNPs. Regional association plots were created using the online web tool LocusZoom24.
We computed gene-based p values using SimpleM25 to evaluate the association evidence at the gene level. SimpleM takes the smallest p value of SNPs within a gene while correcting for the effective number of independent SNPs through Bonferroni correction. The effective number of independent tests was estimated from the correlation matrix of SNPs using PC analysis, so that the corresponding eigenvalues explain 99.5% of the variation for the SNP data. We assigned a SNP to a gene if it was located within the gene, based on hg19 refGene annotation, or within 20 kb upstream or downstream of the gene, to capture regulatory variants.
Expression QTL (eQTL) analysis
To determine whether the lead SNP from association analysis has an effect on regulating gene expression in specific brain regions, we queried the brain eQTL online database (http://www.braineac.org/), which contains eQTL information for ten human brain regions. Full details on the database are available elsewhere26.
Enrichment analysis for ZBTB20 candidate target genes
We examined whether ZBTB20 candidate target genes were enriched for genes associated with SAD. Specifically, a two-sided Fisher’s exact test was used to determine whether there was an overrepresentation of nominally significant genes (p ≤ 0.05) among ZBTB20 candidate targets in comparison to the remaining genes. Candidate target genes of ZBTB20 were derived from a previous study, in which ChIP-Seq combined with microarray gene expression analysis approaches were applied to identify 346 murine genes directly repressed by Zbtb20 during hippocampal CA1 neurogenesis in vivo27. The 346 murine genes were mapped to 342 human orthologous ZBTB20 candidate target genes, of which 330 were assigned a gene-level p value for SAD.
Results
Association analysis
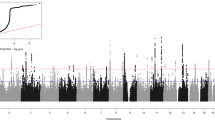
The Manhattan plot for the meta-analysis of GWAS for SAD is illustrated in Figure S1. The QQ plot of p values indicated minimal evidence of population stratification or other systematic bias (lambda = 1.0, Figure S2). Overall, no single SNPs were identified at genome-wide significance (p < 5×10−8). There were 90 SNPs associated with SAD at p < 1×10−5 (Table S1). The strongest association evidence was at an intronic variant (rs139459337) within ZBTB20 (odds ratio (OR) = 1.63, p = 8.4×10−7), which encodes a transcriptional repressor that plays a key role in neurogenesis. Figure 1 shows the regional association plot for variants within and around ZBTB20.

SNPs are plotted with their −log10 (p value) on the y-axis along with their physical position (NCBI build 36) on the x-axis. The SNPs are color coded according to their correlations (r2), with the most significant SNP rs139459337 shown in purple. The light blue line and right y-axis indicate the observed recombination rates in the HapMap CEU samples
Table 2 provides details of the association analysis results for the lead SNP rs139459337. The minor allele “T” was associated with an increased risk for SAD in each sample. We found no significant association for rs139459337 with non-SAD (p = 0.76, Table S1), suggesting the top GWAS signal was specific to SAD. A broader case-only GWAS comparing SAD to non-SAD cases did not reveal compelling signals; however, rs139459337 differed between SAD and non-SAD with OR of 1.47, and p value of 5.9×10−5 (Table S1).
We also evaluated the genetic association evidence at the gene level for the SAD vs. healthy control comparison; however, no genes reached genome-wide significance (p < 2×10−6). The strongest gene-level association evidence was, again, for ZBTB20 (p = 8.0×10−5, Table S2).
eQTL analysis
We queried a brain eQTL database and examined whether the lead SNP rs139459337 regulates ZBTB20 mRNA expression levels in ten brain regions. In this small sample, we observed that the risk allele “T” was associated with lower ZBTB20 mRNA levels in nine of the ten regions, a difference that was nominally significant in the temporal cortex (p = 0.028) (Figure S3). The result does not survive correction for multiple testing.
Enrichment of GWAS signals for ZBTB20 candidate target genes
Because both SNP and gene-level association analyses implicated ZBTB20 as our strongest finding, we tested whether there was an enrichment of SAD GWAS association signals for genes that are candidate targets for the ZBTB20 transcriptional repressor protein. Of the 330 human orthologues of murine genes directly repressed by Zbtb20, we found that 32 were associated with SAD (p < 0.05), representing a significant enrichment as compared to chance expectation (fold enrichment = 1.93, p = 0.001). When we examined the QQ plot of p values, we found an obvious deviation from the uniform expected distribution for p values of the ZBTB20 candidate target gene group, but not for the remaining nontarget genes (Fig. 2). Table S3 provides a list of ZBTB20 targets that showed nominally significant association with SAD (p < 0.05).

The gray shading indicates 95% confidence interval under the null hypothesis of no association
Candidate genes
A number of candidate genes of biological interest have previously been tested for their association with SAD. These include genes related to serotonergic transmission, such as SLC6A46 and HTR2A7,8; circadian genes, such as ARNTL, PER2 and NPAS2; and OPN4, which encodes the photoreceptive opsin protein. Using the number of common SNPs (MAF > 0.01) that appear in the CEU samples of 1000 Genomes as a reference, we first evaluated whether our two genotyping platforms (Affymetrix 6.0 and Illumina Omni1-Quad) provided adequate coverage of SNPs in these candidate genes. Although the two genotyping arrays contained only a limited proportion of SNPs within each candidate gene (ranging from 4 to 19%), imputation greatly improved the coverage of these SNPs (89−97%, Table S4). In our current GWAS, none of these genes yielded SNPs with association p values < 1×10−3. The gene with the strongest association evidence was NPAS2, which included the SNP rs113837129, associated with SAD at p = 0.001. Gene-level analysis of NPAS2 produced a trend toward association (p = 0.09). Gene-level analyses did not show any significant association evidence for other candidate genes (p > 0.3). Table S5 provides the SNP-level analysis results with nominal significance for these candidate genes.
Discussion
In the current study, we conducted a GWAS for SAD in an EA sample. Although no genome-wide significant findings were identified, we report that ZBTB20 is a strong candidate susceptibility gene for SAD based on a convergence of genetic, genomic, and biological evidence. In particular, the lead SNP rs139459337 within ZBTB20 showed modest eQTL evidence for regulating gene expression in temporal cortex. In addition, ZBTB20 candidate targets were enriched for genes nominally associated with SAD.
Intriguingly, prior research suggests there is substantial biological plausibility for ZBTB20 as a candidate gene for SAD. ZBTB20 is a zinc finger transcriptional repressor protein that is particularly abundant in the hippocampus27. The murine Zbtb20 gene is essential for the specification of hippocampal CA1 field identity28. Ectopic expression of Zbtb20 proteins in transgenic mice causes behavioral abnormalities suggestive of deficiency in visual and spatial memory processing29. Most strikingly, a recent study reported that loss of Zbtb20 in mice impairs circadian rhythms, particularly reducing activity in the early evening period of those nocturnal animals30. The Zbtb20-deficient mice also had more difficulty than control mice in entraining to a shortened day.
Haploinsufficiency of ZBTB20 has been reported to be involved in the neurodevelopmental and neuropsychiatric disorders seen in the 3q13.31 microdeletion syndrome31 and in Primrose syndrome32. Interestingly, our eQTL analysis indicated, albeit at a suggestive level, that the risk allele of rs139459337 decreases ZBTB20 expression in temporal cortex, which is in accordance with the haploinsufficiency mechanism of ZBTB20 in neuropsychiatric disorders. The reduced gene expression level of the risk allele also parallels a previous study, which reported that hypermethylation in the ZBTB20 gene was associated with MDD33.
Our analysis has shown significant enrichment of SAD association signals for candidate gene targets of ZBTB20. This result implies that pathways regulated by ZBTB20 might be important in the etiology of SAD. A number of candidate gene targets of ZBTB20 are worth noting, as they have been implicated in various psychiatric disorders and behavioral phenotypes. These include NRXN134, NRXN335, and SYN336. The neurexins are a family of cell adhesion molecules that act predominantly at the presynaptic terminal in neurons and play essential roles in neurotransmission and differentiation of synapses. NRXN1 and NRXN3 have both been strongly implicated in autism, and NRXN1 has been associated with schizophrenia37. There have also been several studies suggesting a relationship of these genes to mood disorders. Deletions in NRXN1 were associated with poorer response to antidepressant medications38. A SNP in NRXN3 was associated, at a genome-wide significant level, with symptom improvement during citalopram/escitalopram treatment, though this did not replicate in additional samples39. Both NRXN1 and NRXN3 have altered expression levels in postmortem brains of suicide completers40. Interestingly, NRXN1 has also been suggested to play a role in circadian rhythm. Diurnal rhythms in mNRXN1 transcription were found in the superchiasmatic nucleus of mice41. SYN3 is a member of the synapsin gene family encoding neuronal phosphoproteins that associate with the cytoplasmic surface of synaptic vesicles. The SYN3 protein has a role in synaptogenesis and the modulation of neurotransmitter release. SYN3 was implicated in a study that examined DNA methylation differences in MZ twins selected based on discordance in cortisol and MRI measures related to anxiety42.
Our analysis of candidate genes yielded little evidence of association with SAD, though it did turn up nominally significant association in NPAS2. Notably, like Zbtb20, Npas2 plays a role in circadian rhythms. It is closely related in primary amino acid sequence to Clock, a transcription factor expressed in the suprachiasmatic nucleus (SCN) that regulates circadian rhythm. Npas2 was identified as an analog of Clock operating as a circadian regulator in the mammalian forebrain43. Later, it was shown that Npas2 can functionally substitute for Clock in the SCN in mice to regulate circadian rhythmicity44. In one study Npas2-deficient mice showed a number of subtle differences in circadian activity as compared to wild-type mice. Under normal 12-h light/12-h dark cycles followed by constant darkness conditions, Npas2 (+/–) and Npas2 (–/–) mice displayed rhythmic locomotor activity largely comparable to that of wild-type littermates. The exception is that Npas2 (–/–) mice postponed or skipped a prominent break present in wild-type mice during the middle of the night45. By comparison, Zbtb20 (–/–) mice, when compared with the Zbtb20 (+/–) and control mice, displayed a dramatic loss of early evening activity along with significantly limited durations in their early morning activity30. In terms of rhythmic oscillation of locomotor activity, it was decreased in Npas2 (–/–) mice compared to the wild-type and (+/–) mice. On the other hand, the intrinsic period was increased in Zbtb20 (–/–) mice compared to the control mice.
This study should be considered in light of several limitations. First, our assessment of seasonality was retrospective. However, given that we were asking about a recurring pattern, rather than a one-time event, recall should be reasonably robust. Retrospective assessment of SAD has also been found to be reliable45. A second limitation lies in our definition of seasonality. We did not use assessment tools such as the Seasonal Pattern Assessment Questionnaire (SPAQ) to quantify seasonality based on the Global Seasonality Score (GSS)46,47. Therefore, we are not able to examine seasonality as a continuous phenotype. Finally, our sample size was small for a GWAS study, and thus our power to detect genome-wide significant associations was limited.
In conclusion, our study identifies ZBTB20 as a candidate susceptibility gene for SAD. Further study in independent samples with the appropriate phenotype data is needed to confirm this association.
References
Golden, R. N. et al. The efficacy of light therapy in the treatment of mood disorders: a review and meta-analysis of the evidence. Am. J. Psychiatry 162, 656–662 (2005).
Thompson, C. & Isaacs, G. Seasonal affective disorder—a British sample. Symptomatology in relation to mode of referral and diagnostic subtype. J. Affect Disord. 14, 1–11 (1988).
Rosenthal, N. E. et al. Seasonal affective disorder: a description of the syndrome and preliminary findings with light therapy. Arch. Gen. Psychiatry 41, 72–80 (1984).
Wirz‐Justice, A. et al. Light treatment of seasonal affective disorder in Switzerland. Acta Psych. Scandin. 74, 193–204 (1986).
Madden, P. A., Heath, A. C., Rosenthal, N. E. & Martin, N. G. Seasonal changes in mood and behavior: the role of genetic factors. Arch. Gen. Psychiatry 53, 47–55 (1996).
Rosenthal, N. E. et al. Role of serotonin transporter promoter repeat length polymorphism (5-HTTLPR) in seasonality and seasonal affective disorder. Mol. Psychiatry 3, 175 (1998).
Enoch, M. et al. Association between seasonal affective disorder and the 5-HT2A promoter polymorphism,-1438G/A. Mol. Psychiatry 4, 89–92 (1999).
Molnar, E. et al. Seasonality and winter-type seasonal depression are associated with the rs731779 polymorphism of the serotonin-2A receptor gene. Eur. Neuropsychopharmacol. 20, 655–662 (2010).
Johansson, C. et al. Circadian clock-related polymorphisms in seasonal affective disorder and their relevance to diurnal preference. Neuropsychopharmacol 28, 734–9 (2003).
Partonen, T. et al. Three circadian clock genes Per2, Arntl, and Npas2 contribute to winter depression. Ann. Med. 39, 229–238 (2007).
Roecklein, K. A. et al. A missense variant (P10L) of the melanopsin (OPN4) gene in seasonal affective disorder. J. Affect Disord. 114, 279–285 (2009).
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Psychiatric GCBDWG. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011).
Wray N.R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Byrne, E. M. et al. Seasonality shows evidence for polygenic architecture and genetic correlation with schizophrenia and bipolar disorder. J. Clin. Psychiatry 76, 128–134 (2015).
Dick, D. M. et al. Genomewide linkage analyses of bipolar disorder: a new sample of 250 pedigrees from the National Institute of Mental Health Genetics Initiative. Am. J. Hum. Genet. 73, 107–114 (2003).
Shi, J. et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiatry 16, 193–201 (2010).
Sanders, A. R. et al. The Internet-based MGS2 control sample: self report of mental illness. Am. J. Psychiatry 167, 854–865 (2010).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Delaneau, O., Marchini, J. & Zagury, J. F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2011).
Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Gao, X., Starmer, J. & Martin, E. R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Gen. Epidemiol. 32, 361–369 (2008).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Nielsen, J. V., Thomassen, M., Mollgard, K., Noraberg, J. & Jensen, N. A. Zbtb20 defines a hippocampal neuronal identity through direct repression of genes that control projection neuron development in the isocortex. Cereb. Cortex 24, 1216–1229 (2014).
Nielsen, J. V., Blom, J. B., Noraberg, J. & Jensen, N. A. Zbtb20-induced CA1 pyramidal neuron development and area enlargement in the cerebral midline cortex of mice. Cereb. Cortex 20, 1904–1914 (2010).
Nielsen, J. V., Nielsen, F. H., Ismail, R., Noraberg, J. & Jensen, N. A. Hippocampus-like corticoneurogenesis induced by two isoforms of the BTB-zinc finger gene Zbtb20 in mice. Development 134, 1133–1140 (2007).
Qu, Z. et al. Loss of ZBTB20 impairs circadian output and leads to unimodal behavioral rhythms. eLife. 5, e17171 (2016).
Rasmussen, M. B. et al. Neurodevelopmental disorders associated with dosage imbalance of ZBTB20 correlate with the morbidity spectrum of ZBTB20 candidate target genes. J. Med. Genet. 51, 605–613 (2014).
Cordeddu, V. et al. Mutations in ZBTB20 cause Primrose syndrome. Nat. Genet. 46, 815–817 (2014).
Davies, M. N. et al. Hypermethylation in the ZBTB20 gene is associated with major depressive disorder. Genome Biol. 15, R56 (2014).
Ching, M. S. et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Neuropsychiatr. Genet. 153B, 937–947 (2010).
Hishimoto, A. et al. Neurexin 3 polymorphisms are associated with alcohol dependence and altered expression of specific isoforms. Hum. Mol. Genet. 16, 2880–2891 (2007).
Tsai, M. T. et al. Mutation analysis of synapsin III gene in schizophrenia. Neuropsychiatr. Genet. 114, 79–83 (2002).
Reissner, C., Runkel, F. & Missler, M. Neurexins. Genome Biol. 14, 213 (2013).
Tansey, K. E. et al. Copy number variants and therapeutic response to antidepressant medication in major depressive disorder. Pharm. J. 14, 395–399 (2014).
Fabbri, C. et al. New insights into the pharmacogenomics of antidepressant response from the GENDEP and STAR*D studies: rare variant analysis and high-density imputation. Pharm. J. 18, 413–421 (2018).
Higgs, B. W., Elashoff, M., Richman, S. & Barci, B. An online database for brain disease research. BMC Genom. 7, 70 (2006).
Shapiro-Reznik, M., Jilg, A., Lerner, H., Earnest, D. J. & Zisapel, N. Diurnal rhythms in neurexins transcripts and inhibitory/excitatory synapse scaffold proteins in the biological clock. PLoS ONE 7, e37894 (2012).
Alisch, R. S. et al. A multi-dimensional characterization of anxiety in monozygotic twin pairs reveals susceptibility loci in humans. Transl. Psychiatry 7, 1282 (2017).
Reick, M., Garcia, J. A., Dudley, C. & McKnight, S. L. NPAS2: an analog of clock operative in the mammalian forebrain. Science 293, 506–509 (2001).
DeBruyne, J. P., Weaver, D. R. & Reppert, S. M. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat. Neurosci. 10, 543–545 (2007).
Dudley, C. A. et al. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science 301, 379–383 (2003).
Mersch, P. P., Middendorp, H. M., Bouhuys, A. L., Beersma, D. G. & van den Hoofdakker, R. H. The prevalence of seasonal affective disorder in The Netherlands: a prospective and retrospective study of seasonal mood variation in the general population. Biol. Psychiatry 45, 1013–1022 (1999).
Rosen, L. N. et al. Prevalence of seasonal affective disorder at four latitudes. Psychiatry Res. 31, 131–144 (1990).
Acknowledgements
The authors wish to thank all of the participants in the study, as without them this work would not have been possible. The project’s funding sources include NIMH grants MH059552 (J.B.P.), MH061686 and MH64197 (D.F.L.), MH060912 (M.M.W.), MH089916 (D.F.L., M.M.W., J.B.P.) and NIAAA grant AA022994 (S.H.). Also supported through the Bipolar Disorder Research Fund at the University of Iowa. Data and biomaterials were collected in four projects that participated in the NIMH Bipolar Disorder Genetics Initiative. From 1991 to 1998, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, U01 MH46282, John Nurnberger, M.D., Ph.D., Marvin Miller, M.D., and Elizabeth Bowman, M.D.; Washington University, St. Louis, MO, U01 MH46280, Theodore Reich, M.D., Allison Goate, Ph.D., and John Rice, Ph.D.; Johns Hopkins University, Baltimore, MD U01 MH46274, J. Raymond DePaulo, Jr., M.D., Sylvia Simpson, M.D., M.P.H., and Colin Stine, Ph.D.; NIMH Intramural Research Program, Clinical Neurogenetics Branch, Bethesda, M.D., Elliot Gershon, M.D., Diane Kazuba, B.A., and Elizabeth Maxwell, M.S.W. Data and biomaterials were collected in ten projects that participated in the NIMH Bipolar Disorder Genetics Initiative. From 1999 to 2007, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, R01 MH59545, John Nurnberger, M.D., Ph.D., Marvin J. Miller, M.D., Elizabeth S. Bowman, M.D., N. Leela Rau, M.D., P. Ryan Moe, M.D., Nalini Samavedy, M.D., Rif El-Mallakh, M.D. (at University of Louisville), Husseini Manji, M.D. (at Wayne State University), Debra A. Glitz, M.D. (at Wayne State University), Eric T. Meyer, M.S., Carrie Smiley, R.N., Tatiana Foroud, Ph.D., Leah Flury, M.S., Danielle M. Dick, Ph.D., Howard J. Edenberg, Ph.D.; Washington University, St. Louis, MO, R01 MH059534, John Rice, Ph.D, Theodore Reich, M.D., Allison Goate, Ph.D., Laura Bierut, M.D.; Johns Hopkins University, Baltimore, MD, R01 MH59533, Melvin McInnis M.D., J. Raymond DePaulo, Jr., M.D., Dean F. MacKinnon, M.D., Francis M. Mondimore, M.D., James B. Potash, M.D., Peter P. Zandi, Ph.D., Dimitrios Avramopoulos, and Jennifer Payne; University of Pennsylvania, PA, R01 MH59553, Wade Berrettini M.D., Ph.D.; University of California at Irvine, CA, R01 MH60068, William Byerley M.D., and Mark Vawter M.D.; University of Iowa, IA, R01 MH059548, William Coryell M.D., and Raymond Crowe M.D.; University of Chicago, IL, R01 MH59535, Elliot Gershon M.D., Judith Badner Ph.D., Francis McMahon M.D., Chunyu Liu Ph.D., Alan Sanders M.D., Maria Caserta, Steven Dinwiddie M.D., Tu Nguyen, Donna Harakal; University of California at San Diego, CA, R01 MH59567, John Kelsoe, M.D., Rebecca McKinney, B.A.; Rush University, IL, R01 MH059556, William Scheftner M.D., Howard M. Kravitz, D.O., M.P.H., Diana Marta, B.S., Annette Vaughn-Brown, M.S.N., R.N., and Laurie Bederow, M.A.; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis J. McMahon, M.D., Layla Kassem, Psy.D., Sevilla Detera-Wadleigh, Ph.D., Lisa Austin, Ph.D., Dennis L. Murphy, M.D. Data and biomaterials from the NIMH GI (Genetics Initiative) MGS2 (Molecular Genetics of Schizophrenia) control sample were collected by NorthShore University HealthSystem, Evanston, IL, R01 MH59571, Pablo V. Gejman, M.D. (Collaboration Coordinator; PI) as part of a collaborative R01 application comprised of ten sites. From 2003 to 2006, the Principal Investigators and Co-Investigators were: NorthShore University HealthSystem, Evanston, IL, R01 MH59571, Pablo V. Gejman, M.D. (Collaboration Coordinator; PI), Alan R. Sanders, M.D.; Emory University School of Medicine, Atlanta, GA, R01 MH59587, Farooq Amin, M.D. (PI); University of California, San Francisco, CA, R01 MH60870, William F. Byerley, M.D. (PI); University of Iowa, Iowa, IA, R01 MH59566, Donald W. Black, M.D. (PI), Raymond R. Crowe, M.D.; Washington University, St. Louis, MO, R01 MH60879, C. Robert Cloninger, M.D. (PI); University of Colorado, Denver, CO, R01 MH59565, Robert Freedman, M.D. (PI), Ann Olincy, M.D.; Stanford University, Palo Alto, CA, R01 MH61675, Douglas F. Levinson, M.D. (PI); Louisiana State University, New Orleans, LA, R01 MH67257, Nancy G. Buccola, A.P.R.N., B.C., M.S.N. (PI); University of Queensland, Brisbane, Queensland, Australia, R01 MH59588, Bryan J. Mowry, M.D. (PI); Mt. Sinai School of Medicine, New York, NY, R01 MH59586, Jeremy M. Silverman, Ph.D. (PI).
Bipolar Genome Study (BiGS) co-authors
John R. Kelsoe, Tiffany A. Greenwood, Caroline M. Nievergelt, Rebecca McKinney, Paul D. Shilling, Erin N. Smith—University of California, San Diego, CA, USA; Nicholas J. Schork, Cinnamon S. Bloss—Scripps Translational Science Institute, La Jolla, CA, USA; John I. Nurnberger, Jr., Howard J. Edenberg, Tatiana Foroud, Daniel L. Koller—Indiana University, Indianapolis, IN, USA; Elliot S. Gershon, Chunyu Liu, Judith A. Badner—University of Chicago, Chicago, IL, USA; William A. Scheftner—Rush University Medical Center, Chicago, IL, USA; William B. Lawson, Evaristus A. Nwulia, Maria Hipolito—Howard University, Washington, D.C., USA; William Coryell, James B. Potash—University of Iowa, Iowa City, IA, USA; John Rice—Washington University, St. Louis, MO, USA; William Byerley—University of California, San Francisco, CA, USA; Francis J. McMahon, Thomas G. Schulze—National Institute of Mental Health Intramural Research Program, Bethesda, MD, USA; Wade H. Berrettini—University of Pennsylvania, Philadelphia, PA, USA; Peter P. Zandi, Pamela B. Mahon—Johns Hopkins School of Medicine, Baltimore, MD, USA; Melvin G. McInnis, Sebastian Zöllner, Peng Zhang—University of Michigan, Ann Arbor, MI, USA; David W. Craig, Szabolcs Szelinger—The Translational Genomics Research Institute, Phoenix, AZ, USA; Thomas B. Barrett—Portland Veterans Affairs Medical Center, Portland, OR, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ho, K.W.D., Han, S., Nielsen, J.V. et al. Genome-wide association study of seasonal affective disorder. Transl Psychiatry 8, 190 (2018). https://doi.org/10.1038/s41398-018-0246-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-018-0246-z
This article is cited by
-
Machine learning analyses reveal circadian clock features predictive of anxiety among UK biobank participants
Scientific Reports (2023)
-
Machine learning and expression analyses reveal circadian clock features predictive of anxiety
Scientific Reports (2022)
-
Genetic Intersections of Language and Neuropsychiatric Conditions
Current Psychiatry Reports (2020)

{kind=link}
{kind=link}
{kind=link}