
Abstract
Genome-wide association studies (GWAS) have identified more than 100 loci that show robust association with schizophrenia risk. However, due to the complexity of linkage disequilibrium and gene regulatory, it is challenging to pinpoint the causal genes at the risk loci and translate the genetic findings from GWAS into disease mechanism and clinical treatment. Here we systematically predicted the plausible candidate causal genes for schizophrenia at genome-wide level. We utilized different approaches and strategies to predict causal genes for schizophrenia, including Sherlock, SMR, DAPPLE, Prix Fixe, NetWAS, and DEPICT. By integrating the results from different prediction approaches, we identified six top candidates that represent promising causal genes for schizophrenia, including CNTN4, GATAD2A, GPM6A, MMP16, PSMA4, and TCF4. Besides, we also identified 35 additional high-confidence causal genes for schizophrenia. The identified causal genes showed distinct spatio-temporal expression patterns in developing and adult human brain. Cell-type-specific expression analysis indicated that the expression level of the predicted causal genes was significantly higher in neurons compared with oligodendrocytes and microglia (P < 0.05). We found that synaptic transmission-related genes were significantly enriched among the identified causal genes (P < 0.05), providing further support for the dysregulation of synaptic transmission in schizophrenia. Finally, we showed that the top six causal genes are dysregulated in schizophrenia cases compared with controls and knockdown of these genes impaired the proliferation of neuronal cells. Our study depicts the landscape of plausible schizophrenia causal genes for the first time. Further genetic and functional validation of these genes will provide mechanistic insights into schizophrenia pathogenesis and may facilitate to provide potential targets for future therapeutics and diagnostics.
Similar content being viewed by others

Introduction
Schizophrenia is a severe mental disorder with complex genetic architectures1. Recent studies have showed that different types of genetic variants (including common variants such as single nucleotide polymorphism, copy number variants, rare structural variants, de novo mutations and rare disruptive variants) are involved in the etiology of schizophrenia2,3,4,5,6,7,8,9,10. Though schizophrenia has a complicated genetic underpinnings, high heritability strongly suggests the pivotal role of inherited variants in genetic predisposition to schizophrenia11. To identify the inherited risk variants for schizophrenia, multiple genome-wide association studies (GWAS) have been performed in different continental populations and numerous risk variants (loci) have been uncovered2,12,13,14,15,16. In 2014, the schizophrenia working group of the psychiatric genomics consortium (PGC2 release) reported the largest GWAS of schizophrenia so far2. Despite the great success of schizophrenia GWAS and more than 100 independent risk loci have been identified, how to translate genetic findings into molecular risk mechanisms remains a major challenge.
Several key steps are needed to elucidate the genetic and pathophysiological mechanisms of schizophrenia. The first step is to identify the genetic risk variants (or loci). The second step is to pinpoint the potential causal gene (or genes) in the identified risk loci. And the third step is to investigate how the causal genes exert their effect on disease susceptibility. During the past decade, great efforts have been made and significant progress has been achieved in identification of risk variants for schizophrenia. To accelerate the discovery of novel risk variants, the PGC conducted large-scale genetic studies2 and over 100 schizophrenia risk loci have been reported. With the rapid increase of sample size, new risk variants (or loci) are being uncovered at an unprecedented rate and the landscape of the inherited genetic risk variants is emerging. Despite the fact that numerous risk loci have been reported, only very limited causal genes were identified in the reported risk loci. For most of the risk loci, the causal gene (or genes) that explains the association signal still remains largely unknown. In contrast to the rapid identification of risk variants, pinpointing the potential causal gene (genes) at the reported risk loci has lagged far behind. To translate the genetic risk loci into molecular risk mechanisms and to facilitate the development of new therapeutic targets, it is of great importance to identify the causal gene (s) that contributes to risk of schizophrenia in the reported risk loci.
Despite its importance in illuminating the genetic and pathogenic mechanisms of schizophrenia, localizing the causal gene remains a major challenge in human genetics. First, the risk loci identified by GWAS usually contain many highly linked genetic variants that usually span large genomic regions (sometimes up to several megabase). For example, among the reported risk loci, the MHC region showed the most significant association with schizophrenia. However, this region encompasses numerous highly linked variants spanning multiple genes (over 100). Due to the complex linkage disequilibrium (LD) between the risk variants, it is difficult to pinpoint the plausible causal gene. Second, in many cases, due to the complexity of gene regulatory17,18,19,20,21, the gene nearest the top associated variants is not necessarily the actual causal gene. Accumulating evidence shows that the risk variants may regulate distal gene expression through long-range chromosomal interactions19,20. These complicated linkage disequilibrium and gene regulatory impede the identification of causal gene in the reported risk loci. In this study, we systematically predicated the plausible candidate causal genes for schizophrenia through comprehensive integrative analyses, including integration of genetic associations from schizophrenia GWAS2 and brain expression quantitative trait (eQTL) data22 and using network-based prioritization approaches (based on brain-specific networks and functionally coherent subnetworks, i.e., shared-function or co-function networks). On the basis of genetic associations from GWAS of schizophrenia, we generated the first landscape of plausible causal genes for schizophrenia. We further showed that the causal genes are highly expressed in neurons and are enriched in synaptic transmission-related pathways. Finally, we found that knockdown of the top six causal genes suppressed the proliferation of neuronal cells. This landscape of plausible causal genes provides a start point to elucidate the genetic and pathophysiological mechanisms underlying schizophrenia.
Materials and methods
GWAS of schizophrenia
Recently, PCG reported the largest GWAS of schizophrenia so far (PGC2 release)2. In the first phase, genome-wide genotypes of 35,476 schizophrenia cases and 46,839 controls were obtained and meta-analyzed. The SNPs with P value smaller than 10−6 were replicated in additional samples. Through combining the results from the first and second phase, more than 100 risk loci were identified (P < 5 × 10−8) and most of them were newly reported. Genome-wide SNPs associations from the first phase (i.e., 35,476 schizophrenia cases and 46,839 controls) of PGC were used in this study. More detailed information about PGC, including sample recruitment and diagnosis, genotyping, quality control, and statistical analysis can be found in the original paper2.
Brain eQTL data
We used the brain eQTL data reported by Myers et al.22 in this study. Briefly, human brain tissues (cortex) of 193 normal human subjects were obtained. All of the individuals were of European of ancestry and had no clinical history of neurologic and neuropsychiatric diseases. DNA and RNA were extracted using standard procedures. Genotyping was performed using Affymetrix GeneChip (Human Mapping 500K Array Set) and gene expression was measured using Illumina HumanRefseq-8 Expression BeadChip. PLINK was used to test the association between the genotyped genetic variants and gene expression using linear regression. More details about sample description, genotyping, expression quantification and statistical analyses can be found in the original paper of Myers et al.22.
Integration of schizophrenia GWAS and brain eQTL data (Sherlock)
Most of the identified schizophrenia risk variants are located in non-coding region, suggesting these variants may exert their effects through regulating gene expression. To infer genes whose expression alteration may contribute to disease risk, He et al. developed a Bayesian statistical method (named Sherlock)23 to identify potential causal genes through combining genetic associations from GWAS and eQTL data. For a given gene, there may be several genetic variants (usually SNPs) that act synergistically to regulate the expression level of this gene (we called these expression-associated SNPs eSNPs). If a gene is not associated with disease, the eSNPs of this gene may not be associated with disease risk. However, if it is a causal gene, genetic variations at these eSNPs may alter its expression level, which may in turn influence disease susceptibility. Thus, the eSNPs of this gene may also be associated with disease as well. Significant overlap between the eQTL of a specific gene and the loci associated with the disease suggests this gene may have a role in disease pathogenesis. To predict the potential causal genes for schizophrenia, we systematically integrated genome-wide SNP associations from PGC22 and brain eQTL from Myers et al.22 using Sherlock statistical framework23. More detailed information about Sherlock statistical inference can be found in the paper of He et al.23.
Integration of schizophrenia GWAS and brain eQTL data (SMR)
In addition to Sherlock, we also used summary data-based Mendelian randomization (SMR)24 developed by Zhu et al24 to predict causal genes for schizophrenia. Similar to Sherlock, SMR predicts causal genes by integrating of summary data from GWAS and eQTL. However, the statistical inference of SMR is different from Sherlock. In this study, we used SMR to infer causal genes through integrating the genome-wide associations from PGC22 and brain eQTL from Myers et al.22. By default, SMR only includes probes with at least one cis-eQTL that has a P < 5 × 10−8. However, due to the relative small sample size used in brain eQTL study, we also performed SMR analysis using lower transcript inclusion threshold (PeQTL < 1 × 10−5). Genes passed SMR and HEIDI tests were inferred as plausible causal genes. More details about SMR method (including statistical inference, distinguishing pleiotropy from linkage and pinpointing functionally relevant genes) can be found in the original paper24.
Predicting causal genes using functionally coherent subnetworks (Prix Fixe)
In contrast to Sherlock and SMR, Prix Fixe uses a different strategy to infer causal genes25. Prix Fixe utilizes shared-function or co-function networks to prioritize candidate causal genes. The prioritization procedure includes several steps. First, disease-associated SNPs (index or lead SNPs) were used to define linkage-disequilibrium windows. SNPs linked with the index SNP (r2 > 0.5) were identified and nearby genes were extracted. Second, Prix Fixe constructs a comprehensive human co-function network through extracting functional relationships between human genes. Third, Prix Fixe identifies the mutually connected (densely interacted) subnetworks using the co-function network. Finally, Prix Fixe prioritizes the candidate genes based on their importance in the subnetworks and a Prix Fixe score (PF score) was obtained for each candidate gene. Prix Fixe evaluates the importance of each gene in the defined LD windows, using a specific parameter, i.e., edge density, the number of edges in the subnetwork. If a gene has no functional interactions (edge connections) with other genes in the subnetworks, the presence or absence of this gene will not change the edge density. However, if a gene has functional relationships with other genes in the subnetwork, the presence or absence of this will affect the edge density. In this study, the top 100 index SNPs from PGC22 were used as input for Prix Fixe. (as Prix Fixe only can accept a maximum of 100 SNPs as input).
Identifying causal genes using brain-specific functional interaction network (NetWAS)
Greene et al. recently generated a genome-scale functional interaction networks for many human tissues (including brain)26. These tissue-specific networks can be used to prioritize potential causal genes27. Briefly, SNP-level association statistics were converted into gene-level statistics (i.e., gene-based P values), which were then integrated with tissue-specific networks to predict the potential causal genes. In this study, we first converted SNP-based summary statistics (SNP P values from PGC2) into gene-based P values using Pascal28. The gene-level P values were then used as input for NetWAS26, and brain-specific functional interaction networks were selected. More detailed information about tissue-specific networks and NetWAS can be found in the original paper26.
Prioritizing causal genes using predicted gene functions (DEPICT)
To identify the genes and pathways that can explain the genome-wide associations, Per et al. developed an integrative tool (DEPICT) to prioritize the most likely causal genes through using predicted gene functions29. Briefly, DEPICT first predicts gene function by using co-regulation of gene expression and previous annotated gene sets. DEPICT then generates a “reconstituted” gene sets, which contain the likelihood of membership of each gene in the reconstituted gene sets. Finally, through using predicted gene function and the statistical significant loci identified by PGC2, DEPICT prioritizes causal genes for schizophrenia. More details about DEPICT can be found in the paper of Pers et al. As Pers et al.30 have predicted the potential causal for schizophrenia using DEPICT, we included their results (i.e., prioritized causal genes) into our study.
Predicting causal genes using protein–protein interaction network (DAPPLE)
Previous studies have shown that proteins encoded by disease-associated genes tend to physically interacted than random expectations31,32, a phenomenon called “guilt by association”33,34. Based on the “guilt by association”, protein interactions can be used to prioritize disease-associated genes35,36,37,38. In this study, we used Disease Association Protein–Protein Link Evaluator (DAPPLE)36 to prioritize the plausible causal genes at the reported loci by using protein–protein interaction data.
Ranking of the prioritized causal genes
We used different approaches to predict the causal genes. A gene may represent a promising candidate if it is predicted by different predicting methods. A cumulative scoring strategy was hence used to rank the casual genes. Briefly, each prioritization approach contributes one point to the total score of the prioritized causal genes. For example, if a gene is only prioritized by one method, the total score of this gene is one point. If a gene is identified by two methods, the total score of this gene is two points. The total score of each gene was calculated and ranked. A higher total score indicates a higher probability that the prioritized gene is causal.
Gene ontology analysis
To investigate whether the predicted causal genes were enriched for specific functional categories, we performed Gene Ontology (GO) analysis using DAVID39,40. Three GO terms, including biological process (BP), cellular component (CC), and molecular function (MF) were used to test whether the prioritized genes are significantly enriched in specific biological processes or pathways. The significance (P values) of the overrepresented GO terms was corrected by the Benjamini–Hochberg procedure.
Spatio-temporal expression pattern analysis of plausible causal genes
RNA sequencing-based expression data from the Brainspan41: Atlas of the developing human brain were used to plot the expression trajectory of the prioritized causal genes. Detailed information about sample collection, quality control, RNA extraction, and quantification can be found in the Brainspan website (http://www.brainspan.org/). We processed the data and plotted the gene expression as previously described8.
Cell-type-specific expression analysis of plausible causal genes
We examined the expression of the prioritized causal genes in different cell types using data from Zhang et al.42. Briefly, different cell types of human brain (including fetal astrocytes, mature astrocytes, neurons, oligodendrocytes and microglia/macrophage cells) were isolated and gene expression was measured by RNA sequencing. The RNA sequencing-based expression values (FPKM, fragments per kilobase of exon per million fragments mapped) were downloaded and processed. As described previously43, the expression level of each gene was expressed as log2(FPKM + 1). In addition, we also examined the expression of the predicted causal genes in human embryonic stem cells (ESCs, line H9) and NSCs using the expression data from Lafaille et al.44. More detailed information can be found in the original papers42,44 and supplementary material. Analysis of variance (ANOVA) was used to test whether the expression level of the prioritized genes was significantly different among different cell types. Test of homogeneity of variances showed lack of homogeneity (P < 0.05). We thus used Dunnett C test45,46 (implemented in SPSS) to compare if the mean expression level of the predicted causal genes was significantly different among different cell types.
Analysis of protein–protein interaction and co-function network
To investigate the physical interaction among the proteins encoded by the predicted causal genes, we extracted the protein–protein interaction (PPI) data from GeneMANIA (http://genemania.org/)47, a well-characterized PPI database that contains high-confidence interaction data. We also explored the functional relationships between the prioritized causal genes using functional-association network (FAN) from study of Tasan et al.25.
Expression analysis of the top causal genes in schizophrenia cases and controls
The expression level of the top causal genes in schizophrenia cases and healthy controls was compared using expression data (GSE53987 and GSE21138) from gene expression omnibus (GEO). GSE53987 contains 15 subjects with schizophrenia and 19 healthy controls. Three brain regions (hippocampus, prefrontal cortex, and straitum) were included in GSE53987. RNA was isolated and gene expression level was quantified using Affymetrix array chips (U133_Plus2). GSE21138 includes 30 schizophrenia cases and 29 healthy controls. Brain tissues from the prefrontal cortex (Brodmann Area 46) were isolated, and gene expression was measured using Human Genome U133 Plus 2.0 array. The raw data of each study were downloaded from GEO, and Bioconductor (‘affy’ package) was used to process the data. We used RMA algorithm48 to normalize the expression data. Expression difference between cases and controls were tested using Student’s t test. More details about RNA isolation, quantification, quality control, and statistical analysis can be found in the original paper49.
Knockdown of top causal genes in SH-SY5Y cell line
Human neuroblastoma SH-SY5Y cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)-F12 (1:1) containing 10% fetal bovine serum (FBS). Short-hairpin RNA (shRNA) sequences targeting to CNTN4, GATAD2A, GPM6A, MMP16, PSMA4, and TCF4 were designed using BLOCK-iT™ RNAi Designer (https://rnaidesigner.thermofisher.com/). Sense and anti-sense oligonucleotides were synthesized, annealed and cloned into the PLKO.1 vector at the AgeI and EcoRI restriction sites. Knockdown efficiency was determined by quantitative PCR (qPCR). The shRNA sequences and primers used for qPCR are listed in Supplementary Material and Supplementary Table S1-S2.
Proliferation assays
SH-SY5Y cells were plated at a density of 1 × 104 cells per well in 96-well plates. After 24 h, cells were transfected with 0.2 μg shRNA constructs using lipofectamine 3000 (Invitrogen). Cell counting kit-8 (CCK8) (Sigma) was used to quantify the cell number, as previously described50. After 72 h transfection, 10 μL CCK8 solutions were added into each well, incubated for 6 h. Then cell amounts through measuring the absorbance at 450 nm using a micro-plate reader.
Results
Causal genes identified by Sherlock
We integrated the SNP associations from schizophrenia GWAS (PGC2)2 and brain eQTL data22 using Sherlock23 and identified 15 potential causal genes whose expression level change may contribute to schizophrenia risk (Bonferroni corrected P < 0.05) (Supplementary Table S3). Of note, Sherlock uses collective information from both cis (located within 1 Mb of the transcription start site of a gene) and trans variants to make statistical inference. Comparing with cis variants, the regulatory effects of trans variants are relatively difficult to investigate. We thus focused on genes supported by cis variants in this study. Among the 15 significant genes, five candidate causal genes were prioritized mainly by cis genetic variants (i.e., cis variants of these genes showed significant association with schizophrenia and gene expression simultaneously), including ALMS1, GLT8D1, ZNF323, CSNK2B, and TBC1D15. These five genes therefore represent the most likely causal genes for schizophrenia.
Causal genes identified by SMR
Through integrating SNP associations from schizophrenia GWAS (PGC2)2 and brain eQTL data22, SMR identified two genes (SULT2B1 and ALMS1) at P < 1.0 × 10−3 (Supplementary Table S4). However, as SULT2B1 did not pass HEIDI test (P < 0.05), only ALMS1 was retained. Intriguingly, Sherlock analysis also suggested that ALMS1 is a causal gene for schizophrenia (Supplementary Table S3). These consistent results strongly suggest that ALMS1 may represent a promising causal gene for schizophrenia.
Causal genes identified by Prix Fixe
We predicted schizophrenia causal genes using Prix Fixe, which utilizes functionally coherent subnetworks to prioritize causal genes. In total, 119 genes (PF score > 0) were prioritized by Prix Fixe. As our goal is to identify the most possible causal genes, we only retained genes with PF score > 0.10. We found that 41 genes have a PF score > 0.10 (Supplementary Table S5). Of note, DRD2 ranked the highest among these genes (Supplementary Table S5). Genes ranked from the second to tenth are as follows: CACNA1C, CACNB2, GRIN2A, CNKSR2, SERPING1, ZNF536, GPM6A, VRK2, and GRIA1. Dysregulation of the dopamine system in the pathophysiology of schizophrenia has been well characterized. In fact, most of antipsychotic drugs exert their effect through blocking dopamine receptors. Thus, the highest PF score of DRD2 strongly suggests the genes prioritized by Prix Fixe may represent promising causal genes for schizophrenia. GO analysis showed that the prioritized causal genes were enriched in synaptic function, neuronal projection, acetylcholine-gated channel complex, and neuronal calcium signaling related categories (Supplementary Table S6).
Causal genes prioritized by NetWAS
Through integrating gene-wide associations2 (SNP-level associations were converted into gene-level associations28) and brain-specific functional interaction network26, we performed genome-wide prediction of causal genes using NetWAS. As our goal is to prioritize the most likely causal genes, top 50 genes were included in this study as promising candidates (Supplementary Table S7). GO analysis showed that neural development and neural projection categories were enriched in the prioritized causal genes (Supplementary Figure S1).
Causal genes prioritized by DAPPLE
Through using DAPPLE, which utilizes PPI to prioritize potential causal genes at the reported risk loci, we identified 83 candidate causal genes (corrected P < 0.01) (Supplementary Table S8). The top prioritized genes include DUS2L, ATXN7, SETD8, CHRNB4, CTNNA1, PCDHA5, KDM4A, TSSK6, EP300, ACD, PCDHA1, PLAA, GATAD2A and PCDHA2. As genes located within the genome-wide significant loci were used as input, in essence, we distilled the promising causal genes at these risk loci. Gene ontology (GO) analysis showed that the prioritized causal genes were enriched in nervous system development (corrected P = 9.9 × 10−3), cell–cell adhesion (corrected P = 2.2 × 10−3) and chromatin organization (corrected P = 4.08 × 10−2) categories (Supplementary Figure S2).
The integrated landscape of causal genes in schizophrenia
We utilized different approaches (including Sherlock, SMR, Prix Fixe, NetWAS, DEPICT and DAPPLE) to prioritize the plausible causal genes for schizophrenia. The overlapping genes represent promising plausible causal genes as they were supported by different methods. We therefore ranked the prioritized causal genes through integrating the results from different prioritization approaches. To obtain the global landscape of plausible causal genes, we also integrated candidate causal genes identified in previous studies. Candidate causal genes from other studies are as follows: (1) causal genes prioritized by Pavlides et al.51. By integrating blood eQTL data52 and GWAS data of schizophrenia (PGC2)2, Pavlides et al. prioritized potential causal genes using SMR24. A total of 17 genes (corrected P < 0.05) were included (Supplementary Table S9). (2) Causal genes predicted by Zhu et al.24. Through integrating brain eQTL data (The Brain eQTL Almanac (Braineac): http://www.braineac.org/) (brain tissues from a total of 134 individuals)53 and GWAS data of schizophrenia (PGC2)2, Zhu et al. used SMR to prioritize causal genes and identified two candidates, SNX19 and NMRAL1. (3) Causal genes identified by Fromer et al.54. Recently, Fromer et al. conducted a large-scale RNA sequencing using brain tissues from schizophrenia cases and normal controls. They generated a comprehensive eQTL resource (from a total of 467 subjects of European ancestry) and prioritized candidate causal genes through integrating eQTL resource and GWAS data of schizophrenia2 (using Sherlock). In total, 33 genes (corrected P < 0.05) were identified (Supplementary Table S10). (4) Causal genes prioritized by DEPICT. Through using DEPICT, Pers et al. predicted the potential causal genes for schizophrenia recently. They predicted a total of 62 plausible candidate causal genes for schizophrenia (FDR < 0.05) (Supplementary Table S11).
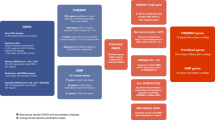
On the basis of their frequency of occurrences in the results of different prioritization approaches, we ranked the prioritized causal genes using a cumulative scoring strategy (Materials and methods) and generated the integrated landscape of causal genes in schizophrenia (Fig. 1). A total of 41 promising causal genes (total score > 2 points) were identified through systematically integrating the prediction results from different methods. Six genes (including CNTN4, GATAD2A, GPM6A, MMP16, PSMA4, and TCF4) have the highest total scores (i.e., 3 points), indicating that at least three different prediction approaches support these genes as plausible causal genes. These six genes thus represent the most promising causal genes for schizophrenia. We therefore called these six genes tier 1 causal genes. In addition, thirty-five genes have a total score of 2 points (these genes were called as tier 2 causal genes). We called these 41 genes high-confidence plausible causal genes as these genes were supported by at least two different prioritization methods.

Through integrating the prediction results from different methods, a total of 41 high-confidence causal genes were identified. CNTN4, GATAD2A, GPM6A, MMP16, PSMA4, and TCF4 have the highest scores, these genes therefore represent the most promising causal genes for schizophrenia
We performed GO analysis and found that “synaptic transmission” and “ion transport” categories are significantly enriched in the 41 causal genes (Fig. 2a), suggesting that synaptic dysfunction may play a key role in schizophrenia pathogenesis. “Transporter complex”, “neuron projection”, and “postsynapse” categories were significantly enriched among the causal genes when cell component (CC) was used as keyword (Fig. 2b). And “ion channel activity” category was highly significantly overrepresented among the causal genes when molecular function (MF) was used as keyword (Fig. 2b). Taken together, these results suggest that synaptic dysfunction has a critical role in the pathophysiology of schizophrenia.

Results from different categories were showed, including biological process (a), cell component (b), and molecular function (c)
Causal genes showed distinct expression pattern in developing human brain
We explored the expression of the 41 high-confidence plausible causal genes in developing human brain and found distinct expression patterns in the prefrontal cortex (Fig. 3). We divided these 41 genes into four classes based on their expression trajectory. Expression levels of genes (e.g., BCL11B and CHRNA5) in the first class (marked by red) are high at early fetal stage, decline at late fetal stage, and then maintain a relative stable level from early infancy to adulthood. In the second class (marked by blue), expression levels of genes gradually increase from early fetal stage to adulthood (e.g., CACNB2 and MAPK3). In the third class (marked by pink), expression levels of genes (e.g., CHRNB4 and PCGH8) gradually increase from early fetal stage to early mid-fetal stage, peaks at late fetal stage, and then decline gradually. Expression levels of genes in the fourth class are relatively stable across entire developing stages (e.g., ZDHHC5). These expression patterns suggest that these genes may have different roles at different developmental stages.

Top causal genes were classified into four categories according to their expression pattern. Genes in the first category (marked by red) are highly expressed at early fetal stage, decline at late fetal stage, then maintain a relative stable level from early infancy to adulthood. Expression level of genes in the second category (marked by blue) gradually increases from early fetal stage to adulthood (e.g., CACNB2 and MAPK3). Expression level of genes in category three (marked by pink) gradually increases from early fetal stage to early mid-fetal stage and peaks at late fetal stage. Expression level of genes in category four (marked by black) is relatively stable at different developing stages
Causal genes are widely expressed in different cell types of CNS
We analyzed the expression of the prioritized causal genes in different cell types of human brain and found that the 41 prioritized causal genes were highly expressed in fetal astrocytes and neurons (Fig. 4a). The average expression level of the prioritized causal genes was higher in neurons compared with other examined cell types (Fig. 4b), suggesting that these genes are mainly involved in neuronal function. Statistical analysis showed that the average expression level of the predicted causal genes was significantly higher in neurons compared with oligodendrocytes and microglia (P < 0.05). In addition, we found that the average expression level of the prioritized causal genes was significantly higher in fetal astrocytes compared with microglia (P < 0.05) (Fig. 4b). We also explored the expression of the prioritized causal genes in human ESCs and NSCs derived from ESCs. Again, we found that the 41 high-confidence causal genes were widely expressed in human ESCs and NSCs (Supplementary Figure S3). Of note, compared with tier 2 causal genes, expression level of tier 1 causal genes (i.e., the six genes with the highest total score) are higher in ESCs and NSCs compared with tier 2 causal genes (Supplementary Figure S3). Collectively, these expression data showed that the prioritized causal genes are abundantly expressed in NSCs, neurons and fetal astrocytes, suggesting that these genes may have pivotal roles in CNS.

a Heatmap showed that the top causal genes are highly expressed in neurons and fetal astrocytes. b Expression level of top causal genes in neurons is significantly higher than other cell types. c Top causal genes encode a densely interconnected PPI network. d Top causal genes form a densely interconnected functional network. *P < 0.05, one-way ANOVA
Causal genes encode a densely interconnected molecular network
Genes usually act synergistically to exert their biological function in cells, and numerous studies have shown that physically interacted genes are more likely to share function, a phenomenon called guilt-by-association34,55,56. We performed network analysis and found that the prioritized causal genes encode a highly interconnected PPI network (Fig. 4c), suggesting these genes may act synergistically in human brain. Using the shared-function (or co-function) network from Tasan et al.25, we further found that the prioritized causal genes form a densely interconnected functional network (Fig. 4d). These results suggest that the prioritized causal genes are likely to share biological function. Dysregulation of any member in this molecular network may lead to similar functional consequences (i.e., increase schizophrenia risk).
Dysregulation of top causal genes in schizophrenia
We identified six top causal genes (CNTN4, GATAD2A, GPM6A, MMP16, PSMA4, and TCF4) through integrating the prediction results from different methods. To further validate the role of these top causal genes in schizophrenia, we compared the expression level of these genes in schizophrenia cases and healthy controls. We found that GATAD2A and TCF4 were significantly upregulated in the hippocampus of schizophrenia cases compared with controls in GSE53987 dataset (P < 0.01) (Fig. 5a). In contrast, PSMA4 was significantly downregulated and CNTN4 showed a trend of downregulation (P = 0.065) in the hippocampus of schizophrenia cases compared with controls (Fig. 5a). In the prefrontal cortex, CNTN4 was significantly downregulated in schizophrenia cases in both GSE53987 (P < 0.01) and GSE21138 (P < 0.01) datasets (Fig. 5b, c). Consistent with the downregulation in the hippocampus, PSMA4 was significantly downregulated in the prefrontal cortex in schizophrenia cases in GSE21138 dataset (P < 0.01) (Fig. 5c). Of note, though it did not reach significance level, TCF4 showed a trend of upregulation in the prefrontal cortex in both GSE53987 and GSE21138 datasets (Fig. 5b, c). Taken together, these results indicate that the top causal genes were dysregulated in schizophrenia cases, supporting that these genes may represent authentic causal genes for schizophrenia.

a Top causal genes were dysregulated in the hippocampus of schizophrenia cases compared with controls. GATAD2A and TCF4 were significantly upregulated in schizophrenia cases, while PSMA4 was significantly downregulated (P < 0.05). Data from GSE53987, which contains 15 schizophrenia cases and 19 controls. bCNTN4 was significantly downregulated in the prefrontal cortex of schizophrenia cases. GATAD2A, PSMA4, and TCF4 also showed a trend of dysregulation (P < 0.10). Data from GSE53987. (c) CNTN4 and PSMA4 were significantly downregulated in the prefrontal cortex of schizophrenia cases. GPM6A and TCF4 also showed a trend of dysregulation (P < 0.10). Data from GSE21138, which contains 30 schizophrenia cases and 29 controls. d Knockdown of CNTN4, GATAD2A, MMP16, PSMA4, and TCF4 impaired proliferation of SH-SY5Y cells
Knockdown of the top causal genes impaired the proliferation of SH-SY5Y cells
Expression analysis showed that the top causal genes were dysregulated in schizophrenia cases (Fig. 5). To explore whether the dysregulation of these top causal genes affects cell proliferation, we transiently knocked down these genes in SH-SY5Y neuroblastoma cells. Reverse transcription PCR (RT-PCR) showed that CNTN4, GATAD2A, MMP16, PSMA4, and TCF4 were expressed in SH-SY5Y cells (Supplementary Figure S4). However, the expression of GMP6A was not detected. We thus knocked down the expression of CNTN4, GATAD2A, MMP16, PSMA4, and TCF4 using shRNAs and conducted proliferation assays. We found that the shRNAs significantly downregulated the expression of CNTN4, GATAD2A, MMP16, PSMA4, and TCF4 genes (Supplementary Figure S5). Interestingly, knockdown of these top causal genes significantly impaired the proliferation of SH-SY5Y cells (Fig. 5d). Of note, previous study also showed that knockdown of TCF4 attenuated the proliferation of cortical progenitor cells. Collectively, these results indicate that dysregulation of top causal genes affects proliferation of neuronal cells, suggesting these genes may play a role in neurodevelopment.
Discussion
Recent GWAS have identified numerous schizophrenia risk loci. However, it is difficult to pinpoint the causal gene as each risk locus usually contains multiple highly linked genetic variants. In addition, gene regulatory is complex. For example, recent studies have showed that genetic variant may regulate the activity of distal gene through long-range chromatin interactions20,57. Thus, in many cases, the gene (s) nearest to the most significant genetic variant (identified by GWAS) may not represent the authentic causal gene (s). Compared with the rapid discovery of schizophrenia risk variants and loci, the identification of causal genes lags far behind. In this study, we systematically predicted the causal genes for schizophrenia by utilizing several well-characterized methods. Through integrating the results from different approaches, we prioritized 41 high-confidence causal genes for schizophrenia. Of note, a recent study also identified GATAD2A, PSMA4, FURIN, and OGFOD2 as schizophrenia risk genes through integrating genetic associations from GWAS and eQTL data from diverse tissues58, further supporting the notation that these genes may have a role in schizophrenia.
Among the 41 prioritized causal genes, CNTN4, GATAD2A, GPM6A, MMP16, PSMA4, and TCF4 represent the most promising causal genes for schizophrenia. CNTN4 encodes contactin-4 protein, a member of the immunoglobulin superfamily. As a neuronal adhesion molecule, contactin-4 plays a role in developing nervous system through influencing axon guidance and fasciculation59,60. In addition to PGC2, previous studies have showed that CNTN4 is associated with schizophrenia61 and disruption of CNTN4 caused developmental delay62. A recent study also showed that dysregulation of CNTN4 impaired proliferation of neural progenitor cells54. Interestingly, several groups reported that CNTN4 is associated with autism63,64, a neurodevelopmental disorder. These results strongly suggest that CNTN4 may play a role in schizophrenia through influencing neurodevelopment. The function of GATAD2A remains largely unknown. However, recent studies showed that GATAD2A is associated with diabetes65 and several types of cancer (breast, ovarian, and prostate cancer)66. Consistent with our observation, Wang et al showed that knockdown of GATAD2A suppressed cell proliferation in thyroid cancer67 and Marino showed that loss of GATAD2A function in mice is embryonic lethal. These results suggest that GATAD2A plays a crucial role in development. Glycoprotein M6A (GPM6A) encodes a transmembrane protein that is abundantly expressed in cell surface of neurons in the CNS68. Multiple studies have showed that GPM6A plays a pivotal role in neurodevelopment through regulating neuronal migration, differentiation, neurite outgrowth, spine formation, and synaptogenesis69,70,71,72. Intriguingly, a recent study showed that altered GPM6A dosage impairs cognition73, a phenotype that is frequently reported to be impaired in schizophrenia. These results strongly suggest that GPM6A may play a role in schizophrenia by affecting brain development. MMP16 encodes matrix mentalloproteinase-16, an enzyme that is response for breakdown of extracellular matrix74. Previous studies have shown that MMPs are involved in invasion75 and migration of cancer cells76,77. PSMA4 encodes proteasome subunit alpha type-478, a member of the 20S proteasome complex. GWAS showed that PSMA4 is associated with lung cancer79 and smoking80. Of note, Han et al. showed that PSMA4 protein is interacted with DTNBP1, a protein encoded by a promising schizophrenia candidate gene81. TCF4 encodes transcription factor 4, a pivotal transcription factor that plays critical role in development. TCF4 is one of the most frequently reported schizophrenia risk genes. Numerous GWASs repeatedly reported the association of TCF4 with schizophrenia2,15,82, strongly suggesting that TCF4 is a causal gene for schizophrenia. Accumulating evidence supports that TCF4 plays pivotal role in neurodevelopment through regulating the columnar distribution of lay2/3 prefrontal pyramidal neurons83, synaptic plasticity and memory function84. Recent studies also showed that TCF4 is associated with cognitive functions in mouse and humans85,86. These lines of evidence suggest that TCF4 may have a crucial role in schizophrenia pathogenesis by modulating neurodevelopment.
The top 41 predicted causal genes are highly expressed in neurons (Fig. 4a, b) and are enriched in synaptic transmission-related pathways (Fig. 2), suggesting these predicted causal genes exert their main functions in the central nervous system (CNS). However, we noticed that several genes (including ATP2A2, PSMA4, PBRM1, SERPING1, and VRK2) were also highly expressed in microglia (Fig. 4a), the resident macrophage cells that act as the first and main form of active immune defense in the CNS87. The high expression level of these genes in microglia implies that these genes may also have a role in neuro-immunity through regulating the function of microglia. Nevertheless, more work is needed to elucidate the role of these genes in neuro-immunity.
There are several limitations of this study. First, only genes supported by at least two different prioritization methods were selected in this study. Though these genes are promising causal genes for schizophrenia, genes supported by individual prediction approach may also have a role in schizophrenia. Second, the prioritization methods used data from PGC2 as a primary source to predict the causal genes for schizophrenia, validation of these genes in independent schizophrenia samples will provide direct support for the involvement of these gene in schizophrenia. Third, though this study identified promising causal genes, further biological experiments are needed to demonstrate the role of the prioritized genes in schizophrenia. Fourth, the number of eQTL datasets used in this study is relatively limited as only three brain eQTL datasets and one blood eQTL dataset22,52,53,54 were included in this study. Accordingly, the number of potential causal genes identified from the four eQTL datasets might be relatively small. Of note, Hauberg et al. integrated large-scale GWAS and multiple eQTL datasets recently and they identified numerous disease-associated genes58. Interestingly, Hauberg et al. showed that eQTLs derived from pathophysiologically relevant tissues play a pivotal role in the identification of disease-associated risk genes. As schizophrenia is a mental disease that mainly originates from abnormal brain function, brain eQTL is more suitable than eQTLs from other tissues. Thus, risk genes identified using brain eQTLs may represent promising candidate genes for schizophrenia. More importantly, as we used lines of convergent evidence to predict the causal genes for schizophrenia, we utilized other prediction methods (such as Prix Fixe and DEPICT) and provided further evidence that support the risk genes identified by integrative analysis (i.e., integrating GWAS signals and eQTLs).
We generated the landscape of causal genes in schizophrenia for the first time. In essence, we distilled the findings of GWAS of schizophrenia. Thus, the identified genes represent the most promising causal genes for schizophrenia. In fact, all of the top six causal genes showed significant association with schizophrenia at genome-wide level (Supplementary Figure S6-S11). GO analysis further showed that identified causal genes were enriched in synaptic signaling pathway, further supporting the notion that synaptic dysregulation may have a key role in schizophrenia. Of note, five (CNTN4, GATAD2A, GPM6A, MMP16, and TCF4) of the top six causal genes are involved in neurodevelopment, further supporting the neurodevelopmental hypothesis of schizophrenia.
References
Sullivan, P. F., Daly, M. J. & O’Donovan, M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet 13, 537–551 (2012).
Schizophrenia Working Group of the Psychiatric Genomics Consortium*. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Xu, B. et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 40, 880–885 (2008).
Vacic, V. et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 471, 499–503 (2011).
Stefansson, H. et al. Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236 (2008).
The International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241 (2008).
Walsh, T. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543 (2008).
Gulsuner, S. et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014).
Purcell, S. M. et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014).
Sullivan, P. F., Kendler, K. S. & Neale, M. C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192 (2003).
Shi, Y. et al. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat. Genet. 43, 1224–1227 (2011).
Yue, W. H. et al. Genome-wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11.2. Nat. Genet. 43, 1228–1231 (2011).
Purcell, S. M. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009).
Stefansson, H. et al. Common variants conferring risk of schizophrenia. Nature 460, 744–747 (2009).
O’Donovan, M. C. et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 40, 1053–1055 (2008).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Miele, A. & Dekker, J. Long-range chromosomal interactions and gene regulation. Mol. Biosyst. 4, 1046–1057 (2008).
Roussos, P. et al. A role for noncoding variation in schizophrenia. Cell Rep. 9, 1417–1429 (2014).
Won, H. et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016).
Sanyal, A., Lajoie, B. R., Jain, G. & Dekker, J. The long-range interaction landscape of gene promoters. Nature 489, 109–113 (2012).
Myers, A. J. et al. A survey of genetic human cortical gene expression. Nat. Genet. 39, 1494–1499 (2007).
He, X. et al. Sherlock: detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am. J. Hum. Genet. 92, 667–680 (2013).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Tasan, M. et al. Selecting causal genes from genome-wide association studies via functionally coherent subnetworks. Nat. Methods 12, 154–159 (2015).
Greene, C. S. et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 47, 569–576 (2015).
Krishnan, A. et al. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 19, 1454–1462 (2016).
Lamparter, D., Marbach, D., Rueedi, R., Kutalik, Z. & Bergmann, S. Fast and rigorous computation of gene and pathway scores from SNP-based summary statistics. PLoS Comput. Biol. 12, e1004714 (2016).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2014).
Pers, T. H. et al. Comprehensive analysis of schizophrenia-associated loci highlights ion channel pathways and biologically plausible candidate causal genes. Hum. Mol. Genet 25, 1247–1254 (2016).
Brunner, H. G. & van Driel, M. A. From syndrome families to functional genomics. Nat. Rev. Genet. 5, 545–551 (2004).
Lim, J. et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125, 801–814 (2006).
Oti, M., Snel, B., Huynen, M. A. & Brunner, H. G. Predicting disease genes using protein-protein interactions. J. Med Genet. 43, 691–698 (2006).
Oti, M. & Brunner, H. G. The modular nature of genetic diseases. Clin. Genet. 71, 1–11 (2007).
Franke, L. et al. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet. 78, 1011–1025 (2006).
Rossin, E. J. et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 7, e1001273 (2011).
Zhang, W., Sun, F. & Jiang, R. Integrating multiple protein-protein interaction networks to prioritize disease genes: a Bayesian regression approach. BMC Bioinform. 12, S11 (2011).
Luo, J. & Liang, S. Prioritization of potential candidate disease genes by topological similarity of protein-protein interaction network and phenotype data. J. Biomed. Inform. 53, 229–236 (2015).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Kang, H. J. et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011).
Zhang, Y. et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016).
Wang, C. et al. Computational inference of mRNA stability from histone modification and transcriptome profiles. Nucleic Acids Res. 40, 6414–6423 (2012).
Lafaille, F. G. et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491, 769–773 (2012).
Dunnett, C. W. A multiple comparison procedure for comparing several treatments with a control. J. Am. Stat. Assoc. 50, 1096–1121 (1955).
Dunnett, C. W. New tables for multiple comparisons with a control. Biometrics 20, 482–491 (1964).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–220 (2010).
Irizarry, R. A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Narayan, S. et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res. 1239, 235–248 (2008).
Zheng, J. et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 48, 747–757 (2016).
Pavlides, J. M. et al. Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome Med. 8, 84 (2016).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
Lee, I., Blom, U. M., Wang, P. I., Shim, J. E. & Marcotte, E. M. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 21, 1109–1121 (2011).
Oliver, S. Guilt-by-association goes global. Nature 403, 601–603 (2000).
Meddens, C. A. et al. Systematic analysis of chromatin interactions at disease associated loci links novel candidate genes to inflammatory bowel disease. Genome Biol. 17, 247 (2016).
Hauberg, M. E. et al. Large-scale identification of common trait and disease variants affecting gene expression. Am. J. Hum. Genet. 100, 885–894 (2017).
Osterhout, J. A., Stafford, B. K., Nguyen, P. L., Yoshihara, Y. & Huberman, A. D. Contactin-4 mediates axon-target specificity and functional development of the accessory optic system. Neuron 86, 985–999 (2015).
Oguro-Ando, A., Zuko, A., Kleijer, K. T. & Burbach, J. P. A current view on contactin-4, -5, and -6: implications in neurodevelopmental disorders. Mol. Cell Neurosci. 81, 72–83 (2016).
Goes, F. S. et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am. J. Med Genet. B Neuropsychiatr. Genet. 168, 649–659 (2015).
Fernandez, T. et al. Disruption of Contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 82, 1385 (2008).
Roohi, J. et al. Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med Genet. 46, 176–182 (2009).
Cottrell, C. E. et al. Contactin 4 as an autism susceptibility locus. Autism Res. 4, 189–199 (2011).
Saxena, R. et al. Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am. J. Hum. Genet 90, 410–425 (2012).
Kar, S. P. et al. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 6, 1052–1067 (2016).
Wang, Z. et al. Knockdown of GATAD2A suppresses cell proliferation in thyroid cancer in vitro. Oncol. Rep. 37, 2147–2152 (2016).
Yan, Y., Lagenaur, C. & Narayanan, V. Molecular cloning of M6: identification of a PLP/DM20 gene family. Neuron 11, 423–431 (1993).
Michibata, H. et al. Inhibition of mouse GPM6A expression leads to decreased differentiation of neurons derived from mouse embryonic stem cells. Stem Cells Dev. 17, 641–651 (2008).
Alfonso, J., Fernandez, M. E., Cooper, B., Flugge, G. & Frasch, A. C. The stress-regulated protein M6a is a key modulator for neurite outgrowth and filopodium/spine formation. Proc. Natl Acad. Sci. USA 102, 17196–17201 (2005).
Michibata, H. et al. Human GPM6A is associated with differentiation and neuronal migration of neurons derived from human embryonic stem cells. Stem Cells Dev. 18, 629–639 (2009).
Mita, S. et al. Transcallosal projections require glycoprotein M6-dependent neurite growth and guidance. Cereb. Cortex 25, 4111–4125 (2015).
Gregor, A. et al. Altered GPM6A/M6 dosage impairs cognition and causes phenotypes responsive to cholesterol in human and Drosophila. Hum. Mutat. 35, 1495–1505 (2014).
Mattei, M. G., Roeckel, N., Olsen, B. R. & Apte, S. S. Genes of the membrane-type matrix metalloproteinase (MT-MMP) gene family, MMP14, MMP15, and MMP16, localize to human chromosomes 14, 16, and 8, respectively. Genomics 40, 168–169 (1997).
Iida, J. et al. Melanoma chondroitin sulfate proteoglycan regulates matrix metalloproteinase-dependent human melanoma invasion into type I collagen. J. Biol. Chem. 276, 18786–18794 (2001).
Xia, H. et al. microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res. 1269, 158–165 (2009).
Anttila, V. et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 45, 912–917 (2013).
Davoli, R. et al. The porcine proteasome subunit A4 (PSMA4) gene: isolation of a partial cDNA, linkage and physical mapping. Anim. Genet. 29, 385–388 (1998).
Amos, C. I. et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 40, 616–622 (2008).
David, S. P. et al. Genome-wide meta-analyses of smoking behaviors in African Americans. Transl. Psychiatry 2, e119–e119 (2012).
Han, M. H. et al. Dysbindin-associated proteome in the p2 synaptosome fraction of mouse brain. J. Proteome Res. 13, 4567–4580 (2014).
Steinberg, S. et al. Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Hum. Mol. Genet. 20, 4076–4081 (2011).
Page, S. C. et al. The schizophrenia- and autism-associated gene, transcription factor 4 regulates the columnar distribution of layer 2/3 prefrontal pyramidal neurons in an activity-dependent manner. Mol. Psychiatry 23, 304–315 (2018).
Kennedy, A. J. et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep. 16, 2666–2685 (2016).
Brzozka, M. M., Radyushkin, K., Wichert, S. P., Ehrenreich, H. & Rossner, M. J. Cognitive and sensorimotor gating impairments in transgenic mice overexpressing the schizophrenia susceptibility gene Tcf4 in the brain. Biol. Psychiatry 68, 33–40 (2010).
Zhu, X. et al. Associations between TCF4 gene polymorphism and cognitive functions in schizophrenia patients and healthy controls. Neuropsychopharmacology 38, 683–689 (2013).
Filiano, A. J., Gadani, S. P. & Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 1617, 18–27 (2015).
Acknowledgements
This study was supported by the National Key Research and Development Program of China (Stem Cell and Translational Research) (2016YFA0100900 to X.J.L.), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13000000 to X.J.L.), the National Nature Science Foundation of China (31722029 to X.J.L.), the Key Research Project of Yunnan Province (2017FA008 to X.J.L.). X.J.L. was supported by the 1000 Young Talents Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, C., Gu, C., Huo, Y. et al. The integrated landscape of causal genes and pathways in schizophrenia. Transl Psychiatry 8, 67 (2018). https://doi.org/10.1038/s41398-018-0114-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-018-0114-x
This article is cited by
-
Exploring the mechanism of artificial selection signature in Chinese indigenous pigs by leveraging multiple bioinformatics database tools
BMC Genomics (2023)
-
Genome-wide Mendelian randomization identifies actionable novel drug targets for psychiatric disorders
Neuropsychopharmacology (2023)
-
Mendelian Randomization Study Using Dopaminergic Neuron-Specific eQTL Identifies Novel Risk Genes for Schizophrenia
Molecular Neurobiology (2023)
-
A polygenic score indexing a DRD2-related co-expression network is associated with striatal dopamine function
Scientific Reports (2022)
-
An analysis of genetically regulated gene expression and the role of co-expression networks across 16 psychiatric and substance use phenotypes
European Journal of Human Genetics (2022)
