
Abstract
In schizophrenia (SCZ) and autism spectrum disorder (ASD), the dysregulation of glutamate transmission through N-methyl-d-aspartate receptors (NMDARs) has been implicated as a potential etiological mechanism. Previous studies have accumulated evidence supporting NMDAR-encoding genes' role in etiology of SCZ and ASD. We performed a screening study for exonic regions of GRIN1, GRIN2A, GRIN2C, GRIN2D, GRIN3A, and GRIN3B, which encode NMDAR subunits, in 562 participates (370 SCZ and 192 ASD). Forty rare variants were identified including 38 missense, 1 frameshift mutation in GRIN2C and 1 splice site mutation in GRIN2D. We conducted in silico analysis for all variants and detected seven missense variants with deleterious prediction. De novo analysis was conducted if pedigree samples were available. The splice site mutation in GRIN2D is predicted to result in intron retention by minigene assay. Furthermore, the frameshift mutation in GRIN2C and splice site mutation in GRIN2D were genotyped in an independent sample set comprising 1877 SCZ cases, 382 ASD cases, and 2040 controls. Both of them were revealed to be singleton. Our study gives evidence in support of the view that ultra-rare variants with loss of function (frameshift, nonsense or splice site) in NMDARs genes may contribute to possible risk of SCZ.
Similar content being viewed by others


Introduction
Schizophrenia (SCZ)and autism spectrum disorder (ASD) both have been implied to a high heritability and a strong genetic basis1,2. SCZ is a common, serious mental disorder which affects nearly 1% people of the world3. Twin studies estimated its heritability to be up to 80%4. ASD is a range of heterogeneous neurodevelopmental conditions which has early-onset deficiency in social communication and interactions, and also behavioral functioning5. The etiology of ASD is strongly impacted by genetics, with heritability estimates of 56–95%6.
Glutamate is one of the most important excitatory neurotransmitter in synapse systems and 40% of all synapses7 are exploiting it. Glutamate is involved in many central nervous system processes and basic neuronal functions8. Thus, abnormal glutamatergic neurotransmission could be a point of convergence to describe the neurocognitive deficits and feature of symptoms presented in nervous system diseases8,9,10. N-methyl-d-aspartate receptors (NMDARs) indicated to be one of the most common glutamate receptors. The pathology of anti-NMDAR encephalitis implies that abnormalities in glutamatergic signaling can result in cognitive impairment, mood changes, and impairment of behavior, which are the symptoms often observed in SCZ/ASD patients11,12. To date, NMDAR subunits have three main family members that have been identified: NR1, NR2, and NR3. Those subunits are encoded by GRIN1, GRIN2A, GRIN2B, GRIN2C, GRIN2D, GRIN3A, and GRIN3B 13,14, respectively.
Resequencing studies suggested that GRIN2A might be a candidate gene for autism and SCZ15,16,17 A genomic data analysis study suggests GRIN2C as another SCZ candidate gene18. An exome sequencing study which sequenced all GRIN genes in SCZ and ASD cases, detected de novo variants in GRIN2B and GRIN2A and loss-of-function (LoF) variants in GRIN2C, GRIN3A, and GRIN3B 19. Novel de novo microduplications in 19q13, where GRIN2D resides in, were identified in ASD20. In addition, a study that identified significant association between GRIN3B and mismatch negativity (MMN) also deserves attention, as decreasing MMN was suggested to be correlated with the pathogenesis of SCZ21. Furthermore, in mouse studies, GRIN2A-null mice exhibit some SCZ-like symptoms22. Studies with GRIN2C knockout mice pointed out NR2C receptors might play a potential role in associative and executive learning23. Further mice studies showed that NR2D subunit incorporates into the NMDARs that mediate excitatory synaptic transmission onto interneurons and influence interneuron function and signal processing24. Altogether, these findings suggested the importance of GRIN genes in the pathogenesis of SCZ and ASD.
Recently, there have been accumulating evidence supporting a role of rare variants in mental disorders causation25,26,27, especially rare LoF (nonsense, splice site or frameshift) variants28,29,30. While frameshift and nonsense mutations are clearly to be LoF, the effect of splice site mutation remains to be defined. To our knowledge, till now, previous papers have not put their attention on GRIN genes splice site mutation in cases of neuropsychiatric disorders. In previous work31, we sequenced GRIN2B in SCZ and ASD, identifying five rare missense mutations. In present study, we performed a mutation screening study for the exonic regions of GRIN1, GRIN2A, GRIN2C, GRIN2D, GRIN3A, and GRIN3B. We assessed the functional impact of splice site mutation by minigene assay, and performed genotyping for LoF mutations in a large sample set.
Methods
Samples
In this study, two independent sample groups were designed (Table 1). The first one, which included 370 SCZ and 192 ASD patients, resequencing for mutation discovery. The second one, with 1877 SCZ cases, 382 ASD cases, and 2040 healthy controls, was used for genotyping of selected mutations identified in the first step. All participates in our study are ethnically Japanese, live on the mainland of Japan. The Hospital of Nagoya University and its co-institutes (Toyama University, Niigata University, Fujita Health University) and co-hospital (Kohnodai Hospital) recruited all the participates. Patients included in the study were diagnosed according to Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria for SCZ or ASD. Controls were recruited from ordinary people and were evaluated with an unstructured interview to ensure that they never suffer from psychiatric disorders, both personal and family history. We explained our study to all participants both verbally and in writing. In addition, if individuals had no capacity to do this alone, their parents or other family members were required to complete written informed consent. The study was supported by Ethics Committees of the Nagoya University Graduate School of Medicine and co-institutes and co-hospital.
Sequencing and data collection
We extracted genomic DNA from whole peripheral blood or saliva by a standard protocol. For covering coding regions of GRIN1, GRIN2A, GRIN2C, GRIN2D, GRIN3A, and GRIN3B (human reference sequence NCBI (build 37)), we designed custom amplification primers by FastPCR (PrimerDigital Ltd, Helsinki, Finland) and NCBI Primer-BLAST. The Ion Library Equalizer Kit Adapters and Ion AmpliSeq Library Kits 2.0 (Thermo Fisher Scientific, Foster City, CA, USA) were used for amplification and equalization. Then Ion Xpress Barcode was used to collect the amplified sequence. We used Ion Torrent PGM™ (Thermo Fisher Scientific) to sequence the products by next-generation sequencing technology. Then we performed an analysis for the resulting data using Ingenuity Variant Analysis (Qiagen Ltd, Hilden, Germany).
Filter conditions and in silico analysis
Rare (minor allele frequency < 1%), nonsynonymous variants, which was located on the functional domain, under the Human Protein Reference Database (http://www.hprd.org), EMBL-EBI (https://www.ebi.ac.uk/), and the existing literatures (Table 2), were selected from the original data for further analysis. These filtered variants were then sequenced for confirming their reliability by Sanger method in a 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
We further analyzed those variants with the following methods: (1) we explored whether they were registered in the NCBI dbSNP database (build 137) (http://www.ncbi.nlm.nih.gov/SNP/), the Exome Aggregation Consortium (http://evs.gs.washington.edu/EVS/), the 1000 Genomes Project (http://www.1000genomes.org/), the Integrative Japanese Genome Variation (https://ijgvd.megabank.tohoku.ac.jp/), or the Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp/index.html); (2) we looked for a possible impact of amino acid substitutions as predicted by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)32, SIFT (http://sift.jcvi.org/)33, and MUTATIONTASTER (http://www.mutationtaster.org/)34; (3) we investigated the conservation status using the PhastCons conservation score and PhyloP scores using the single-nucleotide variants scoring tool Combined Annotation Dependent Depletion35 (Table 2).
Splicing in silico analysis
For the splice site mutation, for predicting splicing outcomes of mutations leading to 5′-splice site splicing defects, we performed the following in silico analysis: SD-Score36, human splice finder37, and MaxEntScan38 (Table 3).
Construction of the plasmid containing the GRIN2D minigene
We constructed the c.1412G>A-GRIN2D minigene in the pcDNA3.1(+) vector by amplifying the 5′-end of exon 4 (starting from the second nucleotide of exon 4 to retain the normal open reading frame) to the 3′-end of exon 6 of GRIN2D from genomic DNA, which was extracted from the c.1412G>A-GRIN2D mutant sample and one healthy sample using the proofreading DNA polymerase KOD-Plus-Neo (Toyobo) (Figure S1). The forward primer 5′-AATCCCAAGCTTCACCATGTACTTCATGAACATCACGTGGGAT-3′ carried a HindIII restriction site at the 3′-end, whereas the reverse primer 5′-GCCTAGTCTAGATCACTCCCCGATCATGCCGTT-3′ had a stop codon and an XbaI restriction site at the 5′-end.
Cell culture and transfection
Dulbecco’s minimum essential medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (Sigma-Aldrich) were used for culturing HEK 293 cells. Cells were plated in 3.5-mm dishes 1 day before the transfection and were transfected with 1 μg minigene construct using FuGENE 6 (Roche) by following the standard method. Forty-eight hours after the transfection, we performed reverse transcription polymerase chain reaction (RT-PCR) for the cells.
RNA extraction and RT-PCR
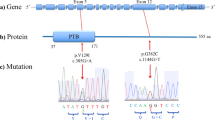
RNeasy Mini Kit (Qiagen) was used to extract total RNA according to the custom methods. Then we synthesized cDNA by using ReverTra Ace reverse transcriptase (Toyobo). GoTaq polymerase (Promega) were used to perform PCR with forward primer designed from exon 4 of GRIN2D (5′-CTTCATGAACATCACGTGGG-3′) and reverse primer from exon 6 (5′-GAGTGGCACCTTCCAGGGTC-3′) (Figure S1). Then, we performed agarose gel electrophoresis of PCR products. The correct size bands were excised from the gel by using PCR Cleanup System (Promega) and Wizard® SV Gel and sequenced using Sanger method to confirm the sequence of every band (Fig. 1).

Mutant GRIN2D caused intron 5 inclusion
Follow-up analysis
The statistical power of effective sample size was processed with website program, Genetic Power Calculator (http://zzz.bwh.harvard.edu/gpc/)39. With the following parameters: risk rare-allele frequency (A) of 0.01, disease prevalence of 0.01, genotype relative risk (Aa) ≥ 2, genotype relative risk (AA) ≥ 4, marker allele frequency (B) of 0.01, number of cases (n = 2259) and controls (n = 2040), and type I error rate of 0.05, we computed the result that our sample size have a statistical power of >80%. Only sequences resulting in possible LoF mutations were selected for genotyping. These included one novel frameshift and one novel splice site mutation. Genotyping of the frameshift mutation was performed using a probe oligo that was designed by and ordered from SIGMA-DLP. Genotyping of the splice site mutation was performed with a probe primer (Thermo Fisher Scientific) and Taqman (Applied Biosystems) standard probes. The 384-well microtiter plates were used for analysis, and every plate contained at least one sample carrying confirmed variant and one non-template control. ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) was used for the following analysis with standard method. Then, we compared the mutations with differences allele and genotype frequencies between patients and controls.
Results
Results of mutation screening
Our sequence data is available with the accession number DRA004490DNA in the Data Bank of Japan databases (http://www.ddbj.nig.ac.jp). Resequencing of the GRIN1, GRIN2A, GRIN2C, GRIN2D, GRIN3A, and GRIN3B coding regions using Ion PGM platform identified 40 rare (minor allele frequency < 1%), nonsynonymous mutations, including 38 missense mutations, one frameshift mutation with 4 bp deletion (P132Fs in GRIN2C), and one splice site mutation (c.1412G > A in GRIN2D). All mutations were confirmed by using the Sanger method, and all of them were heterozygous. Among them, 10 variants had available DNA for both of their parents. All the 10 variants were then identified to be inherited by Sanger sequencing, and no de novo variants were found. We searched five genetic databases (dbSNP build 143, 1000 Genomes Project, ExAC, HGVD, and iJGVD) and identified eight variants to be novel variants including the two LoF mutations (P132Fs in GRIN2C and c.1412G > A in GRIN2D). Then, we conducted in silico analysis for the 40 variants and detected 7 missense variants with deleterious prediction in all of the in silico tools we used (SIFT, PolyPhen-2, MUTATIONTASTER). The details and bioinformatics analysis of all 40 mutations are shown in Table 2.
Results of splice site mutation
The splice site mutation c.1412G > A-GRIN2D revealed a novel G-to-A transition (NM_000836.2: c.1412G > A) at the last nucleotide of exon 5. The predicted results due to splicing according to in silico tools were as follows: SD-Score36 predicted c.1412G > A-GRIN2D to be aberrant, the human splicing finder40 predicted the variant to most probably affect the splicing, and maximum entropy modeling (MaxEntScan)38 assigned the mutation a score of −1.87 points, whereas the normal sequence had a score of 5.49 points (Table 3). Then, we performed a functional splicing reporter minigene assay. In Fig. 1, the image shows the resequencing results of the RT-PCR products. Highest bind from the mutant confirmed to containing exon 4 (114 bp), part of intron 4 (99 bp), exon 5 (212 bp), intron 5 (267 bp), and exon 6 (169 bp); second bind from mutant and wild type (WT) containing exon 4 (114 bp), exon 5 (212 bp), intron 5 (267 bp), and exon 6 (169 bp); lowest bind from WT containing exon 4 (114 bp), exon 5 (212 bp), and exon 6 (169 bp). Thus, we confirmed an intron 5 retention due to the mutation, which result in meeting premature stop codon (Figure S2). As normal GRIN2D were confirmed to have only one isoform (NM_000836.2), we surmise that c.1412G>A-GRIN2D will lead to a truncated, incomplete protein product.
Results of further genotyping
Frameshift mutation P132Fs in GRIN2C and splice site mutation c.1412G>A in GRIN2D were selected for genotyping in an larger sample set which included 1877 SCZ cases, 382 ASD cases, and 2040 controls for association analysis. The result showed that no mutations were found in the sample set used for genotyping (Table 4). Importantly, both mutations, P132Fs in GRIN2C and c.1412G>A in GRIN2D, were only present in a single case, not only among 2821 cases and 2040 controls in this study but also never seen in the following databases: dbSNP build 143, 1000 Genomes Project, ExAC, HGVD, and iJGVD. Thus, we considered them as protein-damaging ultra-rare variants.
Discussion
We performed a systematic work of sequencing the coding regions of NMDARs genes in SCZ and ASD, and detected 40 rare, nonsynonymous mutations in this study. Among them, two LoF mutations in two patients suffering from SCZ were identified: one frameshift mutation (P132Fs in GRIN2C) and one splice site mutation (with intron retaining) (c.1412G>A in GRIN2D). P132Fs in GRIN2C was located in the beginning of the sequence (Figure S3), the 4 bp deletion creating a premature stop codon (p.P132FsX192). Another mutation was c.1412G>A in GRIN2D, with a G-to-A transition in the last nucleotide of exon 5. Minigene assay confirmed that this mutation resulted in intron 5 retention carrying two stop codons (Figure S2), which may lead to the introduction of premature termination codons, and possibly causing nonfunctional NR2D receptor to be created. Notably, frameshift mutations in some genes, such as DISC1 41, NLGN4 42, and UPF3B 43, were identified in SCZ and/or ASD patients, suggesting that frameshift mutations may increase susceptibility to SCZ and ASD. Other studies associate intron retention with the pathogenesis of SCZ44 and other genetic disorders, such as familial partial lipodystrophy type 2 (ref. 45) and limb girdle muscular dystrophy type 1B46, which suggested a role for intron retention in the development of genetic disorders. LoF mutations were often assumed to confer greater disease susceptibility than other missense mutations due to disruption of gene or protein function28. They were identified as having an increased contribution to SCZ and ASD, especially in functional sets that are closely involved in neurodevelopment26. Genome-wide significant association has also been identified between rare LoF mutations and risk for SCZ and other developmental disorders26,29. However, it cannot be ignored that LoF variants were detected also in healthy adults28,47,48 with surprisingly no deleterious consequences.
Furthermore, we conducted association analysis for P132Fs in GRIN2C and c.1412G>A in GRIN2D. Both of the two variants are singleton among 2259 cases and 2040 controls, and also have never been noted in the following databases (dbSNP build 143, 1000 Genomes Project, ExAC, HGVD, and iJGVD), which indicated them to be ultra-rare variants. As LoF ultra-rare variants are suggested to be more abundant among cases with psychiatry disorders than controls49,50,51, the two mutations may confer a strong genetic influence on SCZ risk.
In addition to LoF mutations, we also identified 38 missense mutations in SCZ/ASD patients in GRIN1, GRIN2A, GRIN2C, GRIN2D, GRIN3A, and GRIN3B. Among them, six missense mutations were ultra-rare variants. Seven mutations are predicted to be disruptive using all the three in silico tools we used (SIFT, PolyPhen-2, MUTATIONTASTER). Although in silico predictions are questionable, they are still irreplaceable and used widely to predict the impact of missense variants52,53. Moreover, two missense mutations in GRIN2A (K441N, K590N) and one missense mutation in GRIN2C (E514K) are observed to be positioned on glutamate-binding domain, which is highly conserved in primate. Some studies gave the possibility that the ligand-binding regions were more likely to be disruptive than in other domains54, which also indicate the importance of ligand-binding domain.
There are several limitations of our study that should not be ignored. First, we only sequenced the coding region of GRIN1, GRIN2A, GRIN2C, GRIN2D, GRIN3A, and GRIN3B. We excluded promoter, intronic, and 5′- and 3′-untranslated regions. Second, in the genotyping analysis, no statistical significance were in our study, it may be because the size of our samples had no sufficient power. Future studies should include a larger sample size to identify a wider range of rare mutations. Third, due to the difficulty of collecting samples of family members, we were only able to do pedigree analysis for a few subjects. Fourth, to avoid ambiguous pathogenicity interpretations, we excluded missense variants from the association analysis. This strict exclusion criterion limits the number of potential confounding factors, which may cause potentially important targets to be missed.
In conclusion, we revealed 40 rare variants including 38 missense mutations, one frameshift mutation, and one splice site mutation by screening the exonic regions of GRIN1, GRIN2A, GRIN2C, GRIN2D, GRIN3A, and GRIN3B. Our result might imply that these mutations increase susceptibility to SCZ and ASD. Furthermore, the observation of the two LoF mutations in GRIN2C and GRIN2D supports the hypothesis that an increased burden of ultra-rare deleterious mutations could be observed in SCZ, although statistical significance was not obtained in association analysis. In addition, our data also gave more evidences to support the likely role of NMDARs in SCZ and ASD with a neurodevelopmental origin.
References
Sandin, S. et al. The familial risk of autism. JAMA 311, 1770–1777 (2014).
Giusti-Rodriguez, P. & Sullivan, P. F. The genomics of schizophrenia: update and implications. J. Clin. Invest. 123, 4557–4563 (2013).
Loohuis, L. M. et al. Genome-wide burden of deleterious coding variants increased in schizophrenia. Nat. Commun. 6, 7501 (2015).
Sullivan, P. F., Kendler, K. S. & Neale, M. C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192 (2003).
Lai, M. C., Lombardo, M. V. & Baron-Cohen, S. Autism. Lancet 383, 896–910 (2014).
Colvert, E. et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 72, 415–423 (2015).
Paoletti, P., Bellone, C. & Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400 (2013).
Papenberg, G. et al. Dopamine and glutamate receptor genes interactively influence episodic memory in old age. Neurobiol. Aging 35, e1213–e1218 (2014).
Hansen, K. B. et al. Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-d-aspartate receptors. Mol. Pharmacol. 84, 114–127 (2013).
Meador-Woodruff, J. H., Clinton, S. M., Beneyto, M. & McCullumsmith, R. E. Molecular abnormalities of the glutamate synapse in the thalamus in schizophrenia. Ann. NY Acad. Sci. 1003, 75–93 (2003).
Kayser, M. S. & Dalmau, J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr. Res. 176, 36–40 (2016).
Belforte, J. E. et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 13, 76–83 (2010).
Liu, L. et al. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304, 1021–1024 (2004).
Cull-Candy, S. G. & Leszkiewicz, D. N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, re16 (2004).
Itokawa, M. et al. A microsatellite repeat in the promoter of the N-methyl-d-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 13, 271–278 (2003).
Barnby, G. et al., International Molecular Genetics Study of Autism C. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 76, 950–966 (2005).
Gai, X. et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol. Psychiatry 17, 402–411 (2012).
Forero, D. A. et al. A network of synaptic genes associated with schizophrenia and bipolar disorder. Schizophr. Res. 172, 68–74 (2016).
Tarabeux, J. et al. Rare mutations in N-methyl-d-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 1, e55 (2011).
Perez-Palma, E. et al. Duplications at 19q13. 33 in patients with neurodevelopmental disorders. bioRxiv 130377 (2017).
Lin, Y. T. et al. A recently-discovered NMDA receptor gene, GRIN3B, is associated with duration mismatch negativity. Psychiatry Res. 218, 356–358 (2014).
Sprengel, R. et al. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell 92, 279–289 (1998).
Hillman, B. G., Gupta, S. C., Stairs, D. J., Buonanno, A. & Dravid, S. M. Behavioral analysis of NR2C knockout mouse reveals deficit in acquisition of conditioned fear and working memory. Neurobiol. Learn. Mem. 95, 404–414 (2011).
Perszyk, R. E. et al. GluN2D-containing N-methyl-d-aspartate receptors mediate synaptic transmission in hippocampal interneurons and regulate interneuron activity. Mol. Pharmacol. 90, 689–702 (2016).
Chuang, Y. A. et al. Rare mutations and hypermethylation of the ARC gene associated with schizophrenia. Schizophr. Res. 176, 106–113 (2016).
Singh, T. et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016).
Flint, J. Rare genetic variants and schizophrenia. Nat. Neurosci. 19, 525–527 (2016).
MacArthur, D. G. et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012).
Kataoka, M. et al. Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations. Mol. Psychiatry 21, 885–893 (2016).
Genovese, G. et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 19, 1433–1441 (2016).
Takasaki, Y. et al. Mutation screening of GRIN2B in schizophrenia and autism spectrum disorder in a Japanese population. Sci. Rep. 6, 33311 (2016).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Sahashi, K. et al. In vitro and in silico analysis reveals an efficient algorithm to predict the splicing consequences of mutations at the 5’ splice sites. Nucleic Acids Res. 35, 5995–6003 (2007).
Wang, G. S. & Cooper, T. A. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 8, 749–761 (2007).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Purcell, S., Cherny, S. S. & Sham, P. C. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150 (2003).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Sachs, N. A. et al. A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol. Psychiatry 10, 758–764 (2005).
Yan, J. et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 10, 329–332 (2005).
Addington, A. et al. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol. Psychiatry 16, 238 (2011).
Faul, T. et al. ZDHHC8 as a candidate gene for schizophrenia: analysis of a putative functional intronic marker in case–control and family-based association studies. BMC Psychiatry 5, 35 (2005).
Morel, C. F. et al. A LMNA splicing mutation in two sisters with severe Dunnigan-type familial partial lipodystrophy type 2. J. Clin. Endocrinol. Metab. 91, 2689–2695 (2006).
Muchir, A. et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 9, 1453–1459 (2000).
Burgess, D. J. Human genetics: loss-of-function variants—not always what they seem. Nat. Rev. Genet. 17, 251 (2016).
Narasimhan, V. M. et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science 352, 474–477 (2016).
Genovese, G. et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 2016, 1433–1441 (2016).
Song, W. et al. Identification of high risk DISC1 protein structural variants in patients with bipolar spectrum disorder. Neurosci. Lett. 486, 136–140 (2010).
Epi Kc and Epilepsy Phenome/Genome P. Ultra-rare genetic variation in common epilepsies: a case–control sequencing study. Lancet Neurol. 16, 135–143 (2017).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137 (2015).
Ionita-Laza, I., McCallum, K., Xu, B. & Buxbaum, J. D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 48, 214–220 (2016).
Burnashev, N. & Szepetowski, P. NMDA receptor subunit mutations in neurodevelopmental disorders. Curr. Opin. Pharmacol. 20, 73–82 (2015).
Acknowledgements
The present study was supported by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Ministry of Health, Labor, and Welfare of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, Y., Lin, Y., Takasaki, Y. et al. Rare loss of function mutations in N-methyl-d-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl Psychiatry 8, 12 (2018). https://doi.org/10.1038/s41398-017-0061-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-017-0061-y
This article is cited by
-
Schizophrenia-associated NRXN1 deletions induce developmental-timing- and cell-type-specific vulnerabilities in human brain organoids
Nature Communications (2023)
-
Targeting NMDA receptors in neuropsychiatric disorders by drug screening on human neurons derived from pluripotent stem cells
Translational Psychiatry (2022)
-
Influence of Gestational Chlorpyrifos Exposure on ASD-like Behaviors in an fmr1-KO Rat Model
Molecular Neurobiology (2022)
-
Consequences of NMDA receptor deficiency can be rescued in the adult brain
Molecular Psychiatry (2021)
-
Transcriptome-wide association study of post-trauma symptom trajectories identified GRIN3B as a potential biomarker for PTSD development
Neuropsychopharmacology (2021)
